Trypanocidal Mechanism of Action and in silico Studies of p-Coumaric Acid Derivatives

 ,
, 

Abstract
:1. Introduction
2. Results and Discussion
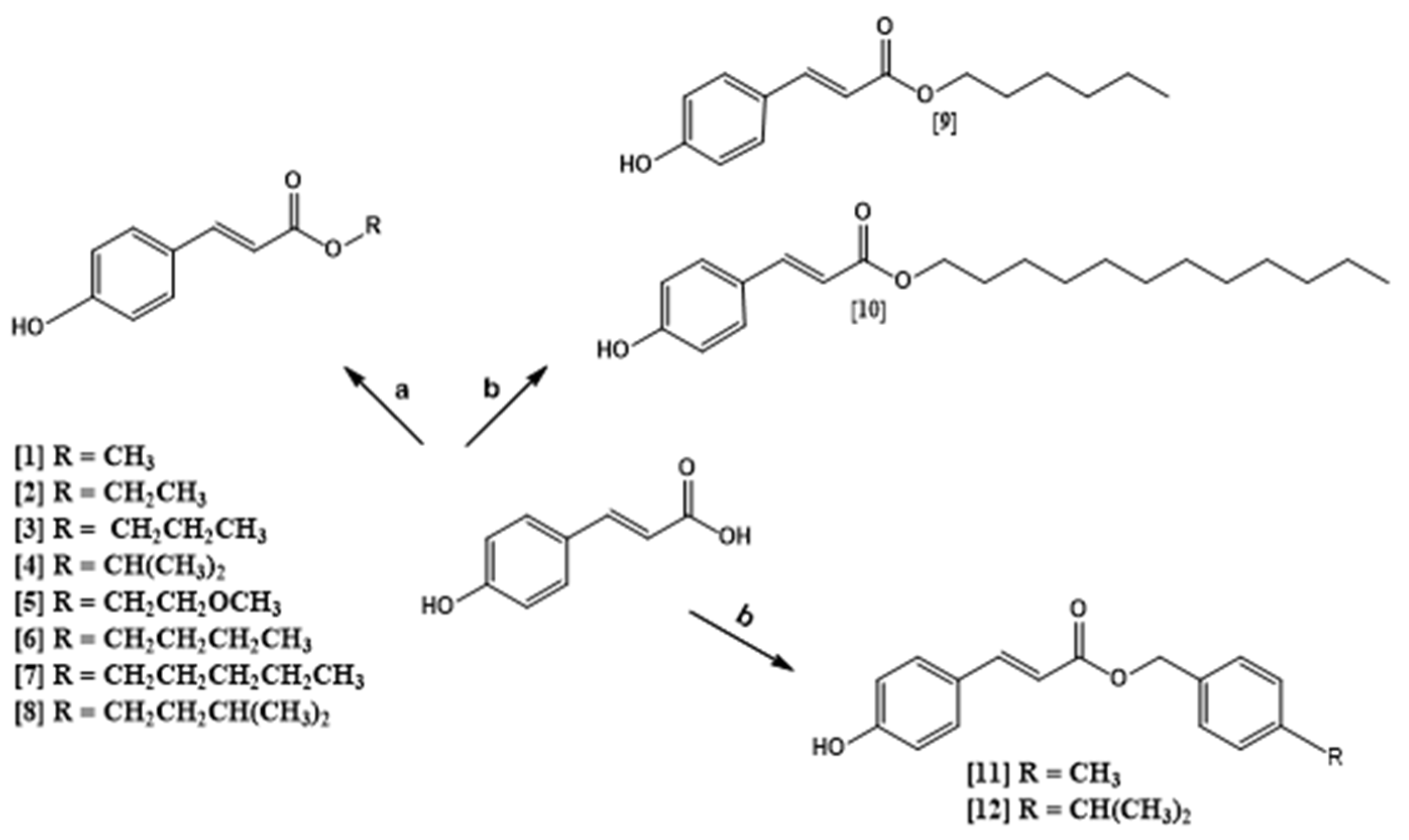
2.1. Chemistry of Compounds 1–12
2.2. Trypanocidal Activity of Compounds 1–12
2.3. Cell Death Profile
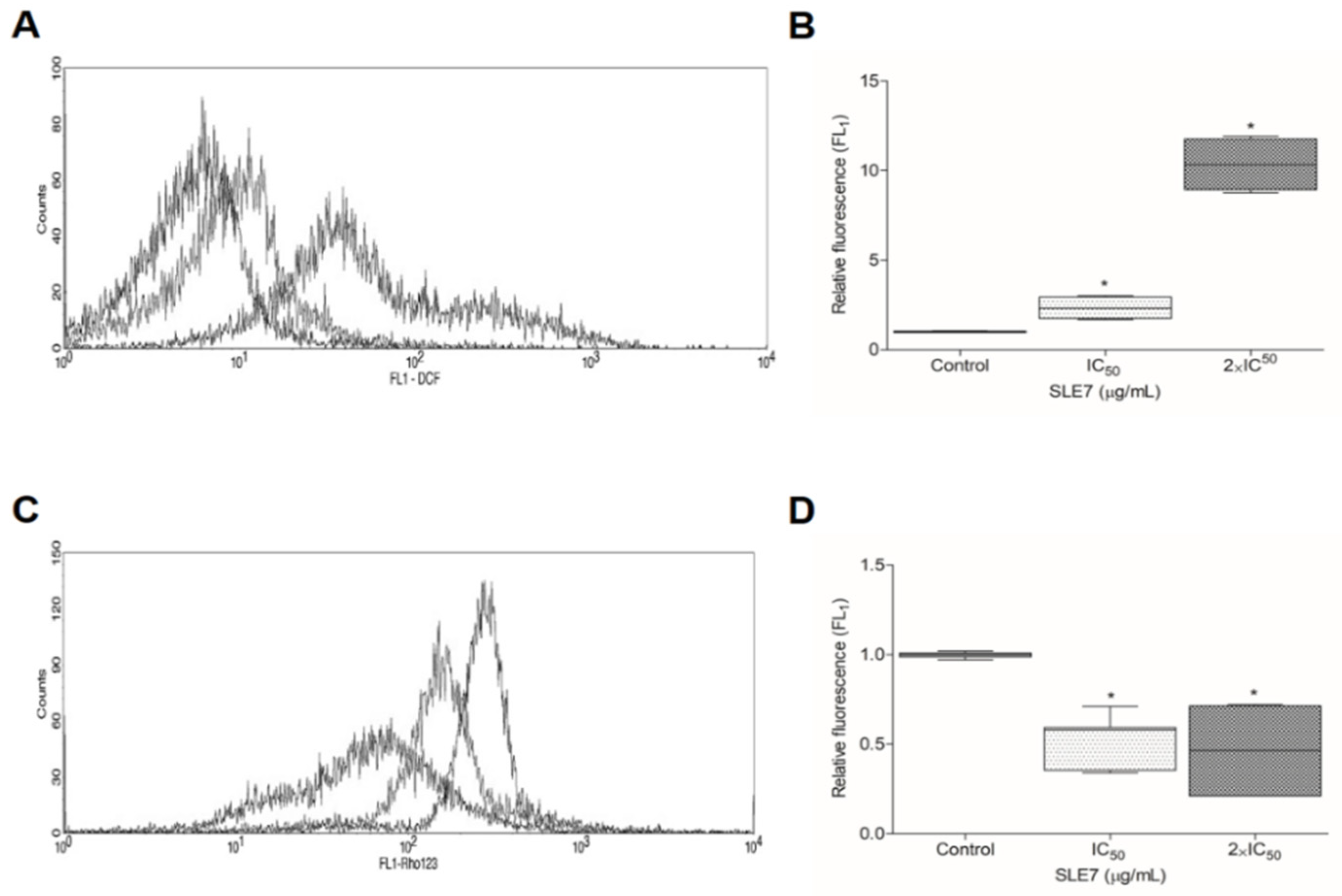
2.4. Analysis of Reactive Oxygen Species
2.5. Mitochondrial Transmembrane Potential
2.6. Scanning Electron Microscopy
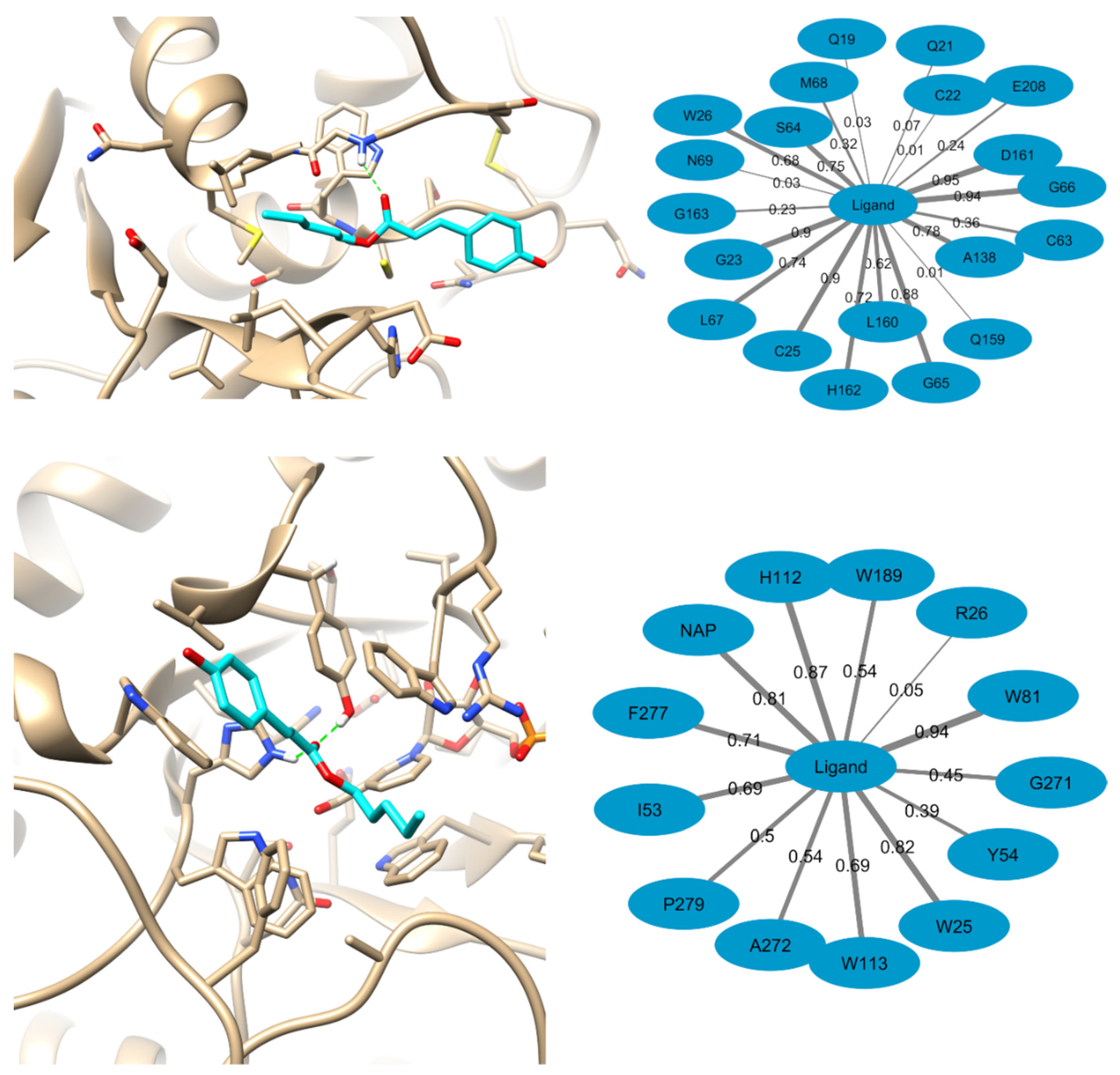
2.7. Computational Methods
3. Conclusions
4. Materials and Methods
4.1. Chemical Characterization and Reagents
4.2. General Procedure for Preparation of Compounds 1–8
4.3. Preparation of Compounds 9–12 by Mitsunobu Reaction
4.4. Chemical Characterization of Compounds 1–12
4.5. Effect of p-Coumaric Derivatives on T. cruzi Epimastigote Forms
4.6. Effect of p-Coumaric Derivatives on T. cruzi Trypomastigote Forms
4.7. Cytotoxicity Evaluation (Mammalian Cells)
4.8. Statistical Analysis
4.9. Evaluation of Cell Death Mechanisms
4.9.1. Principle of the Method
4.9.2. Experimental Procedure
4.10. Analysis of Cytoplasmic Reactive Oxygen Species
4.10.1. Principle of the Method
4.10.2. Experimental Procedure
- mTEST = geometric mean of the group to be analyzed;
- mCONTROL = geometric mean of the control group.
4.11. Assessment of Mitochondrial Transmembrane Potential
4.11.1. Principle of the Method
4.11.2. Experimental Procedure
4.12. Scanning Electron Microscopy
Evaluation of Morphological Changes in T. cruzi Induced by Compound 7
4.13. Computational Methods
4.13.1. General Modeling Workflow
4.13.2. Targets Selection
4.13.3. Molecular Docking
4.13.4. Molecular Dynamics Simulations and MM-PBSA Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pedrique, B.; Strub-Wourgaft, N.; Some, C.; Olliaro, P.; Trouiller, P.; Ford, N.; Bradol, J.-H. The drug and vaccine landscape for neglected diseases (2000–11): A systematic assessment. Lancet Glob. Health. 2013, 1, 371–379. [Google Scholar] [CrossRef]
- Cohen, J.P.; Silva, L.; Cohen, A.; Awatin, J.; Sturgeon, R. Progress Report on Neglected Tropical Disease Drug Donation Programs. Clin. Ther. 2016, 38, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.P.; Sturgeon, G.; Cohen, A. Measuring Progress in Neglected Disease Drug Development. Clin. Ther. 2014, 36, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Acharya, A.S.; Kaur, R.; Goel, A.D. Neglected tropical diseases-Challenges and opportunities in India. PLoS Negl. Trop. Dis. 2007, 8, 102–108. [Google Scholar] [CrossRef]
- Bailey, F.; Eaton, J.; Jidda, M.; Van Brakel, W.H.; Addiss, D.G.; Molyneux, D.H. Neglected Tropical Diseases and Mental Health: Progress, Partnerships, and Integration. Trends Parasitol. 2018, 35, 23–31. [Google Scholar] [CrossRef]
- World Health Organization. Who-Accelerating Work to Overcome the Global Impact of Neglected Tropical Diseases, 1st ed.; World Health Organization: Geneva, Switzerland, 2012; p. 15. [Google Scholar]
- Quansah, E.; Sarpong, E.; Karikari, T.K. Disregard of neurological impairments associated with neglected tropical diseases in Africa. eNeurologicalSci 2016, 3, 11–14. [Google Scholar] [CrossRef]
- Pérez, A.; Prada, Y.A.; Cabanzo, R.; González, C.I.; Mejía-Ospino, E. Diagnosis of chagas disease from human blood serum using surface-enhanced Raman scattering (SERS) spectroscopy and chemometric methods. Sens. BioSens. Res. 2018, 21, 40–45. [Google Scholar]
- Pereira, M.G.; Visbal, G.; Costa, T.F.R.; Frases, S.; De Souza, W.; Atella, G.; Cunha-E-Silva, N. Trypanosoma cruzi epimastigotes store cholesteryl esters in lipid droplets after cholesterol endocytosis. Mol. Biochem. Parasitol. 2018, 224, 6–16. [Google Scholar] [CrossRef]
- Reigada, C.; Phanstiel, O.; Miranda, M.R.; Pereira, C.A. Targeting polyamine transport in Trypanosoma cruzi. Eur. J. Med. Chem. 2018, 147, 1–6. [Google Scholar] [CrossRef]
- Scarim, C.B.; Jornada, D.H.; Mendes Machado, M.G.; Riberio Ferreira, C.M.; Dos Santos, J.L.; Chin, C.M. Thiazole, thio and semicarbazone derivatives against tropical infective diseases: Chagas disease, human African trypanosomiasis (HAT), leishmaniasis, and malaria. Eur. J. Med. Chem. 2018, 155, 824–838. [Google Scholar] [CrossRef]
- Gómez, L.A.; Gutierrez, F.R.S.; Peñuela, O.A. Trypanosoma cruzi infection in transfusion medicine. Hematol. Transfuss Cell Ther. 2019, 41, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Arciniegas, P.; Parra-Henao, G.; Carrión-Bonifacio, Á.; Casas-Cruz, A.; Patiño-Cuellar, A.; Díaz-Rodríguez, K.; Torres-García, O. Trypanosoma cruzi infection in naturally infected dogs from an endemic region of Cundinamarca, Colombia. Vet. Parasitol. Reg. Stud. Rep. 2018, 14, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Scarim, C.B.; Jornada, D.H.; Chelucci, R.C.; De Almeida, L.; Dos Santos, J.L.; Chung, M.C. Current advances in drug discovery for Chagas disease. Eur. J. Med. Chem. 2019, 155, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, L.E.; Morillo, C.A. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. N. Am. 2019, 33, 119–134. [Google Scholar] [CrossRef]
- Dias, J.C.P.; Ramos, A.N.; Gontijo, E.D.; Luquetti, A.; Shikanai-Yasuda, M.A.; Coura, J.R.; Torres, R.M.; Melo, J.R.; Almeida, E.A.; Oliveira, W., Jr.; et al. 2nd Brazilian Consensus on Chagas Disease, 2015. Rev. Soc. Bras. Med. Trop. 2016, 49, 3–60. [Google Scholar] [CrossRef]
- Acuña-Zegarra, M.A.; Olmos-Liceaga, D.; Velasco-Hernández, J.X. The role of animal grazing in the spread of Chagas disease. J. Theor. Biol. 2018, 457, 19–28. [Google Scholar] [CrossRef]
