Differential Signaling Profiles of MC4R Mutations with Three Different Ligands

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
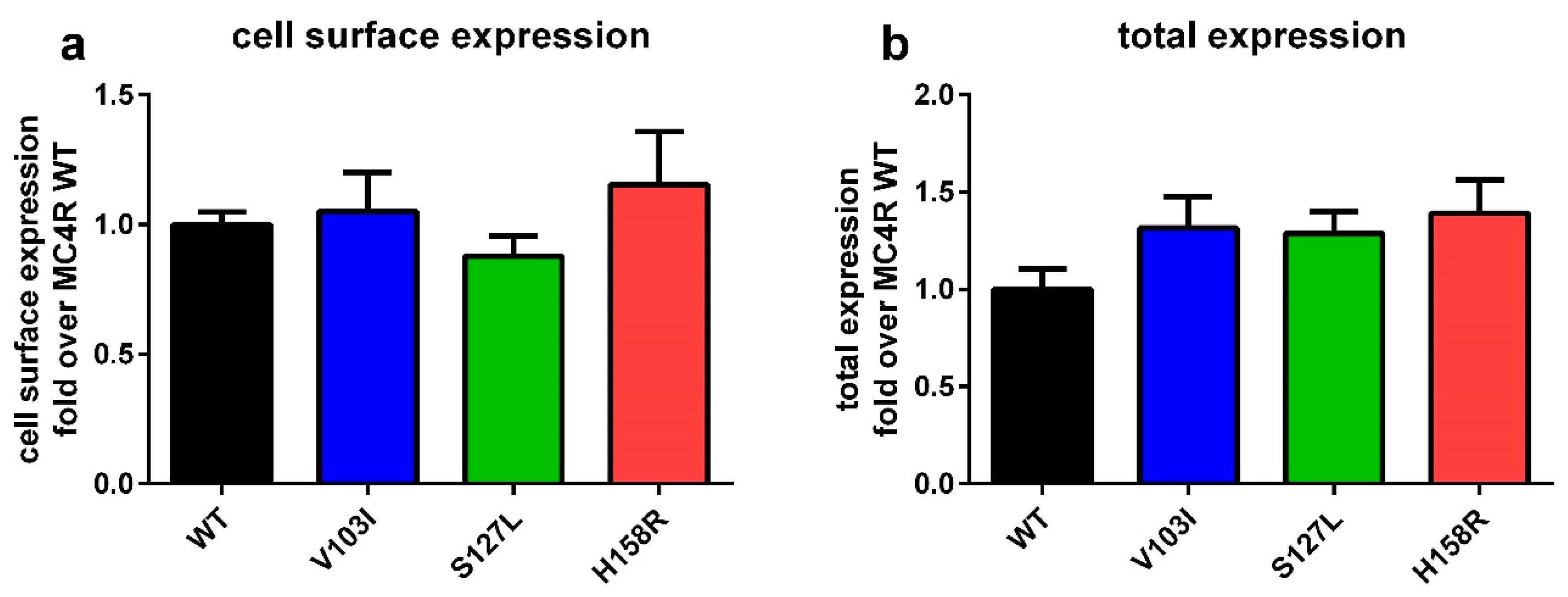
2.1. Total and Cell Surface Expression of the Three MC4R Mutations
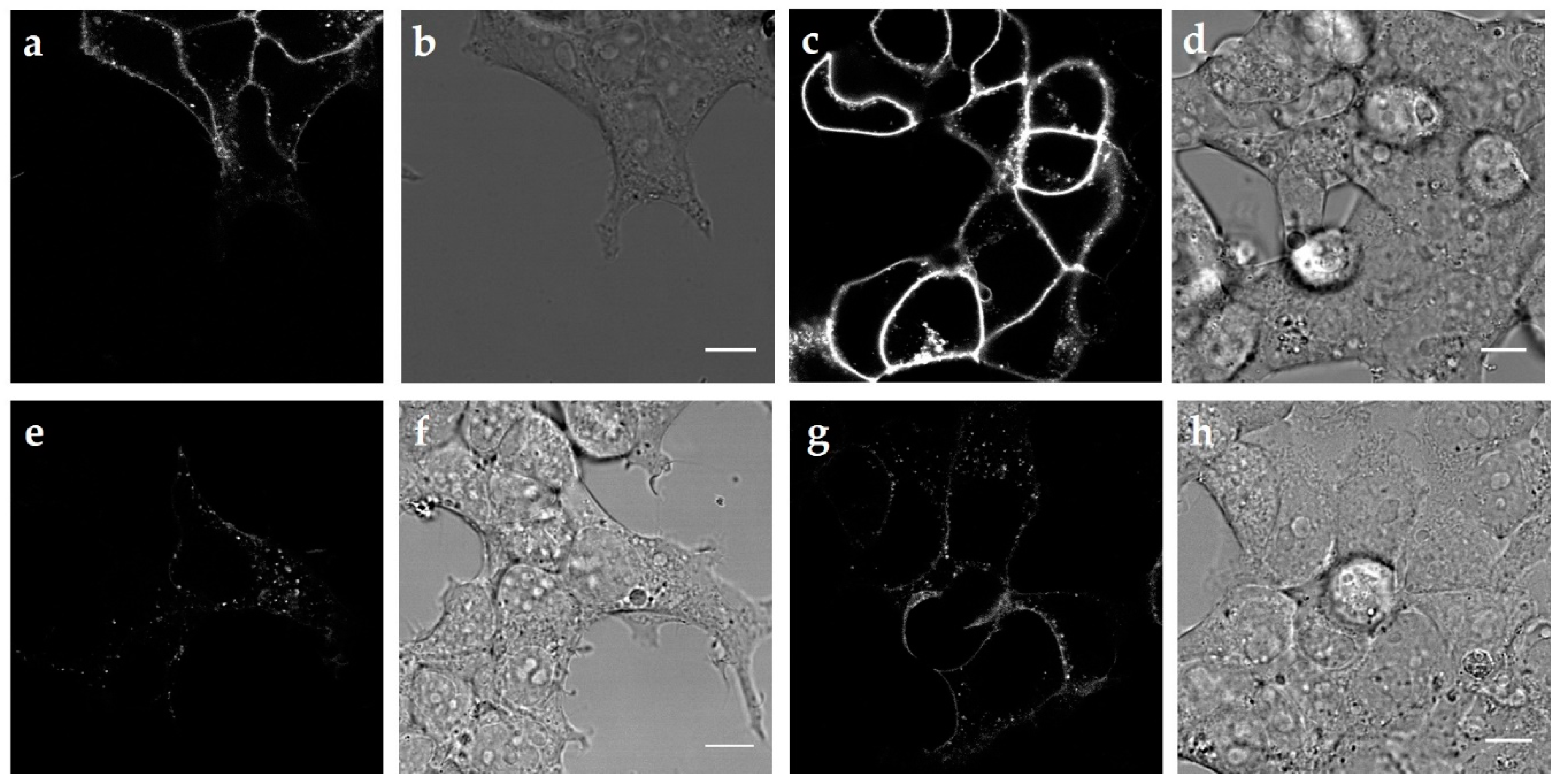
2.2. Internalization of the Three MC4R Mutations
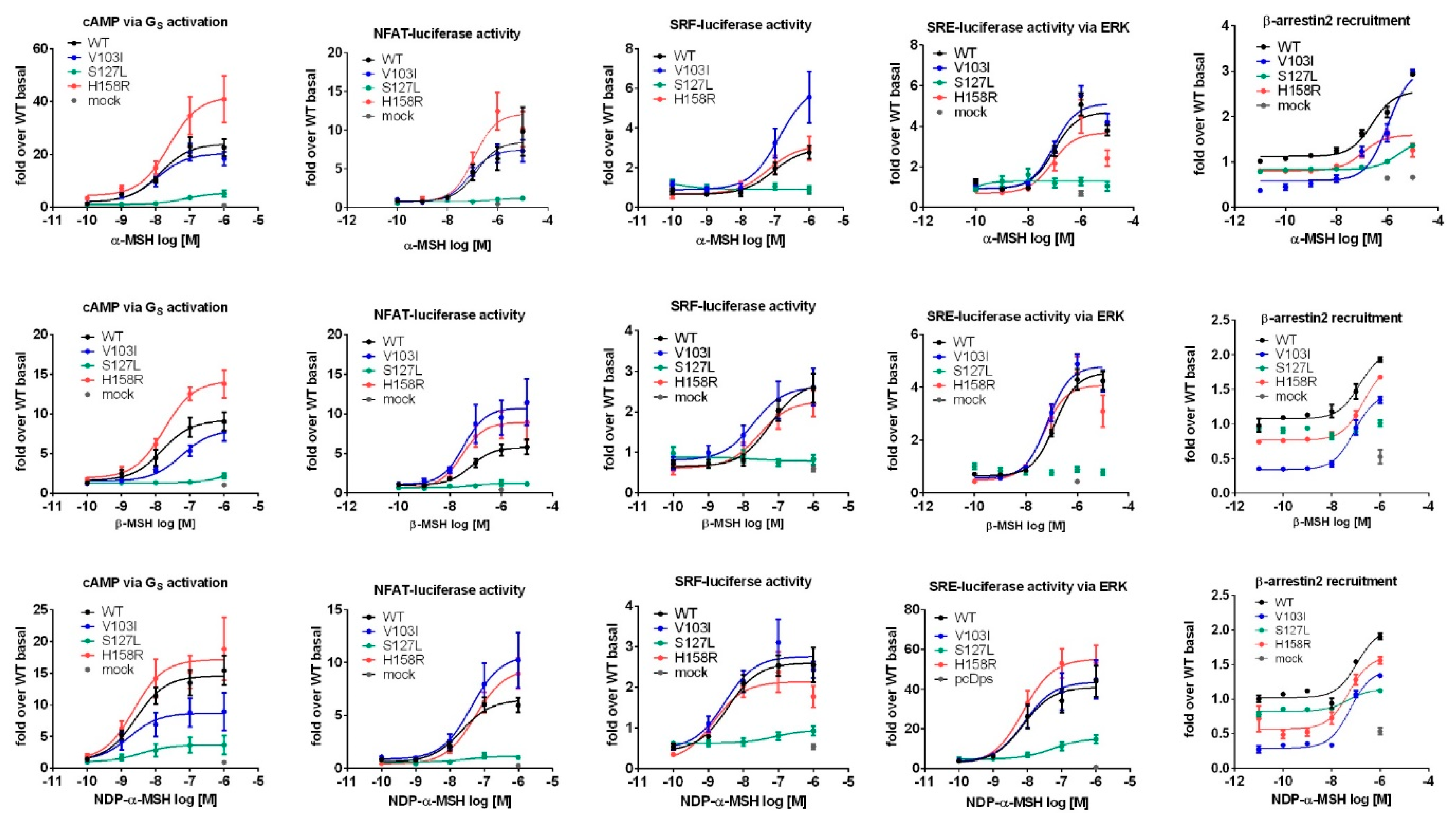
2.3. Comprehensive Functional Characterization of the Three MC4R Mutations
2.3.1. Stimulation with α-MSH Leads to a Strong Signaling Bias towards the Gq/11 Pathway for MC4R Mutations V103I and H158R
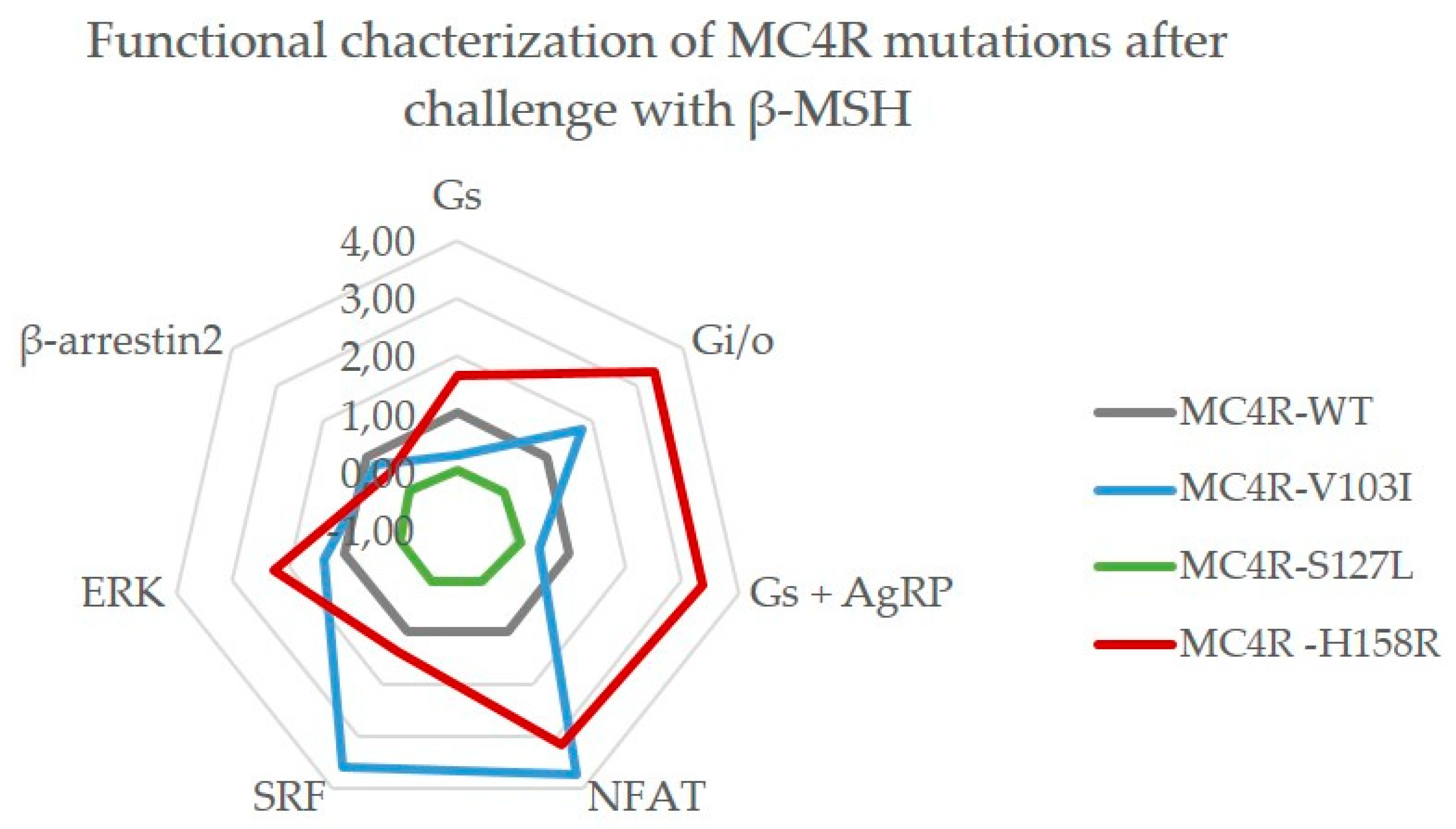
2.3.2. Challenge with β-MSH Results in a Different Activation Pattern Compared to α-MSH for MC4R V103I and H158R Mutations
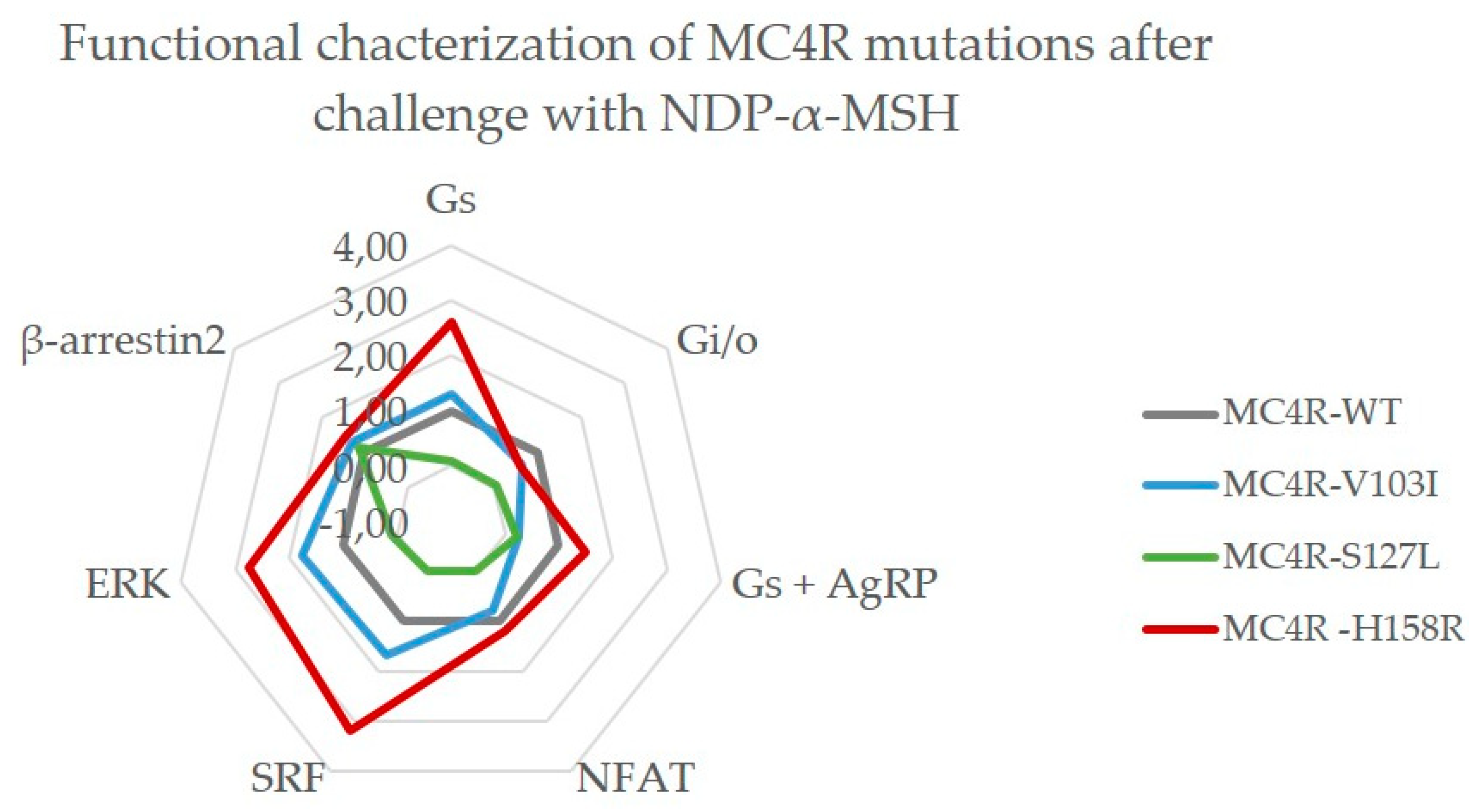
2.3.3. MC4R V103I and H158R Lost Their bias Towards Gq/11 After NDP-α-MSH Stimulation
3. Discussion
3.1. MC4R Mutations are Expressed Equally, but Internalize Differently
3.2. Constitutive Activity in all Tested Pathways Differed for Each Mutation Compared to WT-MC4R
3.3. Signaling Bias towards Gq/11 of Two Mutations When Stimulated with α-MSH
3.4. MC4R Mutations Respond Differently to the Endogenous Melanocortins
3.5. The Potent Synthetic Ligand NDP-α-MSH Changes the Signaling Bias of V103I and H158R
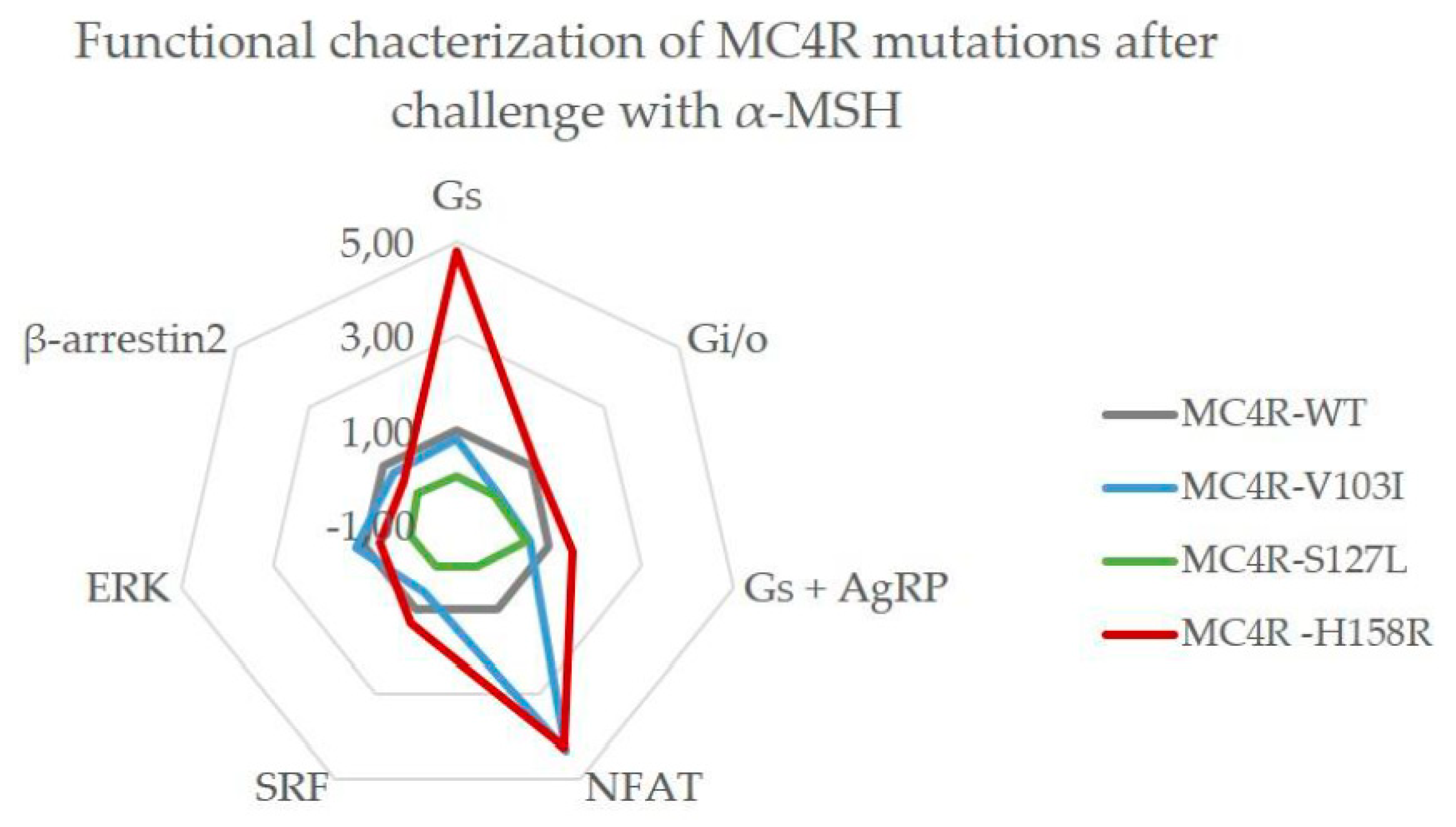
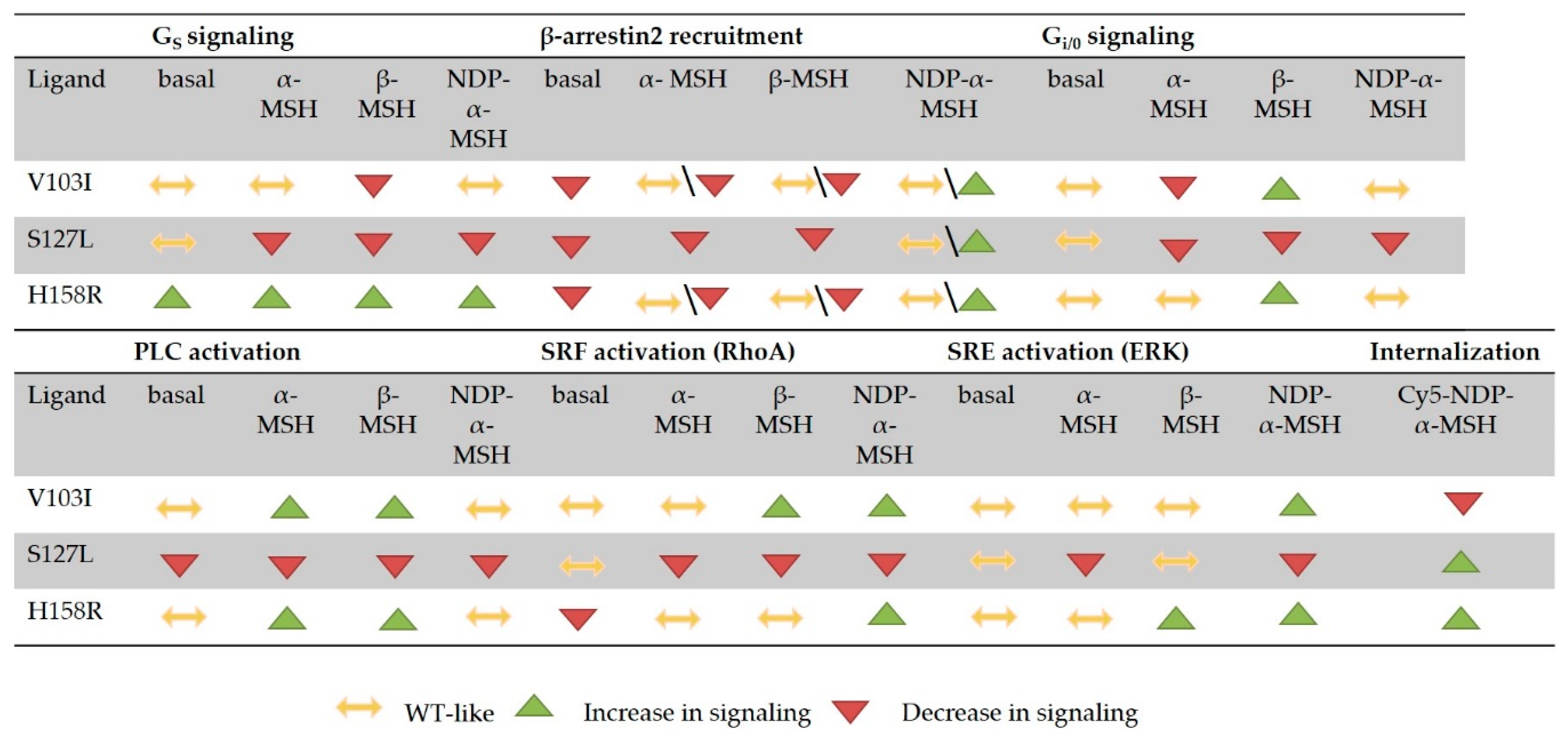
3.6. Summary of the Comprehensive Functional Characterization
4. Materials and Methods
4.1. Cell lines, Cloning and Reagents
4.2. Transfection
4.3. Determination of Total and Cell Surface Expression via HiBiT Assay
4.4. Receptor Staining Using Fluorophor-Tagged Ligand
4.5. Determination of β-Arrestin2 Recruitment via NanoBRET™
4.6. Determination of GS and Gi/o Protein Coupling via Intracellular cAMP Accumulation
4.7. Determination of NFAT, RhoA Activation and ERK Phosphorylation via Reporter Gene Assays
4.8. Mathematical Models and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MC4R | Melanocortin receptor 4 |
| GPCR | G protein coupled receptor |
| MSH | Melanocyte stimulating hormone |
| NDP-α-MSH | (Nle4, d-Phe7)-α-MSH |
| POMC | Proopiomelanocortin |
| AgRP | Agouti related peptide |
| PTX | Pertussis toxin |
| cAMP | Cyclic adenosine monophosphate |
| MAPK | Mitogen-activated kinase |
| ERK1/2 | Extracellular-regulated kinase 1/2 |
| DIC | differential interference contrast |
| NFAT | nuclear factor of activated T-cell |
| SRE | serum response element |
| SRF | serum-response factor |
| PLC | Phospholipase C |
| IBMX | 3-Isobutyl-1-methylxanthine |
| ARRB2 | β-arrestin2 |
| BRET | Bioluminescence resonance energy transfer |
| NL | NanoLuc |
| HT | HaloTag |
References
