Doxorubicin Inhibits Phosphatidylserine Decarboxylase and Modifies Mitochondrial Membrane Composition in HeLa Cells


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. DXR is Incorporated Inside Mitochondria of HeLa Cells
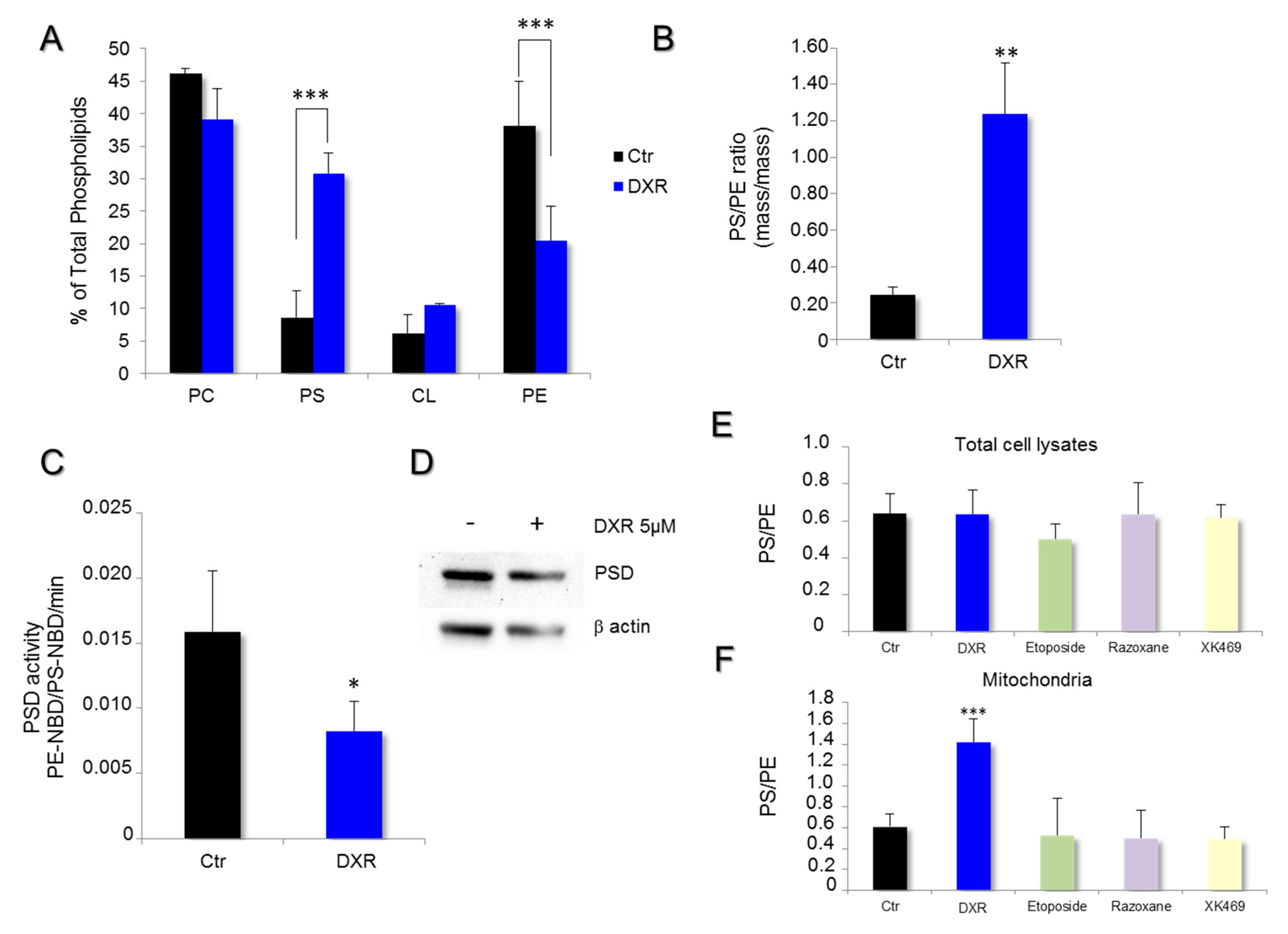
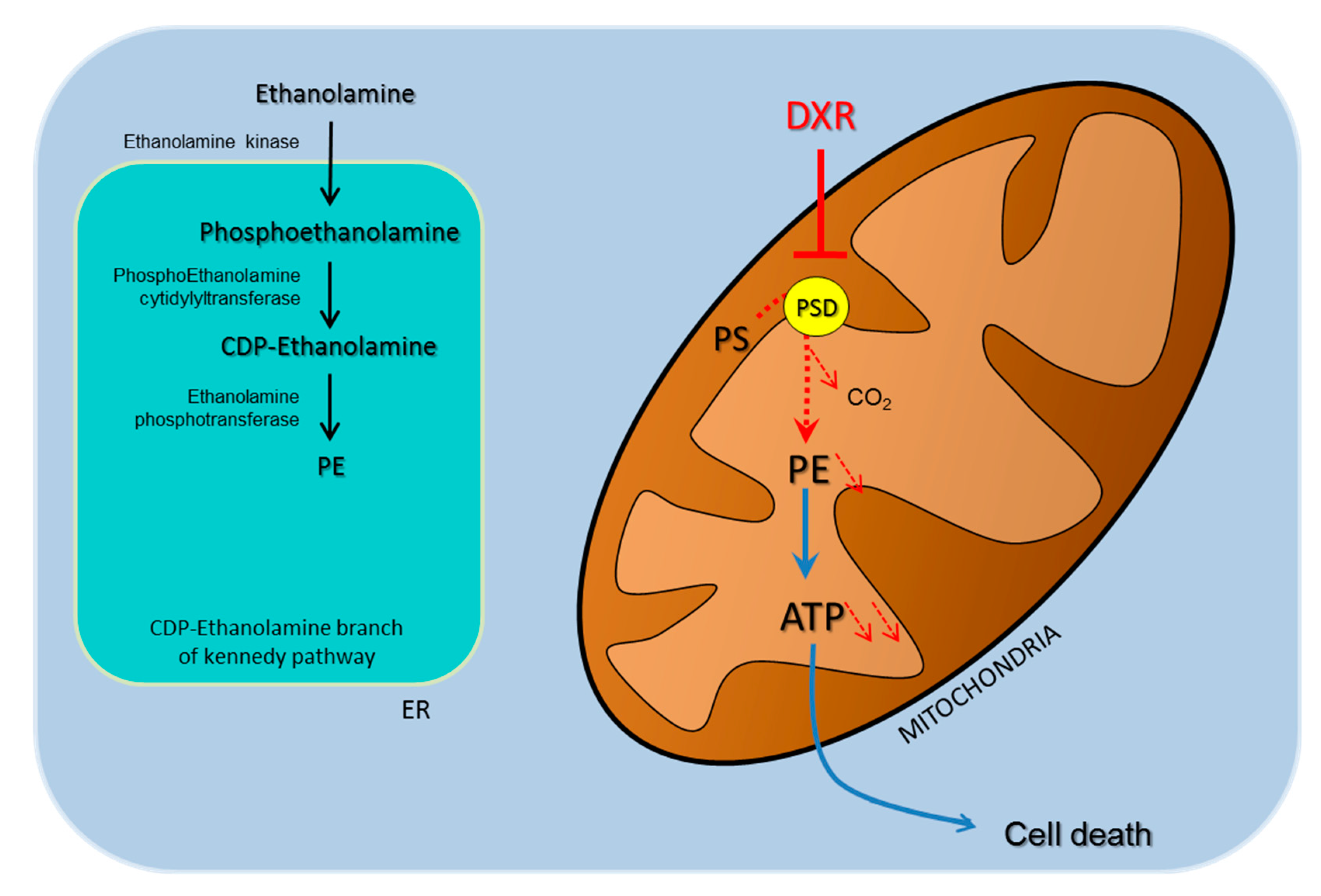
2.2. DXR Modifies the Mitochondrial Membrane Composition via PSD Pathway
2.3. Deleterious Impact of DXR Treatment on Energy Metabolism Depends on the Content of PE
2.4. DXR Sensitivity Depends on PSD Dependency for the Mitochondrial Pool of PE
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Types and Culture Conditions
4.3. Mitochondrial Isolation
4.4. Lipid Composition of Mitochondrial and Cellular Membranes
4.5. Cell Viability Assay
4.6. PSD Activity Fluorimetric Assay
4.7. ATP Measurements and Cell Respiration
4.8. Fluorescence Microscopy
4.9. RT-PCR on Cultured Cells
4.10. Western Blot
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gewirtz, D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Cheung, K.G.; Cole, L.K.; Xiang, B.; Chen, K.; Ma, X.; Myal, Y.; Hatch, G.M.; Tong, Q.; Dolinsky, V.W. Sirtuin-3 (SIRT3) Protein Attenuates Doxorubicin-induced Oxidative Stress and Improves Mitochondrial Respiration in H9c2 Cardiomyocytes. J. Biol. Chem. 2015, 290, 10981–10993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphree, S.A.; Tritton, T.R.; Smith, P.L.; Sartorelli, A.C. Adriamycin-induced changes in the surface membrane of sarcoma 180 ascites cells. Biochim. Biophys. Acta 1981, 649, 317–324. [Google Scholar] [CrossRef]
- Karczmar, G.S.; Tritton, T.R. The interaction of adriamycin with small unilamellar vesicle liposomes. A fluorescence study. Biochim. Biophys. Acta 1979, 557, 306–319. [Google Scholar] [CrossRef]
- Siegfried, J.A.; Kennedy, K.A.; Sartorelli, A.C.; Tritton, T.R. The role of membranes in the mechanism of action of the antineoplastic agent adriamycin. Spin-labeling studies with chronically hypoxic and drug-resistant tumor cells. J. Biol. Chem. 1983, 258, 339–343. [Google Scholar]
- Murphree, S.A.; Cunningham, L.S.; Hwang, K.M.; Sartorelli, A.C. Effects of adriamycin on surface properties of sarcoma 180 ascites cells. Biochem. Pharmacol. 1976, 25, 1227–1231. [Google Scholar] [CrossRef]
