Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity

 and
and
Abstract
:1. Introduction
2. Protein Conformational Disorders (PCDs)
3. Protein Misfolding Generates Neurotoxic Oligomers
4. Cytoplasmic Homeostasis and Neuronal Survival: Loss of Autophagy Efficiency is Associated with Protein Aggregation and Neuronal Loss
5. Potentiation of Autophagy as Neurodegenerative Disease Therapy
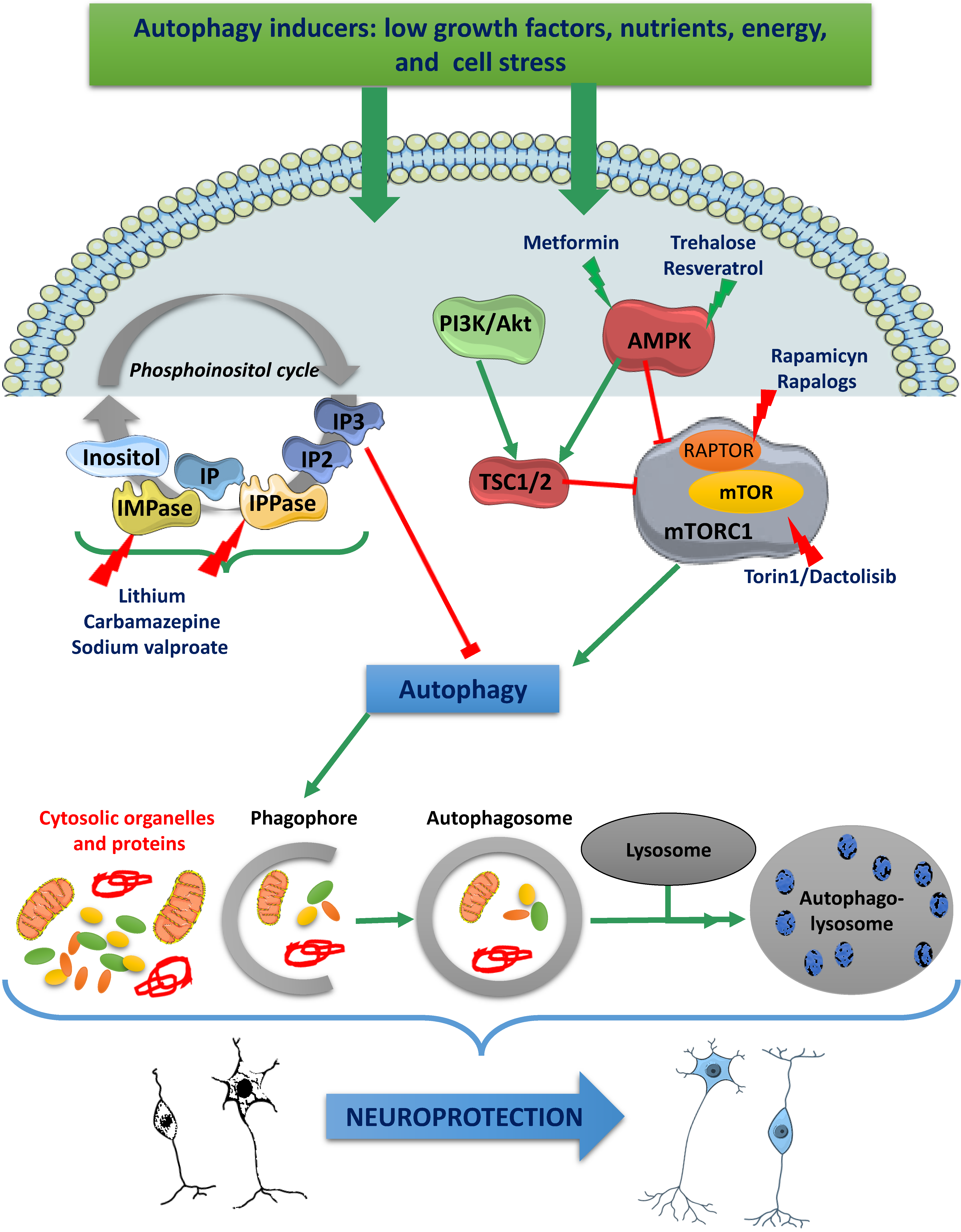
6. Molecular Control of Autophagy
7. Autophagic Structures and Associated Protein Markers
8. Autophagy of Damaged Mitochondria and Lysosomes
9. Aggrephagy is a Form of Macroautophagy Directed Against Misfolded Proteins
10. Pharmacological Control of Autophagy
10.1. Rapamycin and Rapalogues
10.2. ATP Analogues
10.3. AMPK Activators
10.4. Inhibitors of Phosphoinositol Turnover
10.5. Trehalose
10.6. Screenings for Novel Autophagy Enhancer Drugs
11. Future Development of Autophagy Enhancers in Neurodegeneration Therapy
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Alfy | Autophagy-linked FYVE-domain protein |
| AD | Alzheimer’s disease |
| AMPK | AMP-activated protein kinase |
| ALS | Amyotrophic lateral sclerosis |
| Aβ | β-amyloid |
| CBZ | Carbamazepine |
| PrPC | Cellular prion protein |
| CNS | Central nervous system |
| CMA | Chaperone-mediated autophagy |
| DEPTOR | DEP domain-containing mTOR interacting protein |
| 4E-BP1 | EIF4E-binding protein 1 |
| eIF4E | Eukaryotic translation initiation factor 4E |
| GSK-3β | Glycogen synthase kinase-3 β |
| Hsc70 | Heat shock complex 70 |
| HD | Huntington’s disease |
| IMPase | Inositol monophosphatase |
| IPP | Inositol polyphosphatase-1 |
| IP3 | 1,4,5 inositol triphosphate |
| mLST8 | Mammalian lethal with SEC13 protein 8 |
| mTOR | Mammalian target of rapamycin |
| LC3B | Microtubule-associated protein 1 light chain 3 B |
| MTOC | Microtubule organizing center |
| mTORC1 | MTOR enzymatic complex 1 |
| mTORC2 | MTOR enzymatic complex 2 |
| NBR1 | Neighbor of BRCA1 gene1 |
| PD | Parkinson’s disease |
| PIP2 | Phosphatidylinositol 4′,5′-biphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-triphosphate |
| PI3K | Phosphoinositide 3-kinase |
| PIKKs | PI3K-related kinase |
| PrPSc | Prion protein Scrapie |
| PRAS40 | Proline rich AKT substrate 40-kDa |
| PCDs | Protein conformational disorders |
| PINK1 | PTEN-induced putative protein kinase 1 |
| Rheb | RAS homolog enriched in brain |
| RAPTOR | Regulatory associated protein of mTOR |
| ROS | Reactive oxygen species |
| S6K1 | S6 kinase 1 |
| SQSTM1/p62 | Sequestrosome 1/p62 |
| SMERs | Small-molecule enhancers of rapamycin |
| TSE | Transmissible spongiform encephalopathies |
| TSC | Tuberous sclerosis complex |
| UPS | Ubiquitin-proteasome system |
| Ulk1 | Unc-51-like kinase 1 |
| VPA | Valproic acid |
References
- Scott, S.V.; Klionsky, D.J. Delivery of proteins and organelles to the vacuole from the cytoplasm. Curr. Opin. Cell Biol. 1998, 10, 523–529. [Google Scholar] [CrossRef]
- Noda, T.; Suzuki, K.; Ohsumi, Y. Yeast autophagosomes: De novo formation of a membrane structure. Trends Cell Biol. 2002, 12, 231–235. [Google Scholar] [CrossRef]
- Abeliovich, H. Regulation of autophagy by amino acid availability in S. cerevisiae and mammalian cells. Amino Acids 2015, 47, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Eskelinen, E.L.; Deretic, V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes... wait, I’m confused. Autophagy 2014, 10, 549–551. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef]
- Hyttinen, J.M.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef]
- Yamamoto, A.; Simonsen, A. The elimination of accumulated and aggregated proteins: A role for aggrephagy in neurodegeneration. Neurobiol. Dis. 2011, 43, 17–28. [Google Scholar] [CrossRef]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Protein misfolding and disease; protein refolding and therapy. FEBS Lett. 2001, 498, 204–207. [Google Scholar] [CrossRef]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid beta oligomers (AbetaOs) in Alzheimer’s disease. J. Neural Transm. (Vienna) 2018, 125, 177–191. [Google Scholar] [CrossRef]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Esparza, T.J.; Zhao, H.; Cirrito, J.R.; Cairns, N.J.; Bateman, R.J.; Holtzman, D.M.; Brody, D.L. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann. Neurol. 2013, 73, 104–119. [Google Scholar] [CrossRef]
- Wirths, O.; Multhaup, G.; Czech, C.; Blanchard, V.; Moussaoui, S.; Tremp, G.; Pradier, L.; Beyreuther, K.; Bayer, T.A. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 2001, 306, 116–120. [Google Scholar] [CrossRef]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Soto, C. The stress of prion disease. Brain Res. 2016, 1648 Pt B, 553–560. [Google Scholar] [CrossRef]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.C.; Prusiner, S.B. beta-Amyloid Prions and the Pathobiology of Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2018, 8, a023507. [Google Scholar] [CrossRef] [PubMed]
- Woerman, A.L.; Watts, J.C.; Aoyagi, A.; Giles, K.; Middleton, L.T.; Prusiner, S.B. alpha-Synuclein: Multiple System Atrophy Prions. Cold Spring Harb. Perspect. Med. 2018, 8, a024588. [Google Scholar] [CrossRef]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klohn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, A.; Thellung, S.; Villa, V.; Nizzari, M.; Florio, T. Role of prion protein aggregation in neurotoxicity. Int. J. Mol. Sci. 2012, 13, 8648–8669. [Google Scholar] [CrossRef]