- Cone, R.D. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 2005, 8, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Biebermann, H.; Kühnen, P.; Kleinau, G.; Krude, H. The neuroendocrine circuitry controlled by POMC, MSH, and AGRP. In Appetite Control; Springer: Berlin/Heidelberg, Germany, 2012; pp. 47–75. [Google Scholar]
- Kühnen, P.; Krude, H.; Biebermann, H. Melanocortin-4 receptor signalling: Importance for weight regulation and obesity treatment. Trends Mol. Med. 2019, 25, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.S.H.; Farooqi, I.S.; Aminian, S.; Halsall, D.J.; Stanhope, R.G.; O’Rahilly, S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat. Genet. 1998, 20, 111–112. [Google Scholar] [CrossRef] [PubMed]
- Vaisse, C.; Clement, K.; Guy-Grand, B.; Froguel, P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet. 1998, 20, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Gantz, I.; Miwa, H.; Konda, Y.; Shimoto, Y.; Tashiro, T.; Watson, S.; DelValle, J.; Yamada, T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J. Biol. Chem. 1993, 268, 15174–15179. [Google Scholar] [PubMed]
- Daniels, D.; Patten, C.S.; Roth, J.D.; Yee, D.K.; Fluharty, S.J. Melanocortin receptor signaling through mitogen-activated protein kinase in vitro and in rat hypothalamus. Brain Res. 2003, 986, 1–11. [Google Scholar] [CrossRef]
- Vongs, A.; Lynn, N.M.; Rosenblum, C.I. Activation of MAP kinase by MC4-R through PI3 kinase. Regul. Pept. 2004, 120, 113–118. [Google Scholar] [CrossRef]
- Chai, B.; Li, J.-Y.; Zhang, W.; Newman, E.; Ammori, J.; Mulholland, M.W. Melanocortin-4 receptor-mediated inhibition of apoptosis in immortalized hypothalamic neurons via mitogen-activated protein kinase. Peptides 2006, 27, 2846–2857. [Google Scholar] [CrossRef]
- Büch, T.R.; Heling, D.; Damm, E.; Gudermann, T.; Breit, A. Pertussis toxin-sensitive signaling of melanocortin-4 receptors in hypothalamic GT1-7 cells defines agouti-related protein as a biased agonist. J. Biol. Chem. 2009, 284, 26411–26420. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Raimondi, F.; Kadji, F.M.N.; Singh, G.; Kishi, T.; Uwamizu, A.; Ono, Y.; Shinjo, Y.; Ishida, S.; Arang, N. Illuminating G-protein-coupling selectivity of GPCRs. Cell 2019, 177, 1933–1947. [Google Scholar] [CrossRef] [PubMed]
- Clément, K.; Biebermann, H.; Farooqi, I.S.; Van der Ploeg, L.; Wolters, B.; Poitou, C.; Puder, L.; Fiedorek, F.; Gottesdiener, K.; Kleinau, G.; et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat. Med. 2018, 24, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Metzger, P.; Carlson, B.; Sun, H.; Cui, Z.; Gavrilova, O.; Chen, M.; Weinstein, L. OR12-3 Mice with MC4R Site Mutation (F51L) Develop Severe Obesity Independent of Gs-alpha/cAMP Signaling. J. Endocr. Soc. 2019, 3 (Suppl. 1), OR12-3. [Google Scholar] [CrossRef]
