Fluorescent Protein-Based Indicators for Functional Super-Resolution Imaging of Biomolecular Activities in Living Cells

Abstract
:1. Introduction
2. Sense and Sensibility: Anatomy of Genetically Encoded Indicators
3. A Tale of Two Cities: Phototransformable Indicators for Super-Resolution Fluorescence Microscopy
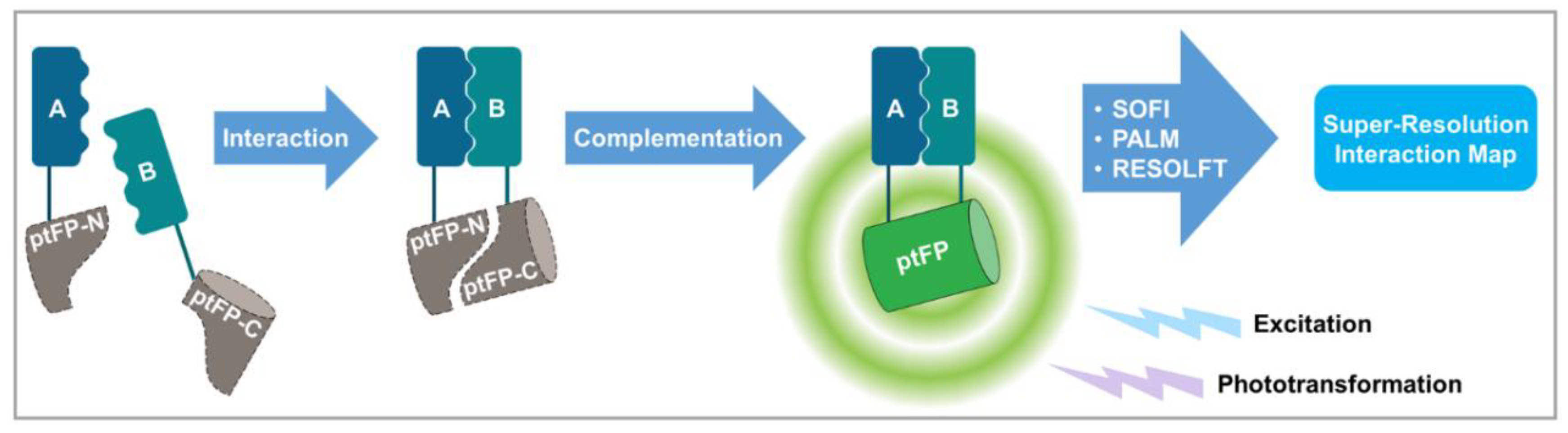
3.1. Diffraction-Unlimited Observation of Protein–Protein interactions by Bimolecular Fluorescence Complementation (BiFC) of Split Phototransformable Fluorescent Proteins
3.2. Diffraction-Unlimited Observation of Biomolecule Interactions by Points Accumulation for Imaging Nanoscale Topography (PAINT) with Photoconvertible Fluorescent Proteins
3.3. Diffraction-Unlimited Observation of Biomolecular Activities Based on Fluorescence Fluctuation Increase by Contact (FLINC)
4. Through the Looking Glass: Exploiting Phototransformation of Fluorescent Proteins for Improved Bioimaging and Biomolecular Sensing
5. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| a.a. | amino acid |
| 3B | Bayesian analysis of the blinking and bleaching |
| BiFC | bimolecular fluorescence complementation |
| cp | circular permutation |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| EGF | epidermal growth factor |
| ESI | entropy-based super-resolution imaging |
| FKBP | FK506-binding protein |
| FLINC | fluorescence fluctuation increase by contact |
| FP | fluorescent protein |
| FRB | FKBP-rapamycin binding domain |
| FRC | Fourier ring correlation |
| FRET | Förster resonance energy transfer |
| FWHM | full width at half maximum |
| GFP | Aequorea Victoria green fluorescence protein |
| HBR | hydroxybenzylidene rhodanine |
| HILO | highly inclined and laminated optical sheet |
| JF646 | Janelia Fluor 646 |
| MUSICAL | multiple signal classification algorithm |
| N/A | not applicable |
| PAINT | points accumulation for imaging nanoscale topography |
| PALM | photo-activated localization microscopy |
| PKA | protein kinase A |
| Pol II | RNA polymerase II |
| PPI | Protein-protein interaction |
| PSF | point spread function |
| psFRET | photoswitching FRET |
| ptFP | phototransformable fluorescent protein |
| PYP | photoactive yellow protein |
| RESOLFT | reversible saturable optical fluorescence transitions |
| S/N | signal-to-noise ratio |
| SCORE | spatial covariance reconstructive |
| SIM | structured illumination microscopy |
| sm-FRET | single-molecule photoactivation FRET |
| SMLM | single-molecule localization microscopy |
| SOFI | super-resolution optical fluctuation imaging |
| SRM | super-resolution light microscopy |
| SRRF | super-resolution radial fluctuations |
| STED | stimulated emission depletion |
| TALEN | transcription activator-like effector nucleases |
| tcPALM | time-correlated photo-activated localization microscopy |
| TFFC | three-fragment fluorescence complementation |
| TIRF | total internal reflection fluorescence |
| Tet | tetracycline |
| UV | ultraviolet |
References
- Bogdanov, A.M.; Mishin, A.S.; Yampolsky, I.V.; Belousov, V.V.; Chudakov, D.M.; Subach, F.V.; Verkhusha, V.V.; Lukyanov, S.; Lukyanov, K.A. Green fluorescent proteins are light-induced electron donors. Nat. Chem. Biol. 2009, 5, 459–461. [Google Scholar] [CrossRef]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Prendergast, F.G.; Mann, K.G. Chemical and physical properties of aequorin and the green fluorescent protein isolated from Aequorea forskalea. Biochemistry 1978, 17, 3448–3453. [Google Scholar] [CrossRef]
- Duwé, S.; Dedecker, P. Optimizing the fluorescent protein toolbox and its use. Curr. Opin. Biotechnol. 2019, 58, 183–191. [Google Scholar] [CrossRef]
- Xue, L.; Karpenko, I.A.; Hiblot, J.; Johnsson, K. Imaging and manipulating proteins in live cells through covalent labeling. Nat. Chem. Biol. 2015, 11, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.H.; Fosbrink, M.D.; Zhang, J. Genetically encodable fluorescent biosensors for tracking signaling dynamics in living cells. Chem. Rev. 2011, 111, 3614–3666. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, E.C.; Mehta, S.; Zhang, J. Genetically encoded fluorescent biosensors illuminate the spatiotemporal regulation of signaling networks. Chem. Rev. 2018, 118, 11707–11794. [Google Scholar] [CrossRef]
- Abbe, E. Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch. Mikrosk. Anat. 1873, 9, 413–468. [Google Scholar] [CrossRef]
- Yan, R.; Wang, B.; Xu, K. Functional super-resolution microscopy of the cell. Curr. Opin. Chem. Biol. 2019, 51, 92–97. [Google Scholar] [CrossRef]
- Gould, T.J.; Gunewardene, M.S.; Gudheti, M.V.; Verkhusha, V.V.; Yin, S.R.; Gosse, J.A.; Hess, S.T. Nanoscale imaging of molecular positions and anisotropies. Nat. Methods 2008, 5, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.A.V.; Shaban, H.A.; Kress, A.; Bertaux, N.; Monneret, S.; Mavrakis, M.; Savatier, J.; Brasselet, S. Quantitative nanoscale imaging of orientational order in biological filaments by polarized superresolution microscopy. Proc. Natl. Acad. Sci. USA 2016, 113, E820–E828. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Schneider, F.; Zilles, V.; Urbančič, I.; Garcia, E.; Waithe, D.; Klymchenko, A.S.; Eggeling, C. Polarity-sensitive probes for superresolution stimulated emission depletion microscopy. Biophys. J. 2017, 113, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, M.N.; Godet, J.; Horrocks, M.H.; Tosatto, L.; Carr, A.R.; Wirthensohn, D.C.; Ranasinghe, R.T.; Lee, J.-E.; Ponjavic, A.; Fritz, J.V.; et al. Multi-dimensional super-resolution imaging enables surface hydrophobicity mapping. Nat. Commun. 2016, 7, 13544. [Google Scholar] [CrossRef]
- Horikawa, K.; Yamada, Y.; Matsuda, T.; Kobayashi, K.; Hashimoto, M.; Matsu-ura, T.; Miyawaki, A.; Michikawa, T.; Mikoshiba, K.; Nagai, T. Spontaneous network activity visualized by ultrasensitive Ca2+ indicators, yellow Cameleon-Nano. Nat. Methods 2010, 7, 729–732. [Google Scholar] [CrossRef]
- Farhana, I.; Hossain, M.N.; Suzuki, K.; Matsuda, T.; Nagai, T. Genetically encoded fluorescence/bioluminescence bimodal indicators for Ca2+ imaging. ACS Sens. 2019, 4, 1825–1834. [Google Scholar] [CrossRef]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005, 23, 102–107. [Google Scholar] [CrossRef]
- Romoser, V.A.; Hinkle, P.M.; Persechini, A. Detection in living cells of Ca2+-dependent changes in the fluorescence emission of an indicator composed of two green fluorescent protein variants linked by a calmodulin-binding sequence. A new class of fluorescent indicators. J. Biol. Chem. 1997, 272, 13270–13274. [Google Scholar] [CrossRef]
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001, 276, 29188–29194. [Google Scholar] [CrossRef]
- Wachter, R.M.; Elsliger, M.A.; Kallio, K.; Hanson, G.T.; Remington, S.J. Structural basis of spectral shifts in the yellow-emission variants of green fluorescent protein. Structure 1998, 6, 1267–1277. [Google Scholar] [CrossRef]
- Tojima, T.; Akiyama, H.; Itofusa, R.; Li, Y.; Katayama, H.; Miyawaki, A.; Kamiguchi, H. Attractive axon guidance involves asymmetric membrane transport and exocytosis in the growth cone. Nat. Neurosci. 2007, 10, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Hell, S.W. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 1999, 24, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Stephan, T.; Roesch, A.; Riedel, D.; Jakobs, S. Live-cell STED nanoscopy of mitochondrial cristae. Sci. Rep. 2019, 9, 12419. [Google Scholar] [CrossRef]
- Kamper, M.; Ta, H.; Jensen, N.A.; Hell, S.W.; Jakobs, S. Near-infrared STED nanoscopy with an engineered bacterial phytochrome. Nat. Commun. 2018, 9, 4762. [Google Scholar] [CrossRef]
- Mishina, N.M.; Mishin, A.S.; Belyaev, Y.; Bogdanova, E.A.; Lukyanov, S.; Schultz, C.; Belousov, V.V. Live-cell STED microscopy with genetically encoded biosensor. Nano Lett. 2015, 15, 2928–2932. [Google Scholar] [CrossRef]
- Boudreau, C.; Wee, T.L.; Duh, Y.R.; Couto, M.P.; Ardakani, K.H.; Brown, C.M. Excitation light dose engineering to reduce photo-bleaching and photo-toxicity. Sci. