Systematical Identification of Breast Cancer-Related Circular RNA Modules for Deciphering circRNA Functions Based on the Non-Negative Matrix Factorization Algorithm

Abstract
:1. Introduction
2. Results
2.1. Identification of Differentially Expressed circRNAs and miRNAs in BC
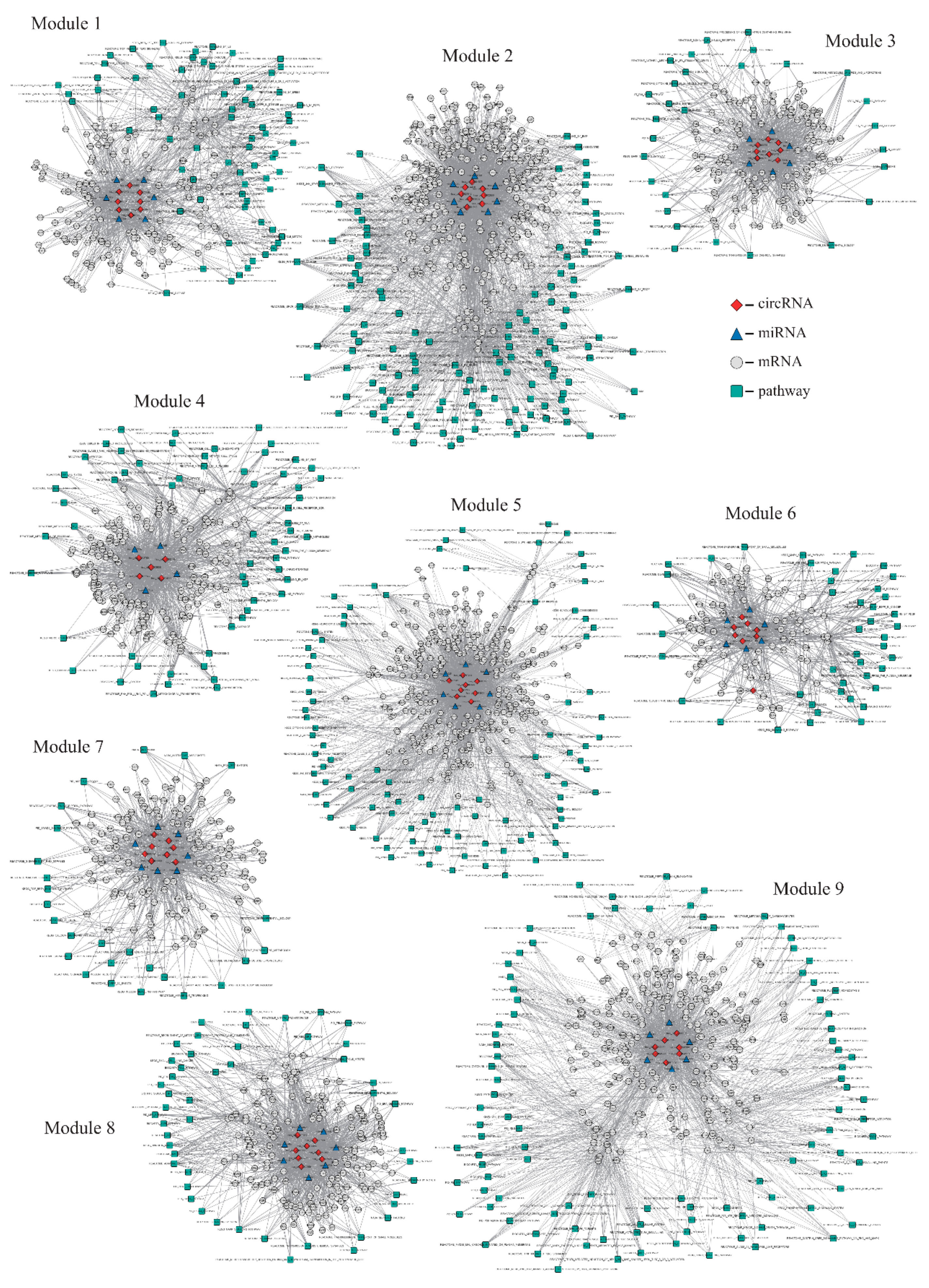
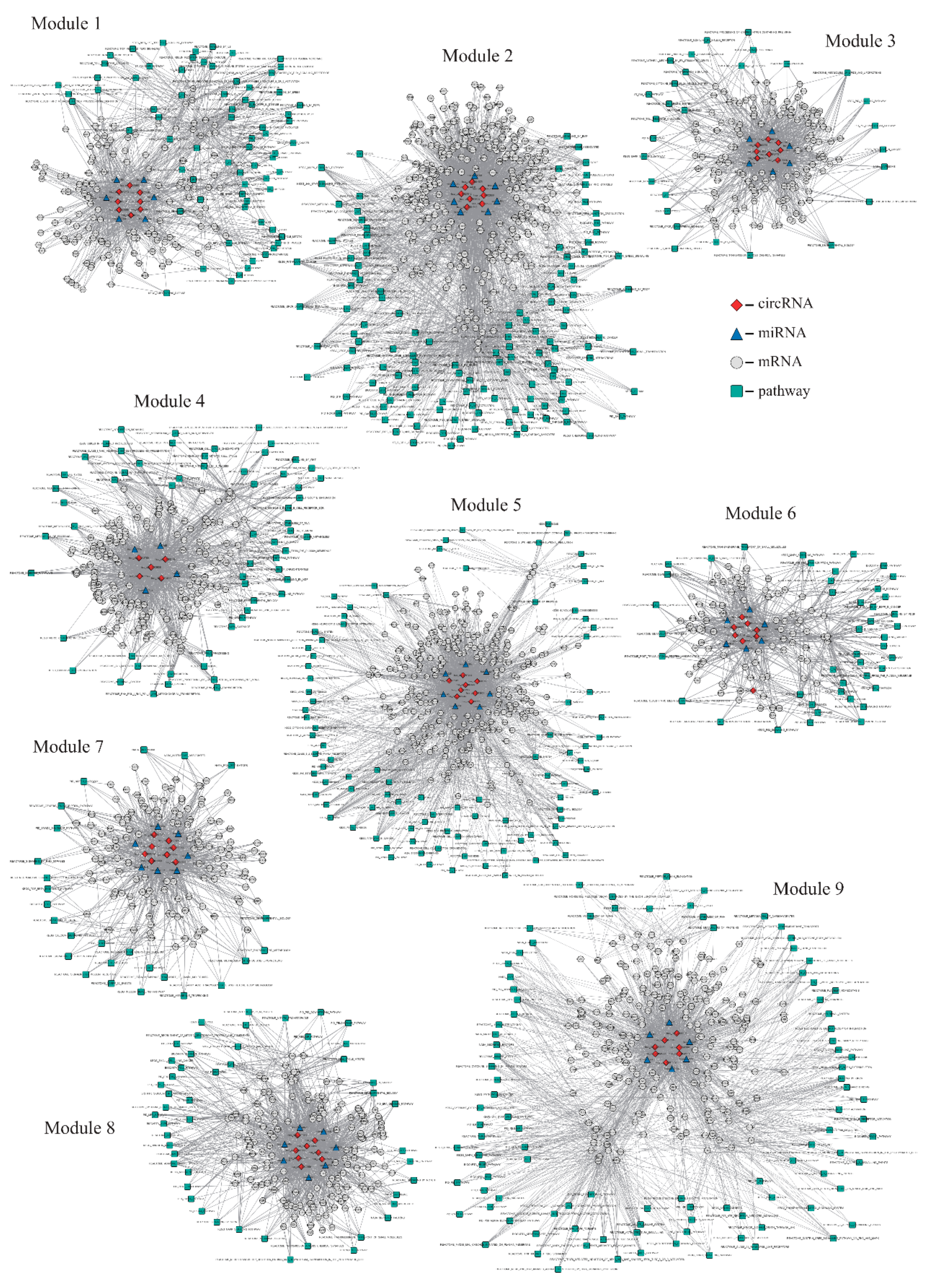
2.2. Identification of circRNA Modules Based on a Non-Negative Matrix Factorization (NMF) Algorithm
2.3. Prediction of Disease Candidate circRNAs in BC
2.4. Comparison with Other circRNA Prioritization Approaches
3. Discussion
4. Materials and Methods
4.1. Data Acquisitions
4.2. Quantification of circRNA-mRNA, miRNA-mRNA, and Pathway-mRNA Binary Matrices
4.3. Construction of circRNA Modules Basing on Non-Negative Matrix Factorization (NMF) Algorithm
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial disparity in mortality by state. CA: Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef]
- Tokunaga, E.; Hisamatsu, Y.; Tanaka, K.; Yamashita, N.; Saeki, H.; Oki, E.; Kitao, H.; Maehara, Y. Molecular mechanisms regulating the hormone sensitivity of breast cancer. Cancer Sci. 2014, 105, 1377–1383. [Google Scholar] [CrossRef] [Green Version]
- Kozlowski, J.; Kozlowska, A.; Kocki, J. Breast cancer metastasis—Insight into selected molecular mechanisms of the phenomenon. Postęp. Hig. Med. Dośw. 2015, 69, 447–451. [Google Scholar] [CrossRef]
- Matsumoto, A.; Jinno, H.; Ando, T.; Fujii, T.; Nakamura, T.; Saito, J.; Takahashi, M.; Hayashida, T.; Kitagawa, Y. Biological markers of invasive breast cancer. Jpn. J. Clin. Oncol. 2016, 46, 99–105. [Google Scholar] [CrossRef]
- Wang, W.; Luo, Y.P. MicroRNAs in breast cancer: oncogene and tumor suppressors with clinical potential. J. Zhejiang Univ. Sci. B 2015, 16, 18–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soudyab, M.; Iranpour, M.; Ghafouri-Fard, S. The Role of Long Non-Coding RNAs in Breast Cancer. Arch. Iran. Med. 2016, 19, 508–517. [Google Scholar] [PubMed]