- Li, Y.-Q.; Shrestha, Y.; Pandey, M.; Chen, M.; Kablan, A.; Gavrilova, O.; Offermanns, S.; Weinstein, L.S. G q/11 α and G s α mediate distinct physiological responses to central melanocortins. J. Clin. Investig. 2016, 126, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Nijenhuis, W.A.; Oosterom, J.; Adan, R.A. AgRP (83–132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol. Endocrinol. 2001, 15, 164–171. [Google Scholar]
- Biebermann, H.; Krude, H.; Elsner, A.; Chubanov, V.; Gudermann, T.; Grüters, A. Autosomal-Dominant Mode of Inheritance of a Melanocortin-4 Receptor Mutation in a Patient with Severe Early-Onset Obesity Is Due to a Dominant-Negative Effect Caused by Receptor Dimerization. Diabetes 2003, 52, 2984–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haskell-Luevano, C.; Monck, E.K. Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regul. Pept. 2001, 99, 1–7. [Google Scholar] [CrossRef]
- Yang, Y.-k.; Thompson, D.A.; Dickinson, C.J.; Wilken, J.; Barsh, G.S.; Kent, S.B.H.; Gantz, I. Characterization of Agouti-Related Protein Binding to Melanocortin Receptors. Mol. Endocrinol. 1999, 13, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Schiöth, H.B.; Muceniece, R.; Wikberg, J.E.S.; Chhajlani, V. Characterisation of melanocortin receptor subtypes by radioligand binding analysis. Eur. J. Pharmacol. Mol. Pharmacol. 1995, 288, 311–317. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243. [Google Scholar] [CrossRef]
- He, S.; Tao, Y.-X. Defect in MAPK signaling as a cause for monogenic obesity caused by inactivating mutations in the melanocortin-4 receptor gene. Int. J. Biol. Sci. 2014, 10, 1128. [Google Scholar] [CrossRef] [Green Version]
- Gillyard, T.; Fowler, K.; Williams, S.Y.; Cone, R.D. Obesity-associated mutant melanocortin-4 receptors with normal Gαs coupling frequently exhibit other discoverable pharmacological and biochemical defects. J. Neuroendocrinol. 2019, 31, e12795. [Google Scholar] [CrossRef] [PubMed]
- Lotta, L.A.; Mokrosiński, J.; de Oliveira, E.M.; Li, C.; Sharp, S.J.; Luan, J.A.; Brouwers, B.; Ayinampudi, V.; Bowker, N.; Kerrison, N. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell 2019, 177, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Tu, Z.; Kleyn, P.W.; Kissebah, A.; Duprat, L.; Lee, J.; Chin, W.; Maruti, S.; Deng, N.; Fisher, S.L. Identification and functional analysis of novel human melanocortin-4 receptor variants. Diabetes 1999, 48, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.; MacKenzie, R.G. Functional characterization of mutations in melanocortin-4 receptor associated with human obesity. J. Biol. Chem. 1999, 274, 35816–35822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovite, V.; Petrovska, R.; Vaivade, I.; Kalnina, I.; Fridmanis, D.; Zaharenko, L.; Peculis, R.; Pirags, V.; Schioth, H.B.; Klovins, J. The role of common and rare MC4R variants and FTO polymorphisms in extreme form of obesity. Mol. Biol. Rep. 2014, 41, 1491–1500. [Google Scholar] [CrossRef]