- Corsaro, A.; Thellung, S.; Bucciarelli, T.; Scotti, L.; Chiovitti, K.; Villa, V.; D’Arrigo, C.; Aceto, A.; Florio, T. High hydrophobic amino acid exposure is responsible of the neurotoxic effects induced by E200K or D202N disease-related mutations of the human prion protein. Int. J. Biochem. Cell Biol. 2011, 43, 372–382. [Google Scholar] [CrossRef]
- Chiovitti, K.; Corsaro, A.; Thellung, S.; Villa, V.; Paludi, D.; D’Arrigo, C.; Russo, C.; Perico, A.; Ianieri, A.; Di Cola, D.; et al. Intracellular accumulation of a mild-denatured monomer of the human PrP fragment 90-231, as possible mechanism of its neurotoxic effects. J. Neurochem. 2007, 103, 2597–2609. [Google Scholar] [CrossRef] [PubMed]
- Thellung, S.; Corsaro, A.; Villa, V.; Simi, A.; Vella, S.; Pagano, A.; Florio, T. Human PrP90-231-induced cell death is associated with intracellular accumulation of insoluble and protease-resistant macroaggregates and lysosomal dysfunction. Cell Death Dis. 2011, 2, e138. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Koffie, R.M.; Meyer-Luehmann, M.; Hashimoto, T.; Adams, K.W.; Mielke, M.L.; Garcia-Alloza, M.; Micheva, K.D.; Smith, S.J.; Kim, M.L.; Lee, V.M.; et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. USA 2009, 106, 4012–4017. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, K.C.; Lacor, P.N.; Pitt, J.; Klein, W.L. Abeta oligomer-induced synapse degeneration in Alzheimer’s disease. Cell. Mol. Neurobiol. 2011, 31, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Cataldo, A.M.; Mathews, P.M. The endosomal-lysosomal system of neurons in Alzheimer’s disease pathogenesis: A review. Neurochem. Res. 2000, 25, 1161–1172. [Google Scholar] [CrossRef]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef]
- Mayer, R.J.; Landon, M.; Laszlo, L.; Lennox, G.; Lowe, J. Protein processing in lysosomes: The new therapeutic target in neurodegenerative disease. Lancet 1992, 340, 156–159. [Google Scholar] [CrossRef]
- Choi, H.S.; Choi, Y.G.; Shin, H.Y.; Oh, J.M.; Park, J.H.; Kim, J.I.; Carp, R.I.; Choi, E.K.; Kim, Y.S. Dysfunction of mitochondrial dynamics in the brains of scrapie-infected mice. Biochem. Biophys. Res. Commun. 2014, 448, 157–162. [Google Scholar] [CrossRef]
- Thellung, S.; Scoti, B.; Corsaro, A.; Villa, V.; Nizzari, M.; Gagliani, M.C.; Porcile, C.; Russo, C.; Pagano, A.; Tacchetti, C.; et al. Pharmacological activation of autophagy favors the clearing of intracellular aggregates of misfolded prion protein peptide to prevent neuronal death. Cell Death Dis. 2018, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Mannini, B.; Mulvihill, E.; Sgromo, C.; Cascella, R.; Khodarahmi, R.; Ramazzotti, M.; Dobson, C.M.; Cecchi, C.; Chiti, F. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem. Biol. 2014, 9, 2309–2317. [Google Scholar] [CrossRef]
- Corsaro, A.; Thellung, S.; Villa, V.; Principe, D.R.; Paludi, D.; Arena, S.; Millo, E.; Schettini, D.; Damonte, G.; Aceto, A.; et al. Prion protein fragment 106-126 induces a p38 MAP kinase-dependent apoptosis in SH-SY5Y neuroblastoma cells independently from the amyloid fibril formation. Ann. N. Y. Acad. Sci. 2003, 1010, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Thellung, S.; Villa, V.; Corsaro, A.; Pellistri, F.; Venezia, V.; Russo, C.; Aceto, A.; Robello, M.; Florio, T. ERK1/2 and p38 MAP kinases control prion protein fragment 90-231-induced astrocyte proliferation and microglia activation. Glia 2007, 55, 1469–1485. [Google Scholar] [CrossRef] [PubMed]
- Thellung, S.; Florio, T.; Corsaro, A.; Arena, S.; Merlino, M.; Salmona, M.; Tagliavini, F.; Bugiani, O.; Forloni, G.; Schettini, G. Intracellular mechanisms mediating the neuronal death and astrogliosis induced by the prion protein fragment 106–126. Int. J. Dev. Neurosci 2000, 18, 481–492. [Google Scholar] [CrossRef]
- Villa, V.; Thellung, S.; Bajetto, A.; Gatta, E.; Robello, M.; Novelli, F.; Tasso, B.; Tonelli, M.; Florio, T. Novel celecoxib analogues inhibit glial production of prostaglandin E2, nitric oxide, and oxygen radicals reverting the neuroinflammatory responses induced by misfolded prion protein fragment 90–231 or lipopolysaccharide. Pharmacol. Res. 2016, 113 Pt A, 500–514. [Google Scholar] [CrossRef]
- Villa, V.; Thellung, S.; Corsaro, A.; Novelli, F.; Tasso, B.; Colucci-D’Amato, L.; Gatta, E.; Tonelli, M.; Florio, T. Celecoxib Inhibits Prion Protein 90-231-Mediated Pro-inflammatory Responses in Microglial Cells. Mol. Neurobiol. 2016, 53, 57–72. [Google Scholar] [CrossRef]
- Rajendran, L.; Paolicelli, R.C. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef]
- Tu, J.; Chen, B.; Yang, L.; Qi, K.; Lu, J.; Zhao, D. Amyloid-beta Activates Microglia and Regulates Protein Expression in a Manner Similar to Prions. J. Mol. Neurosci. 2015, 56, 509–518. [Google Scholar] [CrossRef]
- Thellung, S.; Gatta, E.; Pellistri, F.; Villa, V.; Corsaro, A.; Nizzari, M.; Robello, M.; Florio, T. Different Molecular Mechanisms Mediate Direct or Glia-Dependent Prion Protein Fragment 90–231 Neurotoxic Effects in Cerebellar Granule Neurons. Neurotox. Res. 2017, 32, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Smith, I.F.; Green, K.N.; LaFerla, F.M. A dynamic relationship between intracellular and extracellular pools of Abeta. Am. J. Pathol. 2006, 168, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, R.C.; Villarreal, S.A.; Bowles, N.; Hepler, R.W.; Joyce, J.G.; Shughrue, P.J. Oligomers of beta-amyloid are sequestered into and seed new plaques in the brains of an AD mouse model. Exp. Neurol. 2010, 223, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; DeKosky, S.T.; Galasko, D. Alzheimer’s disease: The right drug, the right time. Science 2018, 362, 1250–1251. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Madeo, F.; Kroemer, G. Autophagy for tissue homeostasis and neuroprotection. Curr. Opin. Cell Biol. 2011, 23, 198–206. [Google Scholar] [CrossRef]