- Olmo, F.; Costa, F.C.; Mann, G.S.; Taylor, M.C.; Kelly, J.M. Optimising genetic transformation of Trypanosoma cruzi using hydroxyurea-induced cell-cycle synchronisation. Mol. Biochem. Parasitol. 2018, 226, 34–36. [Google Scholar] [CrossRef]
- Oliveira De Souza, L.I.; Bezzera-Silva, P.C.; Do Amaral Ferraz Navarro, D.M.; Da Silva, A.G.; Dos Santos Correia, M.T.; Da Silva, M.V.; De Figueiredo, R.C.B.Q. The chemical composition and trypanocidal activity of volatile oils from Brazilian Caatinga plants. Biomed. Pharmacother. 2017, 96, 1055–1064. [Google Scholar] [CrossRef]
- De Oliveira, S.K.; Chiaradia-Delatorre, L.D.; Mascarello, A.; Veleirinho, B.; Ramlov, F.; Kuhnen, S.; Maraschin, M. From Bench to Bedside: Natural Products and Analogs for the Treatment of Neglected Tropical Diseases (NTDs). Studies in Natural Products Chemistry, 1st ed.; Rahman, A.U., Ed.; Elsevier: Florianópolis, Brazil, 2015; Volume 44, pp. 33–92. [Google Scholar]
- Cockram, P.E.; Smith, T.K. Active Natural Product Scaffolds against Trypanosomatid Parasites: A Review. J. Nat. Prod. 2018, 81, 2138–2154. [Google Scholar] [CrossRef]
- Ko, H.C.; Lee, J.Y.; Jang, M.G.; Song, H.; Kim, S.-J. Seasonal variations in the phenolic compounds and antioxidant activity of Sasa quelpaertensis. Ind. Crop. Prod. 2018, 122, 506–512. [Google Scholar] [CrossRef]
- Viñas, P.; Campillo, N. Gas Chromatography: Mass Spectrometry Analysis of Polyphenols in Foods. Polyphenols in Plants, 1st ed.; Watson, R.R., Ed.; Academic Press Inc.: Murcia, Spain, 2014; pp. 103–157. [Google Scholar]
- Nguyen, D.M.T.; Bartley, J.P.; Moghaddam, L.; Doherty, W.O.S. Fenton oxidation products derived from hydroxycinnamic acids increases phenolic-based compounds and organic acid formation in sugar processing. Int. J. Food Sci. Technol. 2018, 53, 1278–1286. [Google Scholar] [CrossRef]
- Song, K.; An, S.M.; Kim, M.; Koh, J.-S.; Boo, Y.C. Comparison of the antimelanogenic effects of p-coumaric acid and its methyl ester and their skin permeabilities. J. Dermatol. Sci. 2011, 63, 17–22. [Google Scholar] [CrossRef]
- Kaur, J.; Katopo, L.; Hung, A.; Ashton, J.; Kasapis, S. Combined spectroscopic, molecular docking and quantum mechanics study of β -casein and p -coumaric acid interactions following thermal treatment. Food Chem. 2018, 252, 163–170. [Google Scholar] [CrossRef]
- Long, R.; Li, T.; Tong, C.; Wu, L.; Shi, S. Molecularly imprinted polymers coated CdTe quantum dots with controllable particle size for fluorescent determination of p-coumaric acid. Talanta 2019, 196, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Song, X.; Li, L.; Sun, J.; Jaiswal, Y.; Huang, J.; Guan, Y. Protective effects of p-coumaric acid against oxidant and hyperlipidemia-an in vitro and in vivo evaluation. Biomed. Pharmacother. 2019, 111, 579–587. [Google Scholar] [CrossRef]
- Wang, S.; Kong, L.; Zhao, Y.; Tan, L.; Zhang, J.; Du, Z.; Zhang, H. Lipophilization and molecular encapsulation of p-coumaric acid by amylose inclusion complex. Food Hydrocoll. 2019, 90, 270–275. [Google Scholar] [CrossRef]
- Li, W.; Yuan, S.; Sun, J.; Li, Q.; Jiang, W.; Cao, J. Ethyl p -coumarate exerts antifungal activity in vitro and in vivo against fruit Alternaria alternata via membrane-targeted mechanism. Int. J. Food Microbiol. 2018, 278, 26–35. [Google Scholar] [CrossRef]
- Moradi-Afrapoli, F.; Yassa, N.; Zimmermann, S.; Saeidnia, S.; Hadjiakhoondia, A.; Ebrahimi, S.N.; Hamburger, M. Cinnamoylphenethyl amides from Polygonum hyrcanicum possess anti-trypanosomal activity. Nat. Prod. Commun. 2012, 7, 753–755. [Google Scholar] [CrossRef]
- Kumar, G.; Degheidy, H.; Casey, B.; Goering, P. Flow cytometry evaluation of in vitro cellular necrosis and apoptosis induced by silver nanoparticles. Food Chem. Toxicol. 2015, 85, 45–51. [Google Scholar] [CrossRef]
- Krysko, D.V.; Berghe, T.V.; D’herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef] [PubMed]
- De Demenezes, R.R.P.P.B.; Sampaio, T.L.; Lima, D.B.; Sousa, P.L.; Azevedo, I.E.P.; Magalhães, E.P.; Tessarolo, L.D.; Marinho, M.M.; Santos, R.P.; Martins, A.M.C. Antiparasitic effect of (-)-α- bisabolol against Tryapanosoma cruzi Y strain forms. Diagn. Microbiol. Infect. Dis. 2019, 95, 114860. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.J.; Supriyanto, E.; Manda, M. Events associated with apoptotic effect of p -Coumaric acid in HCT-15 colon cancer cells. World J. Gastroenterol. 2013, 19, 7726–7734. [Google Scholar]