- Hughes, D.A.; Hinney, A.; Brumm, H.; Wermter, A.-K.; Biebermann, H.; Hebebrand, J.; Stoneking, M. Increased constraints on MC4R during primate and human evolution. Hum. Genet. 2008, 124, 633. [Google Scholar] [CrossRef] [Green Version]
- Melchior, C.; Schulz, A.; Windholz, J.; Kiess, W.; Schöneberg, T.; Körner, A. Clinical and Functional Relevance of Melanocortin-4 Receptor Variants in Obese German Children. Horm. Res. Paediatr. 2012, 78, 237–246. [Google Scholar] [CrossRef]
- Thearle, M.S.; Muller, Y.L.; Hanson, R.L.; Mullins, M.; AbdusSamad, M.; Tran, J.; Knowler, W.C.; Bogardus, C.; Krakoff, J.; Baier, L.J. Greater Impact of Melanocortin-4 Receptor Deficiency on Rates of Growth and Risk of Type 2 Diabetes During Childhood Compared With Adulthood in Pima Indians. Diabetes 2012, 61, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Geller, F.; Reichwald, K.; Dempfle, A.; Illig, T.; Vollmert, C.; Herpertz, S.; Siffert, W.; Platzer, M.; Hess, C.; Gudermann, T.; et al. Melanocortin-4 Receptor Gene Variant I103 Is Negatively Associated with Obesity. Am. J. Hum. Genet. 2004, 74, 572–581. [Google Scholar] [CrossRef] [Green Version]
- Rutanen, J.; Pihlajamäki, J.; Karhapää, P.; Vauhkonen, I.; Kuusisto, J.; Mykkänen, L.M.; Laakso, M. The Val103Ile Polymorphism of Melanocortin-4 Receptor Regulates Energy Expenditure and Weight Gain. Obes. Res. 2004, 12, 1060–1066. [Google Scholar] [CrossRef]
- Heid, I.M.; Vollmert, C.; Hinney, A.; Döring, A.; Geller, F.; Löwel, H.; Wichmann, H.E.; Illig, T.; Hebebrand, J.; Kronenberg, F. Association of the 103I MC4R allele with decreased body mass in 7937 participants of two population based surveys. J. Med. Genet. 2005, 42, e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Z.; Litherland, S.A.; Sorensen, N.B.; Proneth, B.; Wood, M.S.; Shaw, A.M.; Millard, W.J.; Haskell-Luevano, C. Pharmacological Characterization of 40 Human Melanocortin-4 Receptor Polymorphisms with the Endogenous Proopiomelanocortin-Derived Agonists and the Agouti-Related Protein (AGRP) Antagonist. Biochemistry 2006, 45, 7277–7288. [Google Scholar] [CrossRef]
- Young, E.H.; Wareham, N.J.; Farooqi, S.; Hinney, A.; Hebebrand, J.; Scherag, A.; O’Rahilly, S.; Barroso, I.; Sandhu, M.S. The V103I polymorphism of the MC4R gene and obesity: Population based studies and meta-analysis of 29 563 individuals. Int. J. Obes. 2007, 31, 1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutzmann, F.; Vatin, V.; Cauchi, S.; Morandi, A.; Jouret, B.; Landt, O.; Tounian, P.; Levy-Marchal, C.; Buzzetti, R.; Pinelli, L.; et al. Non-synonymous polymorphisms in melanocortin-4 receptor protect against obesity: The two facets of a Janus obesity gene. Hum. Mol. Genet. 2007, 16, 1837–1844. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Ma, J.; Zhang, S.; Hinney, A.; Hebebrand, J.; Wang, Y.; Wang, H.-J. Association of the MC4R V103I Polymorphism with Obesity: A Chinese Case–control Study and Meta-analysis in 55,195 Individuals. Obesity 2010, 18, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Heid, I.M.; Vollmert, C.; Kronenberg, F.; Huth, C.; Ankerst, D.P.; Luchner, A.; Hinney, A.; Brönner, G.; Wichmann, H.E.; Illig, T.; et al. Association of the MC4R V103I Polymorphism With the Metabolic Syndrome: The KORA Study. Obesity 2008, 16, 369–376. [Google Scholar] [CrossRef]
- Elsner, A.; Tarnow, P.; Schaefer, M.; Ambrugger, P.; Krude, H.; Grüters, A.; Biebermann, H. MC4R oligomerizes independently of extracellular cysteine residues. Peptides 2006, 27, 372–379. [Google Scholar] [CrossRef]
- Lubrano-Berthelier, C.; Durand, E.; Dubern, B.; Shapiro, A.; Dazin, P.; Weill, J.; Ferron, C.; Froguel, P.; Vaisse, C. Intracellular retention is a common characteristic of childhood obesity-associated MC4R mutations. Hum. Mol. Genet. 2003, 12, 145–153. [Google Scholar] [CrossRef]
- Valli-Jaakola, K.; Lipsanen-Nyman, M.; Oksanen, L.; Hollenberg, A.N.; Kontula, K.; Bjørbæk, C.; Schalin-Jäntti, C. Identification and Characterization of Melanocortin-4 Receptor Gene Mutations in Morbidly Obese Finnish Children and Adults. J. Clin. Endocrinol. Metab. 2004, 89, 940–945. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.-C.; Tao, Y.-X. Functional characterization and pharmacological rescue of melanocortin-4 receptor mutations identified from obese patients. J. Cell. Mol. Med. 2009, 13, 3268–3282. [Google Scholar] [CrossRef] [Green Version]
- Roubert, P.; Dubern, B.; Plas, P.; Lubrano-Berthelier, C.; Alihi, R.; Auger, F.; Deoliveira, D.B.; Dong, J.Z.; Basdevant, A.; Thurieau, C. Novel pharmacological MC4R agonists can efficiently activate mutated MC4R from obese patient with impaired endogenous agonist response. J. Endocrinol. 2010, 207, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, T.; Hinney, A.; de Sousa, G.; Austrup, F.; Hebebrand, J.; Andler, W. Definable Somatic Disorders in Overweight Children and Adolescents. J. Pediatrics 2007, 150, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Santoro, N.; Cirillo, G.; Xiang, Z.; Tanas, R.; Greggio, N.; Morino, G.; Iughetti, L.; Vottero, A.; Salvatoni, A.; Di Pietro, M.; et al. Prevalence of pathogenetic MC4R mutations in Italian children with early Onset obesity, tall stature and familial history of obesity. BMC Med. Genet. 