- Anderson, A.B.; Xiong, G.; Arriaga, E.A. Doxorubicin accumulation in individually electrophoresed organelles. J. Am. Chem. Soc. 2004, 126, 9168–9169. [Google Scholar] [CrossRef]
- Nicolay, K.; Fok, J.J.; Voorhout, W.; Post, J.A.; de Kruijff, B. Cytofluorescence detection of adriamycin-mitochondria interactions in isolated, perfused rat heart. Biochim. Biophys. Acta 1986, 887, 35–41. [Google Scholar] [CrossRef]
- Buondonno, I.; Gazzano, E.; Jean, S.R.; Audrito, V.; Kopecka, J.; Fanelli, M.; Salaroglio, I.C.; Costamagna, C.; Roato, I.; Mungo, E.; et al. Mitochondria-Targeted Doxorubicin: A New Therapeutic Strategy against Doxorubicin-Resistant Osteosarcoma. Mol. Cancer Ther. 2016, 15, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Ji, X.; Yu, B.; Ji, K.; Gallo, D.; Csizmadia, E.; Zhu, M.; Choudhury, M.R.; De La Cruz, L.K.C.; Chittavong, V.; et al. Enrichment-triggered prodrug activation demonstrated through mitochondria-targeted delivery of doxorubicin and carbon monoxide. Nat. Chem. 2018, 10, 787–794. [Google Scholar] [CrossRef]
- Cheneval, D.; Muller, M.; Toni, R.; Ruetz, S.; Carafoli, E. Adriamycin as a probe for the transversal distribution of cardiolipin in the inner mitochondrial membrane. J. Biol. Chem. 1985, 260, 13003–13007. [Google Scholar] [PubMed]
- Tasseva, G.; Bai, H.D.; Davidescu, M.; Haromy, A.; Michelakis, E.; Vance, J.E. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 2013, 288, 4158–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryal, B.; Rao, V.A. Deficiency in Cardiolipin Reduces Doxorubicin-Induced Oxidative Stress and Mitochondrial Damage in Human B-Lymphocytes. PLoS ONE 2016, 11, e0158376. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. Phosphatidylserine and phosphatidylethanolamine in mammalian cells: Two metabolically related aminophospholipids. J. Lipid Res. 2008, 49, 1377–1387. [Google Scholar] [CrossRef] [Green Version]
- Steenbergen, R.; Nanowski, T.S.; Beigneux, A.; Kulinski, A.; Young, S.G.; Vance, J.E. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 2005, 280, 40032–40040. [Google Scholar] [CrossRef] [Green Version]