- Wurth, R.; Thellung, S.; Bajetto, A.; Mazzanti, M.; Florio, T.; Barbieri, F. Drug-repositioning opportunities for cancer therapy: Novel molecular targets for known compounds. Drug Discov. Today 2016, 21, 190–199. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2018. [Google Scholar] [CrossRef]
- Suresh, S.N.; Verma, V.; Sateesh, S.; Clement, J.P.; Manjithaya, R. Neurodegenerative diseases: Model organisms, pathology and autophagy. J. Genet. 2018, 97, 679–701. [Google Scholar] [CrossRef]
- Liberski, P.P.; Brown, D.R.; Sikorska, B.; Caughey, B.; Brown, P. Cell death and autophagy in prion diseases (transmissible spongiform encephalopathies). Folia Neuropathol. 2008, 46, 1–25. [Google Scholar]
- Hubbard, V.M.; Valdor, R.; Macian, F.; Cuervo, A.M. Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology 2012, 13, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Terman, A. The mitochondrial-lysosomal axis theory of aging: Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur. J. Biochem. 2002, 269, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Terman, A.; Gustafsson, B.; Brunk, U.T. Autophagy, organelles and ageing. J. Pathol. 2007, 211, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Magri, A.; Medina, D.X.; Wisely, E.V.; Lopez-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, D.M.; Lee, J.H.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Hamilton, D.J.; Barnett, J.L.; Paskevich, P.A.; Nixon, R.A. Abnormalities of the endosomal-lysosomal system in Alzheimer’s disease: Relationship to disease pathogenesis. Adv. Exp. Med. Biol. 1996, 389, 271–280. [Google Scholar]
- Cataldo, A.M.; Petanceska, S.; Terio, N.B.; Peterhoff, C.M.; Durham, R.; Mercken, M.; Mehta, P.D.; Buxbaum, J.; Haroutunian, V.; Nixon, R.A. Abeta localization in abnormal endosomes: Association with earliest Abeta elevations in AD and Down syndrome. Neurobiol. Aging 2004, 25, 1263–1272. [Google Scholar] [CrossRef]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef]
- An, W.L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.G.; Winblad, B.; Pei, J.J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, B.J.; Isakson, P.; Lewerenz, J.; Sanchez, H.; Kotzebue, R.W.; Cumming, R.C.; Harris, G.L.; Nezis, I.P.; Schubert, D.R.; Simonsen, A.; et al. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 2011, 7, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Ntsapi, C.; Lumkwana, D.; Swart, C.; du Toit, A.; Loos, B. New Insights Into Autophagy Dysfunction Related to Amyloid Beta Toxicity and Neuropathology in Alzheimer’s Disease. Int. Rev. Cell Mol. Biol. 2018, 336, 321–361. [Google Scholar]
- Orr, M.E.; Salinas, A.; Buffenstein, R.; Oddo, S. Mammalian target of rapamycin hyperactivity mediates the detrimental effects of a high sucrose diet on Alzheimer’s disease pathology. Neurobiol. Aging 2014, 35, 1233–1242. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Molecular pathways of neurodegeneration in Parkinson’s disease. Science 2003, 302, 819–822. [Google Scholar] [CrossRef]
- Engelender, S. Ubiquitination of alpha-synuclein and autophagy in Parkinson’s disease. Autophagy 2008, 4, 372–374. [Google Scholar] [CrossRef]
- Chu, C.T. A pivotal role for PINK1 and autophagy in mitochondrial quality control: Implications for Parkinson disease. Hum. Mol. Genet. 2010, 19, R28–R37. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. alpha-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Sarkar, S.; Perlstein, E.O.; Imarisio, S.; Pineau, S.; Cordenier, A.; Maglathlin, R.L.; Webster, J.A.; Lewis, T.A.; O’Kane, C.J.; Schreiber, S.L.; et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 2007, 3, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2018, 122, 35–40. [Google Scholar] [CrossRef]
- Sikorska, B.; Liberski, P.P.; Giraud, P.; Kopp, N.; Brown, P. Autophagy is a part of ultrastructural synaptic pathology in Creutzfeldt-Jakob disease: A brain biopsy study. Int. J. Biochem. Cell Biol. 2004, 36, 2563–2573. [Google Scholar] [CrossRef]
- Heiseke, A.; Aguib, Y.; Riemer, C.; Baier, M.; Schatzl, H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J. Neurochem. 2009, 109, 25–34. [Google Scholar] [CrossRef]
- Cortes, C.J.; Qin, K.; Norstrom, E.M.; Green, W.N.; Bindokas, V.P.; Mastrianni, J.A. Early Delivery of Misfolded PrP from ER to Lysosomes by Autophagy. Int. J. Cell Biol. 2013, 2013, 560421. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11, eaar4289. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef]
- Mandrioli, J.; D’Amico, R.; Zucchi, E.; Gessani, A.; Fini, N.; Fasano, A.; Caponnetto, C.; Chio, A.; Dalla Bella, E.; Lunetta, C.; et al. Rapamycin treatment for amyotrophic lateral sclerosis: Protocol for a phase II randomized, double-blind, placebo-controlled, multicenter, clinical trial (RAP-ALS trial). Medicine (Baltim.) 2018, 97, e11119. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J. Biol. Chem. 2005, 280, 32081–32089. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef]