2009, 10, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahituv, N.; Kavaslar, N.; Schackwitz, W.; Ustaszewska, A.; Martin, J.; Hébert, S.; Doelle, H.; Ersoy, B.; Kryukov, G.; Schmidt, S. Medical sequencing at the extremes of human body mass. Am. J. Hum. Genet. 2007, 80, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Piechowski, C.L.; Rediger, A.; Lagemann, C.; Muhlhaus, J.; Muller, A.; Pratzka, J.; Tarnow, P.; Gruters, A.; Krude, H.; Kleinau, G. Inhibition of melanocortin-4 receptor dimerization by substitutions in intracellular loop 2. J. Mol. Endocrinol. 2013, 51, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Hinney, A.; Bettecken, T.; Tarnow, P.; Brumm, H.; Reichwald, K.; Lichtner, P.; Scherag, A.; Nguyen, T.T.; Schlumberger, P.; Rief, W.; et al. Prevalence, Spectrum, and Functional Characterization of Melanocortin-4 Receptor Gene Mutations in a Representative Population-Based Sample and Obese Adults from Germany. J. Clin. Endocrinol. Metab. 2006, 91, 1761–1769. [Google Scholar] [CrossRef] [Green Version]
- Hsiung, H.M.; Hertel, J.; Zhang, X.-Y.; Smith, D.P.; Smiley, D.L.; Heiman, M.L.; Yang, D.D.; Husain, S.; Mayer, J.P.; Zhang, L. A novel and selective beta-MSH derived peptide agonist for melanocortin 4 receptor potently decreased food intake and body weight gain in dietinduced obese rats. Endocrinology 2005, 146, 5257–5266. [Google Scholar] [CrossRef]
- Biebermann, H.; Castañeda, T.R.; van Landeghem, F.; von Deimling, A.; Escher, F.; Brabant, G.; Hebebrand, J.; Hinney, A.; Tschöp, M.H.; Grüters, A.; et al. A role for β-melanocyte-stimulating hormone in human body-weight regulation. Cell Metab. 2006, 3, 141–146. [Google Scholar] [CrossRef]
- Sawyer, T.K.; Sanfilippo, P.J.; Hruby, V.J.; Engel, M.H.; Heward, C.B.; Burnett, J.B.; Hadley, M.E. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: A highly potent alpha-melanotropin with ultralong biological activity. Proc. Natl. Acad. Sci. USA 1980, 77, 5754. [Google Scholar] [CrossRef] [Green Version]
- Rached, M.; El Mourabit, H.; Buronfosse, A.; Blondet, A.; Naville, D.; Begeot, M.; Penhoat, A. Expression of the human melanocortin-2 receptor in different eukaryotic cells. Peptides 2005, 26, 1842–1847. [Google Scholar] [CrossRef]
- Blondet, A.; Doghman, M.; Rached, M.; Durand, P.; Bégeot, M.; Naville, D. Characterization of Cell Lines Stably Expressing Human Normal or Mutated EGFP-Tagged MC4R. J. Biochem. 2004, 135, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Sebag, J.A.; Zhang, C.; Hinkle, P.M.; Bradshaw, A.M.; Cone, R.D. Developmental control of the melanocortin-4 receptor by MRAP2 proteins in zebrafish. Science 2013, 341, 278–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonnop, L.; Kleinau, G.; Herrfurth, N.; Volckmar, A.-L.; Cetindag, C.; Müller, A.; Peters, T.; Herpertz, S.; Antel, J.; Hebebrand, J.; et al. Decreased melanocortin-4 receptor function conferred by an infrequent variant at the human melanocortin receptor accessory protein 2 gene. Obesity 2016, 24, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 2018, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Oetjens, M.; Kelly, M.; Sturm, A.; Martin, C.; Ledbetter, D. Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mo, X.-L.; Tao, Y.-X. Activation of MAPK by inverse agonists in six naturally occurring constitutively active mutant human melanocortin-4 receptors. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1939–1948. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Santiago, P.; Lubrano, C.; Vaisse, C.; Conklin, B.R. Engineering the Melanocortin-4 Receptor to Control Constitutive and Ligand-Mediated Gs Signaling In Vivo. PLoS ONE 2007, 2, e668. [Google Scholar] [CrossRef] [Green Version]
- Hinney, A.; Hohmann, S.; Geller, F.; Vogel, C.; Hess, C.; Wermter, A.-K.; Brokamp, B.; Goldschmidt, H.; Siegfried, W.; Remschmidt, H. Melanocortin-4 receptor gene: Case-control study and transmission disequilibrium test confirm that functionally relevant mutations are compatible with a major gene effect for extreme obesity. J. Clin. Endocrinol. Metab. 2003, 88, 4258–4267. [Google Scholar] [CrossRef] [Green Version]
- Josep Agulleiro, M.; Cortés, R.; Fernández-Durán, B.; Navarro, S.; Guillot, R.; Meimaridou, E.; Clark, A.J.L.; Cerdá-Reverter, J.M. Melanocortin 4 Receptor Becomes an ACTH Receptor by Coexpression of Melanocortin Receptor Accessory Protein 2. Mol. Endocrinol. 2013, 27, 1934–1945. [Google Scholar] [CrossRef]
- Newman, E.A.; Chai, B.-X.; Zhang, W.; Li, J.-Y.; Ammori, J.B.; Mulholland, M.W. Activation of the melanocortin-4 receptor mobilizes intracellular free calcium in immortalized hypothalamic neurons. J. Surg. Res. 2006, 132, 201–207. [Google Scholar] [CrossRef]
- Peters, M.F.; Scott, C.W. Evaluating cellular impedance assays for detection of GPCR pleiotropic signaling and functional selectivity. J. Biomol. Screen. 2009, 14, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.Q.; Wang, H.Y.; Friedman, E. Stimulated D1 dopamine receptors couple to multiple Gα proteins in different brain regions. J. Neurochem. 2001, 78, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Heyder, N.; Kleinau, G.; Szczepek, M.; Kwiatkowski, D.; Speck, D.; Soletto, L.; Cerdá-Reverter, J.M.; Krude, H.; Kühnen, P.; Biebermann, H.; et al. Signal Transduction and Pathogenic Modifications at the Melanocortin-4 Receptor: A Structural Perspective. Front. Endocrinol. 