- Selathurai, A.; Kowalski, G.M.; Mason, S.A.; Callahan, D.L.; Foletta, V.C.; Della Gatta, P.A.; Lindsay, A.; Hamley, S.; Kaur, G.; Curtis, A.R.; et al. Phosphatidylserine decarboxylase is critical for the maintenance of skeletal muscle mitochondrial integrity and muscle mass. Mol. Metab. 2019, 27, 33–46. [Google Scholar] [CrossRef]
- Bellance, N.; Benard, G.; Furt, F.; Begueret, H.; Smolkova, K.; Passerieux, E.; Delage, J.P.; Baste, J.M.; Moreau, P.; Rossignol, R. Bioenergetics of lung tumors: Alteration of mitochondrial biogenesis and respiratory capacity. Int. J. Biochem. Cell Biol. 2009, 41, 2566–2577. [Google Scholar] [CrossRef]
- Bellance, N.; Lestienne, P.; Rossignol, R. Mitochondria: From bioenergetics to the metabolic regulation of carcinogenesis. Front. Biosci. 2009, 14, 4015–4034. [Google Scholar]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Young, C.D.; Anderson, S.M. Sugar and fat—That’s where it’s at: Metabolic changes in tumors. Breast Cancer Res. 2008, 10, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, R.; Cunningham, C.; Jester, R.; Waite, M.; Miller, N.; Morris, H.P. Alteration of mitochondrial function and lipid composition in Morris 7777 hepatoma. Cancer Res. 1976, 36, 3246–3254. [Google Scholar] [PubMed]
- Williams, R.D.; Nixon, D.W.; Merrill, A.H., Jr. Comparison of serine palmitoyltransferase in Morris hepatoma 7777 and rat liver. Cancer Res. 1984, 44, 1918–1923. [Google Scholar] [PubMed]
- Pouliquen, D.; Olivier, C.; Debien, E.; Meflah, K.; Vallette, F.M.; Menanteau, J. Changes in liver mitochondrial plasticity induced by brain tumor. BMC Cancer 2006, 6, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiebish, M.A.; Han, X.; Cheng, H.; Chuang, J.H.; Seyfried, T.N. Brain mitochondrial lipid abnormalities in mice susceptible to spontaneous gliomas. Lipids 2008, 43, 951–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelker, D.R. Adriamycin disrupts phosphatidylserine import into the mitochondria of permeabilized CHO-K1 cells. J. Biol. Chem. 1991, 266, 12185–12188. [Google Scholar] [PubMed]
- Voelker, D.R. Phosphatidylserine translocation to the mitochondrion is an ATP-dependent process in permeabilized animal cells. Proc. Natl. Acad. Sci. USA 1989, 86, 9921–9925. [Google Scholar] [CrossRef] [Green Version]