- Cheng, H.; Zheng, R.-R.; Fan, G.-L.; Fan, J.-H.; Zhao, L.-P.; Jiang, X.Y.; Yang, B.; Yu, X.-Y.; Li, S.-Y.; Zhang, X.-Z. Mitochondria and plasma membrane dual-targeted chimeric peptide for single-agent synergistic photodynamic therapy. Biomaterials 2018, 188, 1–11. [Google Scholar] [CrossRef]
- Morais, T.R.; Costa-Silva, T.A.; Ferreira, D.D.; Novais, B.J.; Torrecilhas, A.C.T.; Tempone, A.G.; Lago, J.H.G. Antitrypanosomal activity and effect in plasma membrane permeability of (−)-bornyl p-coumarate isolated from Piper cernuum (Piperaceae). Bioorg Chem. 2019, 89, 103001. [Google Scholar] [CrossRef]
- Amisigo, C.M.; Antwi, C.A.; Adjimani, J.P.; Gwira, T.M. In vitro anti-trypanosomal effects of selected phenolic acids on Trypanosoma brucei. PLoS ONE 2019, 14, 0216078. [Google Scholar] [CrossRef] [Green Version]
- Taladriz, A.; Healy, A.; Flores Pérez, E.J.; Herrero García, V.; Ríos Martínez, C.; Alkhaldi, A.A.M.; Dardonville, C. Synthesis and Structure–Activity Analysis of New Phosphonium Salts with Potent Activity against African Trypanosomes. J. Med. Chem. 2012, 55, 2606–2622. [Google Scholar] [CrossRef] [Green Version]
- Lima, T.C.; Souza, R.J.; Santos, A.D.C.; Moraes, M.H.; Biondo, N.E.; Barison, A.; Biavatti, M.W. Evaluation of leishmanicidal and trypanocidal activities of phenolic compounds from Calea uniflora Less. Nat. Prod. Res. 2015, 30, 551–557. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Subbarao, G.V.; Nakahara, K.; Yoshihashi, T.; Ito, O.; Maeda, I.; Yoshida, M. Nitrification Inhibitors from the root tissues of Brachiaria humidicola, a tropical grass. J. Agric. Food. Chem. 2007, 55, 1385–1388. [Google Scholar] [CrossRef]
- Zhu, F.; Xu, Z.; Yonekura, L.; Yang, R.; Tamura, H. Antiallergic activity of rosmarinic acid esters is modulated by hydrophobicity, and bulkiness of alkyl side chain. Biosci. Biotechnol. Biochem. 2015, 79, 1178–1182. [Google Scholar] [CrossRef]
- Araújo, M.O.; Pessoa, H.L.F.; Lira, A.B.; Castillo, Y.P.; De Sousa, D.P. Synthesis, Antibacterial Evaluation, and QSAR of Caffeic Acid Derivatives. J. Chem. 2019, 2019, 9. [Google Scholar] [CrossRef]
- Lira, A.B.; Montenegro, C.A.; Oliveira, K.M.; Filho, A.A.O.; Paz, A.R.; Araújo, M.O.; Sousa, D.M.; Almeida, C.L.F.; Silva, T.G.; Lima, C.M.B.L.; et al. Isopropyl Caffeate: A Caffeic Acid Derivative-Antioxidant Potential and Toxicity. Oxid. Med. Cell Longev. 2018, 2018, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gu, S.-S.; Pang, N.; Wang, F.Q.; Pang, F.; Cui, H.S.; Wu, F.A. Alkyl Caffeates Improve the Antioxidant Activity, Antitumor Property and Oxidation Stability of Edible Oil. PLoS ONE 2014, 9, 95909. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D.; Da Nóbrega, F.R.; Rushworth, S.A.; De Sousa, D.P. Trypanocidal and cysteine protease inhibitory activity of isopentyl caffeate is not linked in Trypanosoma brucei. Parasitol Res. 2016, 115, 4397–4403. [Google Scholar] [CrossRef] [PubMed]
- Andréo, R.; Regasini, L.O.; Petrônio, M.S.; Chiari-Andréo, B.G.; Tansini, A.; Silva, D.H.S.; Cicarelli, R.M.C. Toxicity and Loss of Mitochondrial Membrane Potential Induced by Alkyl Gallates in Trypanosoma cruzi. Int Sch Res Notices 2015, 2015, 7. [Google Scholar] [CrossRef]
- Serafim, R.A.M.; De Oliveira, T.F.; Loureiro, A.P.M.; Krogh, R.; Andricopulo, A.D.; Dias, L.C.; Ferreira, E.I. Molecular modeling and structure–activity relationships studies of bioisoster hybrids of N-acylhydrazone and furoxan groups on cruzain. Med. Chem. Res. 2017, 26, 760–769. [Google Scholar] [CrossRef]
- Meira, C.S.; Guimarães, E.T.; Bastos, T.M.; Moreira, D.R.; Tomassini, T.C.; Ribeiro, I.M.; Dos Santos, R.R.; Soares, M.B. Physalins B and F, seco-steroids isolated from Physalis angulate L., strongly inhibit proliferation, ultrastructure and infectivity of Trypanosoma cruzi. Parasitology 2013, 140, 1811–1821. [Google Scholar]