2019, 10, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caverzasio, J.; Palmer, G.; Suzuki, A.; Bonjour, J.-P. Evidence for the Involvement of Two Pathways in Activation of Extracellular Signal-Regulated Kinase (Erk) and Cell Proliferation by Gi and Gq Protein-Coupled Receptors in Osteoblast-Like Cells. J. Bone Miner. Res. 2000, 15, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Vaisse, C.; Clement, K.; Durand, E.; Hercberg, S.; Guy-Grand, B.; Froguel, P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J. Clin. Investig. 2000, 106, 253–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutigliano, G.; Bräunig, J.; Del Grande, C.; Carnicelli, V.; Masci, I.; Merlino, S.; Kleinau, G.; Tessieri, L.; Pardossi, S.; Paisdzior, S. Non-Functional Trace Amine-Associated Receptor 1 Variants in Patients With Mental Disorders. Front. Pharmacol. 2019, 10, 1027. [Google Scholar] [CrossRef]
- Bräunig, J.; Dinter, J.; Höfig, C.S.; Paisdzior, S.; Szczepek, M.; Scheerer, P.; Rosowski, M.; Mittag, J.; Kleinau, G.; Biebermann, H. The Trace Amine-Associated Receptor 1 Agonist 3-Iodothyronamine Induces Biased Signaling at the Serotonin 1b Receptor. Front. Pharmacol. 2018, 9, 222. [Google Scholar] [CrossRef] [Green Version]
- Kenakin, T. A scale of agonism and allosteric modulation for assessment of selectivity, bias, and receptor mutation. Mol. Pharmacol. 2017, 92, 414–424. [Google Scholar] [CrossRef]
- Winpenny, D.; Clark, M.; Cawkill, D. Biased ligand quantification in drug discovery: From theory to high throughput screening to identify new biased μ opioid receptor agonists. Br. J. Pharmacol. 2016, 173, 1393–1403. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paisdzior, S.; Dimitriou, I.M.; Schöpe, P.C.; Annibale, P.; Scheerer, P.; Krude, H.; Lohse, M.J.; Biebermann, H.; Kühnen, P. Differential Signaling Profiles of MC4R Mutations with Three Different Ligands. Int. J. Mol. Sci. 2020, 21, 1224. https://doi.org/10.3390/ijms21041224
Paisdzior S, Dimitriou IM, Schöpe PC, Annibale P, Scheerer P, Krude H, Lohse MJ, Biebermann H, Kühnen P. Differential Signaling Profiles of MC4R Mutations with Three Different Ligands. International Journal of Molecular Sciences. 2020; 21(4):1224. https://doi.org/10.3390/ijms21041224
Chicago/Turabian StylePaisdzior, Sarah, Ioanna Maria Dimitriou, Paul Curtis Schöpe, Paolo Annibale, Patrick Scheerer, Heiko Krude, Martin J. Lohse, Heike Biebermann, and Peter Kühnen. 2020. "Differential Signaling Profiles of MC4R Mutations with Three Different Ligands" International Journal of Molecular Sciences 21, no. 4: 1224. https://doi.org/10.3390/ijms21041224