- Dupont, N.; Chauhan, S.; Arko-Mensah, J.; Castillo, E.F.; Masedunskas, A.; Weigert, R.; Robenek, H.; Proikas-Cezanne, T.; Deretic, V. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr. Biol. 2014, 24, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Haobam, B.; Nozawa, T.; Minowa-Nozawa, A.; Tanaka, M.; Oda, S.; Watanabe, T.; Aikawa, C.; Maruyama, F.; Nakagawa, I. Rab17-mediated recycling endosomes contribute to autophagosome formation in response to Group A Streptococcus invasion. Cell Microbiol. 2014, 16, 1806–1821. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Tooze, S.A. Recycling endosomes contribute to autophagosome formation. Autophagy 2012, 8, 1682–1683. [Google Scholar] [CrossRef]
- Orsi, A.; Polson, H.E.; Tooze, S.A. Membrane trafficking events that partake in autophagy. Curr. Opin. Cell Biol. 2010, 22, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Vicinanza, M.; Rubinsztein, D.C. Phagophores evolve from recycling endosomes. Autophagy 2018, 14, 1475–1477. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [PubMed]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef] [PubMed]
- Jahreiss, L.; Menzies, F.M.; Rubinsztein, D.C. The itinerary of autophagosomes: From peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008, 9, 574–587. [Google Scholar] [CrossRef]
- Kochl, R.; Hu, X.W.; Chan, E.Y.; Tooze, S.A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic 2006, 7, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Ariosa, A.; Mari, M.; Klionsky, D.J. A newly characterized vacuolar serine carboxypeptidase, Atg42/Ybr139w, is required for normal vacuole function and the terminal steps of autophagy in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2018, 29, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Epple, U.D.; Eskelinen, E.L.; Thumm, M. Lysis of autophagic vesicles depends on the putative lipase Aut5p. Yeast 2001, 18, S249. [Google Scholar]
- Teter, S.A.; Eggerton, K.P.; Scott, S.V.; Kim, J.; Fischer, A.M.; Klionsky, D.J. Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J. Biol. Chem. 2001, 276, 2083–2087. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. Vacuolar hydrolysis and efflux: Current knowledge and unanswered questions. Autophagy 2018, 1–16. [Google Scholar] [CrossRef]
- Sekito, T.; Chardwiriyapreecha, S.; Sugimoto, N.; Ishimoto, M.; Kawano-Kawada, M.; Kakinuma, Y. Vacuolar transporter Avt4 is involved in excretion of basic amino acids from the vacuoles of Saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 2014, 78, 969–975. [Google Scholar] [CrossRef]
- Yang, Z.F.; Huang, J.; Geng, J.F.; Nair, U.; Klionsky, D.J. Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol. Biol. Cell 2006, 17, 5094–5104. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Stout, A.K.; Lund, S.; Baptista, M.; Panov, A.V.; Cookson, M.R.; Greenamyre, J.T. An in vitro model of Parkinson’s disease: Linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci. 2002, 22, 7006–7015. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Richardson, J.R.; Testa, C.M.; Seo, B.B.; Panov, A.V.; Yagi, T.; Matsuno-Yagi, A.; Miller, G.W.; Greenamyre, J.T. Mechanism of toxicity of pesticides acting at complex I: Relevance to environmental etiologies of Parkinson’s disease. J. Neurochem. 2007, 100, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Abdulwahid Arif, I.; Ahmad Khan, H. Environmental toxins and Parkinson’s disease: Putative roles of impaired electron transport chain and oxidative stress. Toxicol. Ind. Health 2010, 26, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Koyama-Honda, I.; Mizushima, N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J. Cell Sci. 2012, 125 Pt 6, 1488–1499. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Rodolfo, C.; Campello, S.; Cecconi, F. Mitophagy in neurodegenerative diseases. Neurochem. Int. 2018, 117, 156–166. [Google Scholar] [CrossRef]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Bove, J.; Martinez-Vicente, M.; Dehay, B.; Perier, C.; Recasens, A.; Bombrun, A.; Antonsson, B.; Vila, M. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy 2014, 10, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef]
- Hung, Y.H.; Chen, L.M.; Yang, J.Y.; Yang, W.Y. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat. Commun. 2013, 4, 2111. [Google Scholar] [CrossRef] [PubMed]
- Thurston, T.L.M.; Wandel, M.P.; von Muhlinen, N.; Foeglein, A.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, C.; Meyer, H. Detection and Clearance of Damaged Lysosomes by the Endo-Lysosomal Damage Response and Lysophagy. Curr. Biol. 2017, 27, R1330–R1341. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Kumar, S.; Jain, A.; Ponpuak, M.; Mudd, M.H.; Kimura, T.; Choi, S.W.; Peters, R.; Mandell, M.; Bruun, J.A.; et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 2016, 39, 13–27. [Google Scholar] [CrossRef]
- Jagust, W. Imaging the evolution and pathophysiology of Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 687–700. [Google Scholar] [CrossRef]
- Sengupta, I.; Udgaonkar, J.B. Structural mechanisms of oligomer and amyloid fibril formation by the prion protein. Chem. Commun. 2018, 54, 6230–6242. [Google Scholar] [CrossRef]
- Overbye, A.; Fengsrud, M.; Seglen, P.O. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy 2007, 3, 300–322. [Google Scholar] [CrossRef] [PubMed]
- Fortun, J.; Dunn, W.A. Jr.; Joy, S.; Li, J.; Notterpek, L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef] [PubMed]