- Li, S.; Li, B.; Zheng, Y.; Li, M.; Shi, L.; Pu, X. Exploring functions of long noncoding RNAs across multiple cancers through co-expression network. Sci. Rep. 2017, 7, 754. [Google Scholar] [CrossRef]
- Nogales-Cadenas, R.; Cai, Y.; Lin, J.R.; Zhang, Q.; Zhang, W.; Montagna, C.; Zhang, Z.D. MicroRNA expression and gene regulation drive breast cancer progression and metastasis in PyMT mice. Breast Cancer Res. BCR 2016, 18, 75. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Ganesan, K.; Miller, L.D.; Tan, P. A modular analysis of breast cancer reveals a novel low-grade molecular signature in estrogen receptor-positive tumors. Clin. Cancer Res Off. J. Am. Assoc. Cancer Res. 2006, 12, 3288–3296. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed]
- Danan, M.; Schwartz, S.; Edelheit, S.; Sorek, R. Transcriptome-wide discovery of circular RNAs in Archaea. Nucleic Acids Res. 2012, 40, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.W.; Yang, W.; Liu, E.; Yang, Z.; Dhaliwal, P.; Yang, B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016, 44, 2846–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarnerio, J.; Bezzi, M.; Jeong, J.C.; Paffenholz, S.V.; Berry, K.; Naldini, M.M.; Lo-Coco, F.; Tay, Y.; Beck, A.H.; Pandolfi, P.P. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 2016, 166, 1055–1056. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Du, W.W.; Li, X.; Yee, A.J.; Yang, B.B. Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene 2016, 35, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.F.; Zhang, X.Z.; Liu, B.G.; Jia, G.T.; Li, W.L. Circular RNA circ-ABCB10 promotes breast cancer proliferation and progression through sponging miR-1271. Am. J. Cancer Res. 2017, 7, 1566–1576. [Google Scholar] [PubMed]
- Fang, L.; Du, W.W.; Lyu, J.; Dong, J.; Zhang, C.; Yang, W.; He, A.; Kwok, Y.S.S.; Ma, J.; Wu, N.; et al. Enhanced breast cancer progression by mutant p53 is inhibited by the circular RNA circ-Ccnb1. Cell Death Differ. 2018, 25, 2195–2208. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.N.; Xia, S.Q.; Zhang, Y.Y.; Zheng, J.H.; Li, W. Circular RNAs: A novel type of biomarker and genetic tools in cancer. Oncotarget 2017, 8, 64551–64563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.D.; Jiang, L.H.; Sun, D.W.; Hou, J.C.; Ji, Z.L. CircRNA: A novel type of biomarker for cancer. Breast Cancer 2018, 25, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Zhou, H.; Feng, Z.; Xu, Z.; Tang, Y.; Li, P.; Wu, M. CircRNA: Functions and properties of a novel potential biomarker for cancer. Mol. Cancer 2017, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, T.; Xiao, J. Circular RNAs: Promising Biomarkers for Human Diseases. EBioMedicine 2018, 34, 267–274. [Google Scholar] [CrossRef]
- Lu, L.; Sun, J.; Shi, P.; Kong, W.; Xu, K.; He, B.; Zhang, S.; Wang, J. Identification of circular RNAs as a promising new class of diagnostic biomarkers for human breast cancer. Oncotarget 2017, 8, 44096–44107. [Google Scholar]
- Chen, B.; Wei, W.; Huang, X.; Xie, X.; Kong, Y.; Dai, D.; Yang, L.; Wang, J.; Tang, H.; Xie, X. circEPSTI1 as a Prognostic Marker and Mediator of Triple-Negative Breast Cancer Progression. Theranostics 2018, 8, 4003–4015. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Beck, D.; Thoms, J.A.I.; Liu, L.; Zhao, W.; Pimanda, J.E.; Zhou, X. Annotating function to differentially expressed LincRNAs in myelodysplastic syndrome using a network-based method. Bioinformatics 2017, 33, 2622–2630. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Michailidis, G. A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinformatics 2016, 32, 1–8. [Google Scholar] [CrossRef]
- Abdellatif, M. Differential expression of microRNAs in different disease states. Circ. Res. 2012, 110, 638–650. [Google Scholar] [CrossRef]
- Greene, J.; Baird, A.M.; Brady, L.; Lim, M.; Gray, S.G.; McDermott, R.; Finn, S.P. Circular RNAs: Biogenesis, Function and Role in Human Diseases. Front. Mol. Biosci. 2017, 4, 38. [Google Scholar] [CrossRef]
- Gao, D.; Zhang, X.; Liu, B.; Meng, D.; Fang, K.; Guo, Z.; Li, L. Screening circular RNA related to chemotherapeutic resistance in breast cancer. Epigenomics 2017, 9, 1175–1188. [Google Scholar] [CrossRef]