- Henry, N.; Fantine, E.O.; Bolard, J.; Garnier-Suillerot, A. Interaction of adriamycin with negatively charged model membranes: Evidence of two types of binding sites. Biochemistry 1985, 24, 7085–7092. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Theophilidis, G.; Thomopoulos, G.N.; Tsiftsoglou, A.S. Structural and functional impairment of mitochondria in adriamycin-induced cardiomyopathy in mice: Suppression of cytochrome c oxidase II gene expression. Biochem. Pharmacol. 1999, 57, 481–489. [Google Scholar] [CrossRef]
- Tokarska-Schlattner, M.; Lucchinetti, E.; Zaugg, M.; Kay, L.; Gratia, S.; Guzun, R.; Saks, V.; Schlattner, U. Early effects of doxorubicin in perfused heart: Transcriptional profiling reveals inhibition of cellular stress response genes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1075–R1088. [Google Scholar] [CrossRef] [Green Version]
- Lombes, A.; Mendell, J.R.; Nakase, H.; Barohn, R.J.; Bonilla, E.; Zeviani, M.; Yates, A.J.; Omerza, J.; Gales, T.L.; Nakahara, K.; et al. Myoclonic epilepsy and ragged-red fibers with cytochrome oxidase deficiency: Neuropathology, biochemistry, and molecular genetics. Ann. Neurol. 1989, 26, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Hansson, A.; Hance, N.; Dufour, E.; Rantanen, A.; Hultenby, K.; Clayton, D.A.; Wibom, R.; Larsson, N.G. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc. Natl. Acad. Sci. USA 2004, 101, 3136–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wredenberg, A.; Wibom, R.; Wilhelmsson, H.; Graff, C.; Wiener, H.H.; Burden, S.J.; Oldfors, A.; Westerblad, H.; Larsson, N.G. Increased mitochondrial mass in mitochondrial myopathy mice. Proc. Natl. Acad. Sci. USA 2002, 99, 15066–15071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Forbes, C.D.; Toth, J.G.; Özbal, C.C.; Lamarr, W.A.; Pendleton, J.A.; Rocks, S.; Gedrich, R.W.; Osterman, D.G.; Landro, J.A.; Lumb, K.J. High-throughput mass spectrometry screening for inhibitors of phosphatidylserine decarboxylase. J. BioMol. Screen. 2007, 12, 628–634. [Google Scholar] [CrossRef] [Green Version]
- Trounce, I.A.; Kim, Y.L.; Jun, A.S.; Wallace, D.C. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996, 264, 484–509. [Google Scholar]