- Corboy, M.J.; Thomas, P.J.; Wigley, W.C. Aggresome formation. Methods Mol. Biol. 2005, 301, 305–327. [Google Scholar] [PubMed]
- Dehvari, N.; Mahmud, T.; Persson, J.; Bengtsson, T.; Graff, C.; Winblad, B.; Ronnback, A.; Behbahani, H. Amyloid precursor protein accumulates in aggresomes in response to proteasome inhibitor. Neurochem. Int. 2012, 60, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Song, G.; Panoutsopoulos, A.; Riyadh, M.A.; Kaushik, G.; Halmai, J.; Levenson, R.; Zarbalis, K.S.; Giulivi, C. Beyond autophagy: A novel role for autism-linked Wdfy3 in brain mitophagy. Sci. Rep. 2018, 8, 11348. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Overvatn, A.; Stenmark, H.; Bjorkoy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 2010, 38, 265–279. [Google Scholar] [CrossRef]
- Yamamoto, A.; Simonsen, A. Alfy-dependent elimination of aggregated proteins by macroautophagy: Can there be too much of a good thing? Autophagy 2011, 7, 346–350. [Google Scholar] [CrossRef]
- Young, J.E.; Martinez, R.A.; La Spada, A.R. Nutrient deprivation induces neuronal autophagy and implicates reduced insulin signaling in neuroprotective autophagy activation. J. Biol. Chem. 2009, 284, 2363–2373. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef]
- Kuhn, J.G.; Chang, S.M.; Wen, P.Y.; Cloughesy, T.F.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.L.; Robins, H.I.; Mehta, M.; et al. Pharmacokinetic and tumor distribution characteristics of temsirolimus in patients with recurrent malignant glioma. Clin. Cancer Res. 2007, 13, 7401–7406. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, C.; Oberbauer, R. The future role of target of rapamycin inhibitors in renal transplantation. Curr. Opin. Urol. 2002, 12, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Bissler, J.J.; Kingswood, J.C.; Radzikowska, E.; Zonnenberg, B.A.; Belousova, E.; Frost, M.D.; Sauter, M.; Brakemeier, S.; de Vries, P.J.; Berkowitz, N.; et al. Everolimus long-term use in patients with tuberous sclerosis complex: Four-year update of the EXIST-2 study. PLoS ONE 2017, 12, e0180939. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, F.; Albertelli, M.; Grillo, F.; Mohamed, A.; Saveanu, A.; Barlier, A.; Ferone, D.; Florio, T. Neuroendocrine tumors: Insights into innovative therapeutic options and rational development of targeted therapies. Drug Discov. Today 2014, 19, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Nakagaki, T.; Satoh, K.; Ishibashi, D.; Fuse, T.; Sano, K.; Kamatari, Y.O.; Kuwata, K.; Shigematsu, K.; Iwamaru, Y.; Takenouchi, T.; et al. FK506 reduces abnormal prion protein through the activation of autolysosomal degradation and prolongs survival in prion-infected mice. Autophagy 2013, 9, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.H.; Zheng, X.F. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharmacol. Sin. 2015, 36, 1163–1169. [Google Scholar] [CrossRef]
- Menzies, F.M.; Huebener, J.; Renna, M.; Bonin, M.; Riess, O.; Rubinsztein, D.C. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010, 133 Pt 1, 93–104. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, M.S.; Wang, H.F.; Cao, L.; Zhang, Q.Q.; Shi, J.Q.; Gao, L.; Qin, H.; et al. Temsirolimus promotes autophagic clearance of amyloid-beta and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol. Res. 2014, 81, 54–63. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Wang, H.F.; Tan, M.S.; Gao, Q.; Qin, H.; Zhang, Y.D.; et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 2014, 85, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Frederick, C.; Ando, K.; Leroy, K.; Heraud, C.; Suain, V.; Buee, L.; Brion, J.P. Rapamycin ester analog CCI-779/Temsirolimus alleviates tau pathology and improves motor deficit in mutant tau transgenic mice. J. Alzheimer’s Dis. JAD 2015, 44, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Paterniti, I.; Cordaro, M.; Crupi, R.; Bruschetta, G.; Campolo, M.; Cuzzocrea, S.; Esposito, E. Neuroprotective Effects of Temsirolimus in Animal Models of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 2403–2419. [Google Scholar] [CrossRef] [PubMed]
- Cassano, T.; Magini, A.; Giovagnoli, S.; Polchi, A.; Calcagnini, S.; Pace, L.; Lavecchia, M.A.; Scuderi, C.; Bronzuoli, M.R.; Ruggeri, L.; et al. Early intrathecal infusion of everolimus restores cognitive function and mood in a murine model of Alzheimer’s disease. Exp. Neurol. 2019, 311, 88–105. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Bellozi, P.M.; Lima, I.V.; Doria, J.G.; Vieira, E.L.; Campos, A.C.; Candelario-Jalil, E.; Reis, H.J.; Teixeira, A.L.; Ribeiro, F.M.; de Oliveira, A.C. Neuroprotective effects of the anticancer drug NVP-BEZ235 (dactolisib) on amyloid-beta 1-42 induced neurotoxicity and memory impairment. Sci. Rep. 2016, 6, 25226. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.S.; Zhang, J.M.; Gao, Y.; Reichling, L.J.; Sim, T.B.; Sabatini, D.M.; Gray, N.S. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Mason, J.S.; Wileman, T.; Chapman, T. Lifespan extension without fertility reduction following dietary addition of the autophagy activator Torin1 in Drosophila melanogaster. PLoS ONE 2018, 13, e0190105. [Google Scholar] [CrossRef]
- Arbelaez-Quintero, I.; Palacios, M. To Use or Not to Use Metformin in Cerebral Ischemia: A Review of the Application of Metformin in Stroke Rodents. Stroke Res. Treat. 2017, 2017, 9756429. [Google Scholar] [CrossRef]