- Mistry, M.; Pavlidis, P. Gene Ontology term overlap as a measure of gene functional similarity. BMC Bioinform. 2008, 9, 327. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.; Pavlopoulos, G.A.; Ouzounis, C.A.; Kyrpides, N.C.; Buluc, A. HipMCL: A high-performance parallel implementation of the Markov clustering algorithm for large-scale networks. Nucleic Acids Res. 2018, 46, e33. [Google Scholar] [CrossRef] [PubMed]
- Krejci, A.; Hupp, T.R.; Lexa, M.; Vojtesek, B.; Muller, P. Hammock: A hidden Markov model-based peptide clustering algorithm to identify protein-interaction consensus motifs in large datasets. Bioinformatics 2016, 32, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Walsh, S.; McDermott, E.W.; Crown, J. Biomarkers in Breast Cancer: Where Are We and Where Are We Going? Adv. Clin. Chem. 2015, 71, 1–23. [Google Scholar] [PubMed]
- Banin Hirata, B.K.; Oda, J.M.; Losi Guembarovski, R.; Ariza, C.B.; de Oliveira, C.E.; Watanabe, M.A. Molecular markers for breast cancer: prediction on tumor behavior. Dis. Markers 2014, 2014, 513158. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.D.; He, H.J.; Wang, L. Breast cancer biomarker measurements and standards. Proteomics. Clin. Appl. 2013, 7, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yin, J.; Qu, J.; Huang, L. MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 14, e1006418. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L. LRSSLMDA: Laplacian Regularized Sparse Subspace Learning for MiRNA-Disease Association prediction. PLoS Comput. Biol. 2017, 13, e1005912. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.H.; Liu, H. BNPMDA: Bipartite Network Projection for MiRNA-Disease Association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L.; Xie, D.; Zhao, Q. EGBMMDA: Extreme Gradient Boosting Machine for MiRNA-Disease Association prediction. Cell Death Dis. 2018, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Song, X.; Zhang, N.; Han, P.; Moon, B.S.; Lai, R.K.; Wang, K.; Lu, W. Circular RNA profile in gliomas revealed by identification tool UROBORUS. Nucleic Acids Res. 2016, 44, e87. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, Y.; Hu, Q.; Li, S. Systematic analysis of new drug indications by drug-gene-disease coherent subnetworks. CPT: Pharmacomet. Syst. Pharmacol. 2014, 3, e146. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Association Matrix | #(circRNA/miRNA/patnway) | #(mRNA) | Dimensions |
|---|---|---|---|
| circRNA-mRNA matrix | 80 | 2703 | 80 × 2703 |
| miRNA-mRNA matrix | 63 | 2703 | 63 × 2703 |
| pathway-mRNA matrix | 1318 | 2703 | 1318 × 2703 |
| Modules | Nodes | CircRNAs | mRNAs | miRNAs | Pathways | Edges |
|---|---|---|---|---|---|---|
| 1 | 222 | 8 | 136 | 6 | 72 | 1069 |
| 2 | 415 | 8 | 271 | 7 | 129 | 3299 |
| 3 | 172 | 8 | 129 | 6 | 29 | 864 |
| 4 | 233 | 5 | 163 | 4 | 61 | 1375 |
| 5 | 382 | 8 | 271 | 7 | 96 | 2708 |
| 6 | 141 | 8 | 83 | 6 | 44 | 665 |
| 7 | 171 | 8 | 130 | 7 | 26 | 827 |
| 8 | 216 | 8 | 152 | 7 | 49 | 1237 |
| 9 | 331 | 7 | 217 | 6 | 101 | 2054 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Xia, P.; Zhang, L.; Yu, L.; Liu, H.; Meng, Q.; Liu, S.; Li, J.; Song, Q.; Wu, J.; et al. Systematical Identification of Breast Cancer-Related Circular RNA Modules for Deciphering circRNA Functions Based on the Non-Negative Matrix Factorization Algorithm. Int. J. Mol. Sci. 2019, 20, 919. https://doi.org/10.3390/ijms20040919
Wang S, Xia P, Zhang L, Yu L, Liu H, Meng Q, Liu S, Li J, Song Q, Wu J, et al. Systematical Identification of Breast Cancer-Related Circular RNA Modules for Deciphering circRNA Functions Based on the Non-Negative Matrix Factorization Algorithm. International Journal of Molecular Sciences. 2019; 20(4):919. https://doi.org/10.3390/ijms20040919
Chicago/Turabian StyleWang, Shuyuan, Peng Xia, Li Zhang, Lei Yu, Hui Liu, Qianqian Meng, Siyao Liu, Jie Li, Qian Song, Jie Wu, and et al. 2019. "Systematical Identification of Breast Cancer-Related Circular RNA Modules for Deciphering circRNA Functions Based on the Non-Negative Matrix Factorization Algorithm" International Journal of Molecular Sciences 20, no. 4: 919. https://doi.org/10.3390/ijms20040919