- Ardail, D.; Gasnier, F.; Lerme, F.; Simonot, C.; Louisot, P.; Gateau-Roesch, O. Involvement of mitochondrial contact sites in the subcellular compartmentalization of phospholipid biosynthetic enzymes. J. Biol. Chem. 1993, 268, 25985–25992. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, F.; Zanetta, J.P. Thin-layer chromatography of phospholipids. J. Chromatogr. 1978, 166, 637–640. [Google Scholar] [CrossRef]
- Macala, L.J.; Yu, R.K.; Ando, S. Analysis of brain lipids by high performance thin-layer chromatography and densitometry. J. Lipid Res. 1983, 24, 1243–1250. [Google Scholar] [PubMed]
- Borenfreund, E.; Puerner, J.A. Toxicity determined in vitro by morphological alterations and neutral red absorption. Toxicol. Lett. 1985, 24, 119–124. [Google Scholar] [CrossRef]
- Nouette-Gaulain, K.; Bellance, N.; Prévost, B.; Passerieux, E.; Pertuiset, C.; Galbes, O.; Smolkova, K.; Masson, F.; Miraux, S.; Delage, J.P.; et al. Erythropoietin protects against local anesthetic myotoxicity during continuous regional analgesia. Anesthesiology 2009, 110, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellance, N.; Furt, F.; Melser, S.; Lalou, C.; Thoraval, D.; Maneta-Peyret, L.; Lacombe, D.; Moreau, P.; Rossignol, R. Doxorubicin Inhibits Phosphatidylserine Decarboxylase and Modifies Mitochondrial Membrane Composition in HeLa Cells. Int. J. Mol. Sci. 2020, 21, 1317. https://doi.org/10.3390/ijms21041317
Bellance N, Furt F, Melser S, Lalou C, Thoraval D, Maneta-Peyret L, Lacombe D, Moreau P, Rossignol R. Doxorubicin Inhibits Phosphatidylserine Decarboxylase and Modifies Mitochondrial Membrane Composition in HeLa Cells. International Journal of Molecular Sciences. 2020; 21(4):1317. https://doi.org/10.3390/ijms21041317
Chicago/Turabian StyleBellance, Nadège, Fabienne Furt, Su Melser, Claude Lalou, Didier Thoraval, Lilly Maneta-Peyret, Didier Lacombe, Patrick Moreau, and Rodrigue Rossignol. 2020. "Doxorubicin Inhibits Phosphatidylserine Decarboxylase and Modifies Mitochondrial Membrane Composition in HeLa Cells" International Journal of Molecular Sciences 21, no. 4: 1317. https://doi.org/10.3390/ijms21041317