- Hsu, C.C.; Wahlqvist, M.L.; Lee, M.S.; Tsai, H.N. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J. Alzheimer’s Dis. JAD 2011, 24, 485–493. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed]
- Rizos, C.V.; Elisaf, M.S. Metformin and cancer. Eur. J. Pharmacol. 2013, 705, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Wahlqvist, M.L.; Lee, M.S.; Hsu, C.C.; Chuang, S.Y.; Lee, J.T.; Tsai, H.N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat. Disord. 2012, 18, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Wurth, R.; Barbieri, F.; Florio, T. New molecules and old drugs as emerging approaches to selectively target human glioblastoma cancer stem cells. BioMed Res. Int. 2014, 2014, 126586. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.R.; Logie, L.; Patel, K.; Erhardt, S.; Bacon, S.; Middleton, P.; Harthill, J.; Forteath, C.; Coats, J.T.; Kerr, C.; et al. Metformin selectively targets redox control of complex I energy transduction. Redox Biol. 2018, 14, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef]
- Tamargo-Gomez, I.; Marino, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef]
- Zhang, C.S.; Lin, S.C. AMPK Promotes Autophagy by Facilitating Mitochondrial Fission. Cell Metab. 2016, 23, 399–401. [Google Scholar] [CrossRef]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Curry, D.W.; Stutz, B.; Andrews, Z.B.; Elsworth, J.D. Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinson’s Disease. J. Parkinson’s Dis. 2018, 8, 161–181. [Google Scholar] [CrossRef]
- Lu, M.; Su, C.J.; Qiao, C.; Bian, Y.Q.; Ding, J.H.; Hu, G. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol. 2016, 19, pyw047. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Park, C.; Jeong, J.W. AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem. Biophys. Res. Commun. 2010, 391, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Tayara, K.; Espinosa-Oliva, A.M.; Garcia-Dominguez, I.; Ismaiel, A.A.; Boza-Serrano, A.; Deierborg, T.; Machado, A.; Herrera, A.J.; Venero, J.L.; de Pablos, R.M. Divergent Effects of Metformin on an Inflammatory Model of Parkinson’s Disease. Front. Cell. Neurosci. 2018, 12, 440. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.M.; Mechanic-Hamilton, D.; Xie, S.X.; Combs, M.F.; Cappola, A.R.; Xie, L.; Detre, J.A.; Wolk, D.A.; Arnold, S.E. Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data From a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 2017, 31, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, A.A.; Das, D.K. Grapes, wines, resveratrol, and heart health. J. Cardiovasc. Pharmacol. 2009, 54, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.L.; Zhou, Y.; Yi, L.; Gao, Y.X.; Ran, L.; Chen, S.H.; Zhang, T.; Zhou, X.; Zou, D.; et al. Resveratrol improves hepatic steatosis by inducing autophagy through the cAMP signaling pathway. Mol. Nutr. Food Res. 2015, 59, 1443–1457. [Google Scholar] [CrossRef]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 902–915. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, T.; Li, W.; Gao, N.; Zhang, T. Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer’s disease. Toxicol. Lett. 2018, 282, 100–108. [Google Scholar] [CrossRef]
- Hashimoto, R.; Hough, C.; Nakazawa, T.; Yamamoto, T.; Chuang, D.M. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: Involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J. Neurochem. 2002, 80, 589–597. [Google Scholar] [CrossRef]
- Hou, L.; Xiong, N.; Liu, L.; Huang, J.; Han, C.; Zhang, G.; Li, J.; Xu, X.; Lin, Z.; Wang, T. Lithium protects dopaminergic cells from rotenone toxicity via autophagy enhancement. BMC Neurosci. 2015, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Hough, C.J.; Chuang, D.M. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc. Natl. Acad. Sci. USA 1998, 95, 2642–2647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, F.; Zhai, X.; Li, X.H.; He, X.J. Lithium promotes recovery of neurological function after spinal cord injury by inducing autophagy. Neural Regen. Res. 2018, 13, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.; Sugars, K.L.; Bao, Y.P.; Rubinsztein, D.C. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J. Biol. Chem. 2002, 277, 33791–33798. [Google Scholar] [CrossRef] [PubMed]
- Motoi, Y.; Shimada, K.; Ishiguro, K.; Hattori, N. Lithium and autophagy. ACS Chem. Neurosci. 2014, 5, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Motoi, Y.; Ishiguro, K.; Kambe, T.; Matsumoto, S.E.; Itaya, M.; Kunichika, M.; Mori, H.; Shinohara, A.; Chiba, M.; et al. Long-term oral lithium treatment attenuates motor disturbance in tauopathy model mice: Implications of autophagy promotion. Neurobiol. Dis. 2012, 46, 101–108. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Diniz, B.S.; Radanovic, M.; Santos, F.S.; Talib, L.L.; Gattaz, W.F. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: Randomised controlled trial. Br. J. Psychiatry 2011, 198, 351–356. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Criollo, A.; Kroemer, G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010, 29, 515–516. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef]