- Bombaça, A.C.S.; Von Dossow, D.; Barbosa, J.M.C.; Paz, C.; Burgos, V.; Menna-Barreto, R.F.S. Trypanocidal Activity of Natural Sesquiterpenoids Involves Mitochondrial Dysfunction, ROS Production and Autophagic Phenotype in Trypanosoma cruzi. Molecules 2018, 23, 2800. [Google Scholar] [CrossRef] [Green Version]
- Bertho, Á.L.; Santiago, M.A.; Coutinho, S.G. Flow cytometry in the study of cell death. Mem. Inst. Oswaldo Cruz. 2000, 95, 429–433. [Google Scholar] [CrossRef] [Green Version]
- Mello, C.P.; Lima, D.B.; Menezes, R.R.P.P.B.; Bandeira, I.C.J.; Tessarolo, L.D.; Sampaio, T.L.; Martins, A.M.C. Evaluation of the antichagasic activity of batroxicidin, a cathelicidin-related antimicrobial peptide found in Bothrops atrox venom gland. Toxicon 2017, 130, 56–62. [Google Scholar] [CrossRef]
- Ladame, S.; Castilho, M.S.; Silva, C.H.; Denier, C.; Hannaert, V.; Périé, J. Crystal structure of Trypanosoma cruzi glyceraldehyde-3-phosphate dehydrogenase complexed with an analogue of 1,3-bisphospho-d-glyceric acid. Eur. J. Biochem. 2003, 270, 4574–4586. [Google Scholar] [CrossRef] [PubMed]
- Pavão, F.; Castilho, M.S.; Pupo, M.T.; Dias, R.L.A.; Correa, A.G.; Fernandes, J.B.; da Silva, M.F.; Mafezoli, J.; Vieira, P.C.; Oliva, G. Structure of Trypanosoma cruzi glycosomal glyceraldehyde-3-phosphate dehydrogenase complexed with chalepin, a natural product inhibitor, at 1.95 Å resolution. FEBS Lett. 2002, 520, 13–17. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, A.; Truppo, E.; Supuran, C.T.; Alterio, V.; Dathan, N.; Bootorabi, F.; Parkkila, S.; Monti, S.M.; De Simone, G. Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorg. Med. Chem. 2010, 20, 5023–5026. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O.; Ruiz, F.X.; Ardèvol, A.; Domínguez, M.; Alvarez, R.; Lera, A.R. Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc. Natl. Acad. Sci. USA 2007, 104, 20764–20769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liping, Z.; Xuehua, Z.; Shangke, C.; Jing, Z. Crystal Structure of AKR1B10 Complexed with NADP+ and Caffeic Acid Phenethyl Ester. 2012. Available online: https://www.rcsb.org/structure/4GQ0 (accessed on 22 August 2012).
- Choe, Y.; Brinen, L.S.; Price, M.S.; Engel, J.C.; Lange, M.; Grisostomi, C.; Weston, S.G.; Pallai, P.V.; Cheng, H.; Hardy, L.W.; et al. Development of α-keto-based inhibitors of cruzain, a cysteine protease implicated in Chagas disease. Bioorg Med. Chem. 2005, 13, 2141–2156. [Google Scholar] [CrossRef] [PubMed]
- Persch, E.; Bryson, S.; Todoroff, N.K.; Eberle, C.; Thelemann, J.; Dirdjaja, N.; Kaiser, M.; Weber, M.; Derbani, H.; Brun, R.; et al. Binding to large enzyme pockets: Small-molecule inhibitors of trypanothione reductase. Chem. Med. Chem. 2014, 9, 1880–1891. [Google Scholar] [CrossRef]
- Khatkar, A.; Nanda, A.; Kumar, P.; Narasimhan, B. Synthesis, antimicrobial evaluation and QSAR studies of p-coumaric acid derivaties. Arab. J. Chem. 2017, 10, 3804–3815. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Takenaka, Y.; Kishi, M.; Tanahashi, T.; Hiromi Yoshida, H.; Okuda, C.; Mizushin, Y. Synthesis and DNA Polymerase α and β Inhibitory Activity of Alkyl p-Coumarates and Related Compounds. Chem. Pharm. Bull. 2009, 57, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.H.S.; Ueda-Nakamura, T.; Correâ, A.G.; Sangi, D.P.; Nakamura, C.V. A Quinoxaline Derivative as a Potent Chemotherapeutic Agent, Alone or in Combination with Benznidazole, against Trypanosoma cruzi. PLoS ONE 2014, 9, 85706. [Google Scholar] [CrossRef] [Green Version]
- Meira, C.S.; Guimarães, E.T.; Dos Santos, J.A.F.; Moreira, D.R.M.; Nogueira, R.C.; Tomassini, T.C.B.; Soares, M.B.P. In vitro and in vivo antiparasitic activity of Physalis angulata L. concentrated ethanolic extract against Trypanosoma cruzi. Phytomedicine 2015, 22, 969–974. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and citotoxicity. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Lecoeur, H.; De Oliveira-Pinto, L.M.; Gougeon, M.L. Multiparametric flow cytometric analysis of biochemical and functional events associated with apoptosis and oncosis using the 7-aminoactinomycin D assay. J. Immunol. Methods 2002, 265, 81–96. [Google Scholar] [CrossRef]