- Hampel, H.; Ewers, M.; Burger, K.; Annas, P.; Mortberg, A.; Bogstedt, A.; Frolich, L.; Schroder, J.; Schonknecht, P.; Riepe, M.W.; et al. Lithium trial in Alzheimer’s disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 2009, 70, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, A.; Briggs, K.; Poppe, M.; Higgins, A.; Velayudhan, L.; Lovestone, S. A feasibility and tolerability study of lithium in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2008, 23, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.V.; Forlenza, O.V.; Gattaz, W.F. Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br. J. Psychiatry 2007, 190, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Busceti, C.L.; Biagioni, F.; Riozzi, B.; Battaglia, G.; Storto, M.; Cinque, C.; Molinaro, G.; Gradini, R.; Caricasole, A.; Canudas, A.M.; et al. Enhanced tau phosphorylation in the hippocampus of mice treated with 3,4-methylenedioxymethamphetamine (“Ecstasy”). J. Neurosci. 2008, 28, 3234–3245. [Google Scholar] [CrossRef] [PubMed]
- Dill, J.; Wang, H.; Zhou, F.; Li, S. Inactivation of glycogen synthase kinase 3 promotes axonal growth and recovery in the CNS. J. Neurosci. 2008, 28, 8914–8928. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.L.; Leng, Y.; Ma, C.H.; Zhang, J.; Ren, M.; Chuang, D.M. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008, 155, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Mora, G. The final chapter of the ALS lithium saga. Lancet Neurol. 2013, 12, 324–325. [Google Scholar] [CrossRef]
- Fornai, F.; Longone, P.; Ferrucci, M.; Lenzi, P.; Isidoro, C.; Ruggieri, S.; Paparelli, A. Autophagy and amyotrophic lateral sclerosis: The multiple roles of lithium. Autophagy 2008, 4, 527–530. [Google Scholar] [CrossRef]
- Kim, Y.H.; Rane, A.; Lussier, S.; Andersen, J.K. Lithium protects against oxidative stress-mediated cell death in alpha-synuclein-overexpressing in vitro and in vivo models of Parkinson’s disease. J. Neurosci. Res. 2011, 89, 1666–1675. [Google Scholar] [CrossRef]
- Williams, R.S.; Cheng, L.; Mudge, A.W.; Harwood, A.J. A common mechanism of action for three mood-stabilizing drugs. Nature 2002, 417, 292–295. [Google Scholar] [CrossRef]
- Xiong, N.; Jia, M.; Chen, C.; Xiong, J.; Zhang, Z.; Huang, J.; Hou, L.; Yang, H.; Cao, X.; Liang, Z.; et al. Potential autophagy enhancers attenuate rotenone-induced toxicity in SH-SY5Y. Neuroscience 2011, 199, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, J.Y.; Weng, L.H.; Li, X.X.; Yu, L.J.; Xu, Y. Valproic acid protects against MPP(+)-mediated neurotoxicity in SH-SY5Y Cells through autophagy. Neurosci. Lett. 2017, 638, 60–68. [Google Scholar] [CrossRef]
- Li, L.; Zhang, S.; Zhang, X.; Li, T.; Tang, Y.; Liu, H.; Yang, W.; Le, W. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Zhou, Q.M.; Chen, S.; Le, W.D. Repurposing carbamazepine for the treatment of amyotrophic lateral sclerosis in SOD1-G93A mouse model. CNS Neurosci. Ther. 2018, 24, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saiki, S.; O’Kane, C.J.; Rubinsztein, D.C. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum. Mol. Genet. 2008, 17, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Arguelles, J.C. Physiological roles of trehalose in bacteria and yeasts: A comparative analysis. Arch. Microbiol. 2000, 174, 217–224. [Google Scholar]
- Kaushik, J.K.; Bhat, R. Why is trehalose an exceptional protein stabilizer? An analysis of the thermal stability of proteins in the presence of the compatible osmolyte trehalose. J. Biol. Chem. 2003, 278, 26458–26465. [Google Scholar] [CrossRef]
- Lee, H.J.; Yoon, Y.S.; Lee, S.J. Mechanism of neuroprotection by trehalose: Controversy surrounding autophagy induction. Cell Death Dis. 2018, 9, 712. [Google Scholar] [CrossRef]
- Wolkers, W.F.; Walker, N.J.; Tablin, F.; Crowe, J.H. Human platelets loaded with trehalose survive freeze-drying. Cryobiology 2001, 42, 79–87. [Google Scholar] [CrossRef]
- Arora, A.; Ha, C.; Park, C.B. Inhibition of insulin amyloid formation by small stress molecules. FEBS Lett. 2004, 564, 121–125. [Google Scholar] [CrossRef]
- Liu, R.; Barkhordarian, H.; Emadi, S.; Park, C.B.; Sierks, M.R. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol. Dis. 2005, 20, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Casarejos, M.J.; Solano, R.M.; Gomez, A.; Perucho, J.; de Yebenes, J.G.; Mena, M.A. The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochem. Int. 2011, 58, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; van Zundert, B.; Hetz, C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navarro, J.A.; Rodriguez, L.; Casarejos, M.J.; Solano, R.M.; Gomez, A.; Perucho, J.; Cuervo, A.M.; Garcia de Yebenes, J.; Mena, M.A. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol. Dis. 2010, 39, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2018, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zheng, G.; Feng, Z.; Chen, Y.; Lou, Y.; Wang, C.; Zhang, X.; Zhang, Y.; Xu, H.; Shang, P.; et al. Trehalose ameliorates oxidative stress-mediated mitochondrial dysfunction and ER stress via selective autophagy stimulation and autophagic flux restoration in osteoarthritis development. Cell Death Dis. 2017, 8, e3081. [Google Scholar] [CrossRef] [PubMed]
- Mizunoe, Y.; Kobayashi, M.; Sudo, Y.; Watanabe, S.; Yasukawa, H.; Natori, D.; Hoshino, A.; Negishi, A.; Okita, N.; Komatsu, M.; et al. Trehalose protects against oxidative stress by regulating the Keap1-Nrf2 and autophagy pathways. Redox Biol. 2018, 15, 115–124. [Google Scholar] [CrossRef]
- DeBosch, B.J.; Heitmeier, M.R.; Mayer, A.L.; Higgins, C.B.; Crowley, J.R.; Kraft, T.E.; Chi, M.; Newberry, E.P.; Chen, Z.; Finck, B.N.; et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci. Signal. 2016, 9, ra21. [Google Scholar] [CrossRef]