- Atale, N.; Gupta, S.; Yadav, U.C.; Rani, V. Cell-death assessment by fluorescent and nonfluorescent cytosolic and nuclear staining techniques. J. Microsc. 2014, 255, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: Forty years of application and controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef]
- Kessler, R.L.; Soares, M.J.; Probst, C.M.; Krieger, M.A. Trypanosoma cruzi Response to Sterol Biosynthesis Inhibitors: Morphophysiological Alterations Leading to Cell Death. PLoS ONE 2013, 8, 55497. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.V.; Walsh, M.L.; Chen, L.B. Localization of mitochondria in living cells with rhodamine 123. Proc. Natl. Acad. Sci. USA 1980, 77, 990–994. [Google Scholar] [CrossRef] [Green Version]
- Pokorný, J.; Pokorný, J.; Kobilková, J.; Jandová, A.; Vrba, J.J. Targeting mitochondria for cancer treatment-two types of mitochondrial dysfunction. Prague Med. Rep. 2014, 115, 104–119. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Zhang, C.; Su, Y.; Cheng, T.; Shi, C. Newly developed strategies for multifunctional mitochondria-targeted agents in cancer therapy. Drug. Discov. Today 2011, 16, 140–146. [Google Scholar] [CrossRef]
- Sampaio, T.L.; Menezes, R.R.P.P.B.; Da Costa, M.F.B.; Meneses, G.C.; Arrieta, M.C.V.; Chaves Filho, A.J.M.; de Morais, G.B.; Libório, A.B.; Alves, R.S.; Evangelista, J.S.; et al. Nephroprotective effects of (−)-α-bisabolol against ischemic-reperfusion acute kidney injury. Phytomedicine 2016, 23, 1843–1852. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Peón, A.; Naulaerts, S.; Ballester, P.J. Predicting the Reliability of Drug-target Interaction Predictions with Maximum Coverage of Target Space. Sci. Rep. 2017, 7, 3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, J.; Gohlke, B.-O.; Erehman, J.; Banerjee, P.; Rong, W.W.; Goede, A.; Preissner, R. SuperPred: Update on drug classification and target prediction. Nucl. Acids Res. 2014, 42, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucl. Acids Res. 2019, 47, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chenm, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucl. Acids Res. 2019, 47, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl. Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Bienert, S.; Waterhouse, A.; De Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository-new features and functionality. Nucl. Acids Res. 2017, 45, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. OMEGA [Internet]. Santa Fe, NM: OpenEye Scientific Software. Available online: http://www.eyesopen.com (accessed on 10 April 2019).
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking11Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.I.Y.; Ben-Shalom, S.R.; Brozell, D.S.; Cerutti, T.E.; Cheatham, V.W.D.; Cruzeiro, T.A.; Darden, R.E.; Duke, D.; Ghoreishi, M.K.; Gilson, H.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p-Coumaric Esters | |||||
|---|---|---|---|---|---|
| Y strain | Y strain | ||||
| (Epimastigotes) | (Tripomastigotes) | LLC-MK2 | |||
| IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | |
| 1 | 601.06 ± 249.17 | >561.21 | |||
| 2 | 216.06 ± 109.25 | 443.41 ± 109.25 | |||
| 3 | 16.76 ± 1.93 | 26.55 | 117.09 ± 59.15 | 3.8 | 445.55 ± 187.16 |
| 4 | 334.90 ± 54.30 | 289.71 ± 48.97 | |||
| 5 | 32.03 ± 5.39 | >14.05 | 360.42 ± 112.49 | >1.25 | >449.96 |
| 6 | 154.99 ± 33.14 | 273.89 ± 46.76 | |||
| 7 | 5.16 ± 1.28 | 23.86 | 61.63 ± 28.59 | 2 | 123.26 ± 22.62 |
| 8 | 13.23 ± 2.56 | 9.1 | 111.31 ± 38.70 | 1.08 | 120.32 ± 19.20 |
| 9 | 68.86 ± 18.12 | 131.32 ± 16.91 | |||
| 10 | 50.55 ± 18.94 | 141.39 ± 15.18 | |||
| 11 | 75.47 ± 13.41 | 160.18 ± 97.64 | |||
| 12 | 40.05 ± 10.12 | 2.74 ± 9.11 | |||
| Target | Conformer | CHEMPLP | GoldScore | ChemScore | ASP | Consensus Z-Score | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Score | Z-Score | Score | Z-Score | Score | Z-Score | Score | Z-Score | |||
| CA | 1 | 49.10 | 2.02 | 20.96 | 1.55 | 19.49 | 1.33 | 21.82 | −0.24 | 1.17 |
| AKR | 1 | 56.39 | 2.66 | 29.48 | 2.02 | 23.39 | 1.07 | 29.91 | −0.64 | 1.28 |
| 2 | 52.95 | 1.25 | 29.35 | 1.98 | 21.63 | −0.19 | 35.77 | 1.98 | 1.25 | |
| 3 | 53.55 | 1.50 | 23.25 | 0.13 | 23.85 | 1.40 | 34.20 | 1.28 | 1.08 | |
| CZ | 1 | 48.74 | 2.44 | 25.33 | 1.60 | 12.71 | 0.21 | 16.45 | 0.83 | 1.27 |
| 2 | 44.60 | 0.58 | 20.03 | −0.23 | 14.08 | 1.18 | 20.10 | 2.55 | 1.02 | |
| SOD-A | 1 | 40.44 | 1.43 | 22.70 | 1.56 | 14.33 | 1.24 | 25.08 | 1.61 | 1.46 |
| 2 | 39.44 | 1.01 | 19.10 | 0.50 | 15.20 | 1.88 | 25.10 | 1.62 | 1.25 | |
| SOD-B | 1 | 45.11 | 2.83 | 13.65 | 0.27 | 14.59 | 0.80 | 22.45 | 0.54 | 1.11 |
| TR | 1 | 48.30 | 1.53 | 24.73 | 1.31 | 17.34 | 1.76 | 23.78 | 1.25 | 1.46 |
| 2 | 45.41 | 0.57 | 24.56 | 1.26 | 15.73 | 0.25 | 26.48 | 2.44 | 1.13 | |
| GAPDH (a) | 1 | 42.90 | 2.35 | 20.61 | 1.36 | 13.26 | 1.80 | 13.79 | −0.01 | 1.37 |
| 2 | 40.97 | 1.56 | 21.16 | 1.56 | 13.01 | 1.60 | 14.72 | 0.26 | 1.24 | |
| 3 | 41.83 | 1.92 | 18.52 | 0.59 | 12.89 | 1.50 | 14.47 | 0.19 | 1.05 | |
| GAPDH+NAD (b) | 1 | 44.37 | 2.47 | 23.51 | 0.99 | 13.25 | 1.34 | 15.69 | 0.53 | 1.33 |
| Target | Conformer | MM-PBSA Component | ΔG Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Vdwaals | EEL | EPB | ENPOLAR | EDISPER | ΔG Gas | ΔG Solv | |||
| CA | 1 | −21.95 | −8.52 | 20.85 | −19.61 | 32.47 | −30.47 | 33.71 | 3.24 |
| AKR | 1 | −18.68 | −3.84 | 13.79 | −15.23 | 25.38 | −22.52 | 23.95 | 1.43 |
| 2 | −22.98 | −13.56 | 24.72 | −19.13 | 31.65 | −36.54 | 37.24 | 0.70 | |
| 3 | −25.04 | −8.37 | 17.98 | −20.41 | 32.43 | −33.41 | 30.00 | −3.42 | |
| CZ | 1 | −28.47 | −4.95 | 15.34 | −21.01 | 34.94 | −33.42 | 29.27 | −4.15 |
| 2 | −26.87 | −4.96 | 21.43 | −19.48 | 32.47 | −31.83 | 34.43 | 2.60 | |
| SOD-A | 1 | −34.61 | −14.76 | 35.23 | −25.32 | 42.74 | −49.37 | 52.64 | 3.27 |
| 2 | −33.36 | −13.14 | 33.30 | −24.53 | 42.19 | −46.51 | 50.96 | 4.45 | |
| SOD-B | 1 | −32.10 | −22.17 | 47.67 | −25.49 | 43.36 | −54.27 | 65.54 | 11.27 |
| TR | 1 | −24.51 | −9.66 | 21.91 | −20.20 | 32.74 | −34.17 | 34.45 | 0.28 |
| 2 | −22.75 | −3.72 | 15.00 | −18.36 | 30.81 | −26.47 | 27.45 | 0.97 | |
| CZ | AKR | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Residue | Van der Waals | Electrostatic | Polar Solvation | ΔG TOTAL | Residue | Van der Waals | Electrostatic | Polar Solvation | ΔG TOTAL |
| W26 | −0.98 | −0.65 | 0.52 | −1.11 | W81 | −1.45 | −0.28 | 0.34 | −1.39 |
| G65 | −0.76 | −1.33 | 1.17 | −0.92 | W25 | −1.84 | 0.33 | 0.24 | −1.28 |
| H162 | −1.39 | −0.27 | 0.84 | −0.82 | P279 | −0.97 | 0.09 | −0.11 | −1.00 |
| L160 | −0.97 | 0.10 | 0.25 | −0.62 | F277 | −1.31 | 0.08 | 0.26 | −0.97 |
| A138 | −0.45 | 0.06 | −0.15 | −0.54 | W189 | −0.74 | −0.11 | 0.01 | −0.84 |
| L67 | −0.79 | 0.24 | 0.03 | −0.52 | I53 | −0.95 | −0.25 | 0.50 | −0.70 |
| C25 | −0.76 | −0.05 | 0.31 | −0.50 | W113 | −0.97 | −0.97 | 1.54 | −0.41 |
| M68 | −0.54 | 0.03 | 0.09 | −0.42 | A272 | −0.46 | 0.03 | 0.03 | −0.41 |
| G23 | −0.97 | 0.16 | 0.40 | −0.42 | H112 | −0.51 | −1.70 | 1.97 | −0.24 |
| G163 | −0.48 | 0.06 | 0.05 | −0.37 | Y54 | −0.36 | −0.76 | 1.20 | 0.08 |
| S64 | −0.90 | −0.16 | 0.75 | −0.32 | R26 | −0.21 | 0.27 | 0.05 | 0.11 |
| C22 | −0.23 | −0.11 | 0.06 | −0.28 | G271 | −0.44 | 0.05 | 0.77 | 0.38 |
| E208 | −0.36 | −0.17 | 0.39 | −0.14 | |||||
| Q159 | −0.11 | −0.04 | 0.06 | −0.09 | |||||
| D161 | −1.55 | −0.54 | 2.06 | −0.03 | |||||
| N69 | −0.04 | 0.00 | 0.06 | 0.01 | |||||
| Q21 | −0.12 | −0.05 | 0.19 | 0.01 | |||||
| C63 | −0.39 | −0.15 | 0.62 | 0.08 | |||||
| Q19 | −0.17 | 0.21 | 0.15 | 0.19 | |||||
| G66 | −0.80 | −0.46 | 2.02 | 0.76 | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, S.P.; Castillo, Y.P.; Monteiro, M.L.; Menezes, R.R.P.P.B.d.; Almeida, R.N.; Martins, A.M.C.; Sousa, D.P.d. Trypanocidal Mechanism of Action and in silico Studies of p-Coumaric Acid Derivatives. Int. J. Mol. Sci. 2019, 20, 5916. https://doi.org/10.3390/ijms20235916
Lopes SP, Castillo YP, Monteiro ML, Menezes RRPPBd, Almeida RN, Martins AMC, Sousa DPd. Trypanocidal Mechanism of Action and in silico Studies of p-Coumaric Acid Derivatives. International Journal of Molecular Sciences. 2019; 20(23):5916. https://doi.org/10.3390/ijms20235916
Chicago/Turabian StyleLopes, Susiany P., Yunierkis P. Castillo, Marilia L. Monteiro, Ramon R. P. P. B. de Menezes, Reinaldo N. Almeida, Alice M. C. Martins, and Damião P. de Sousa. 2019. "Trypanocidal Mechanism of Action and in silico Studies of p-Coumaric Acid Derivatives" International Journal of Molecular Sciences 20, no. 23: 5916. https://doi.org/10.3390/ijms20235916