- Mardones, P.; Rubinsztein, D.C.; Hetz, C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci. Signal. 2016, 9, fs2. [Google Scholar] [CrossRef]
- Aguib, Y.; Heiseke, A.; Gilch, S.; Riemer, C.; Baier, M.; Schatzl, H.M.; Ertmer, A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009, 5, 361–369. [Google Scholar] [CrossRef]
- Pupyshev, A.B.; Tikhonova, M.A.; Akopyan, A.A.; Tenditnik, M.V.; Dubrovina, N.I.; Korolenko, T.A. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2018, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Rubinsztein, D.C. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol. Biosyst. 2008, 4, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, J.; Pan, H.; Hu, P.; Hao, Y.; Cai, W.; Zhu, H.; Yu, A.D.; Xie, X.; Ma, D.; et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc. Natl. Acad. Sci. USA 2007, 104, 19023–19028. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Escobar, K.A.; Cole, N.H.; Mermier, C.M.; VanDusseldorp, T.A. Autophagy and aging: Maintaining the proteome through exercise and caloric restriction. Aging Cell 2019, 18, e12876. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Grune, T. Oxidative stress, aging and the proteasomal system. Biogerontology 2000, 1, 31–40. [Google Scholar] [CrossRef]
- Kastle, M.; Grune, T. Protein oxidative modification in the aging organism and the role of the ubiquitin proteasomal system. Curr. Pharm. Des. 2011, 17, 4007–4022. [Google Scholar] [CrossRef]
- Shetty, A.K.; Kodali, M.; Upadhya, R.; Madhu, L.N. Emerging Anti-Aging Strategies—Scientific Basis and Efficacy. Aging Dis. 2018, 9, 1165–1184. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Tettamanti, M.; Lucca, U.; Albanese, Y.; Quaglio, E.; Chiesa, R.; Erbetta, A.; Villani, F.; Redaelli, V.; Tagliavini, F.; et al. Preventive study in subjects at risk of fatal familial insomnia: Innovative approach to rare diseases. Prion 2015, 9, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Macchi, G.; Porro, M.; Giaccone, G.; Bugiani, M.; Scarpini, E.; Scarlato, G.; Molini, G.E.; Sasanelli, F.; Bugiani, O.; et al. Fatal familial insomnia: Genetic, neuropathologic, and biochemical study of a patient from a new Italian kindred. Neurology 1998, 50, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Kahan, B. Toxicity spectrum of inhibitors of mammalian target of rapamycin in organ transplantation: Etiology, pathogenesis and treatment. Expert Opin. Drug Saf. 2011, 10, 727–749. [Google Scholar] [CrossRef] [PubMed]
- Wermke, M.; Schuster, C.; Nolte, F.; Al-Ali, H.K.; Kiewe, P.; Schonefeldt, C.; Jakob, C.; von Bonin, M.; Hentschel, L.; Klut, I.M.; et al. Mammalian-target of rapamycin inhibition with temsirolimus in myelodysplastic syndromes (MDS) patients is associated with considerable toxicity: Results of the temsirolimus pilot trial by the German MDS Study Group (D-MDS). Br. J. Haematol. 2016, 175, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Huang, Y.; Zheng, W.; Yan, J.; Cheng, M.; Zhao, R.; Chen, L.; Hu, C.; Jia, W. Resveratrol reduces intracellular reactive oxygen species levels by inducing autophagy through the AMPK-mTOR pathway. Front. Med. 2018, 12, 697–706. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Drug | Clinical Indications | Pro-Autophagic Mechanism | References |
|---|---|---|---|
| mTOR inhibitors | Anti-fungal, immunosuppressant, anti-cancer | Destabilization of mTOR-RAPTOR complex | [157,164,165] |
| Rapamycin (Sirolimus) | |||
| Temsirolimus | |||
| Tacrolimus | |||
| Everolimus | |||
| ATP analogues | |||
| Torin 1/2 | Anti-cancer | Blockade of ATP binding site on mTOR | [168] |
| Dactolisib | [166] | ||
| Phosphoinositide cycle inhibitors | |||
| Lithium | Mood stabilizers | IP3 depletion, GSK-3β inhibition | [195,197,198,210] |
| Valproic acid | Anti-epileptic, mood stabilizers | ||
| Carbamazepine | Anti-epileptic, mood stabilizers, analgesic | ||
| AMPK activators | |||
| Metformin | Antidiabetic | AMPK activation | [173,176] |
| Trehalose * | Antioxidant | GLUT blockade | [221,222,223,224,225,226,227,228,229,230,231] |
| Resveratrol * | Antioxidant | AMPK activation | [188,189] |
| Ca2+ antagonists | |||
| Verapamil | Anti-arrhythmic and antihypertensive | IP3 depletion, calpain inhibition | [233,234,235] |
| Nimodipine | |||
| Nitrendipine |
| NCT Code | Drug Name | Disease | Stage of Disease | Purpose | Phase | Duration of Treatment | Estimated Study Completion Date |
|---|---|---|---|---|---|---|---|
| 03185208 | Lithium | AD | MCI * | Delay passage to overt dementia | 4 | 2 years | March 2022 |
| 02336633 | Resveratrol | HD | Early phase | Delay caudate atrophy | NA | 1 year | Jan 2019 |
| 03359538 | Rapamycin | ALS | all | Improvement of ALSFRS ** | 2 | 18 weeks | Apr 2019 |
| 03272503 | Pimozide | ALS | all | Delay Progression | 2 | 22 weeks | Dec 2019 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity. Int. J. Mol. Sci. 2019, 20, 901. https://doi.org/10.3390/ijms20040901
Thellung S, Corsaro A, Nizzari M, Barbieri F, Florio T. Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity. International Journal of Molecular Sciences. 2019; 20(4):901. https://doi.org/10.3390/ijms20040901
Chicago/Turabian StyleThellung, Stefano, Alessandro Corsaro, Mario Nizzari, Federica Barbieri, and Tullio Florio. 2019. "Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity" International Journal of Molecular Sciences 20, no. 4: 901. https://doi.org/10.3390/ijms20040901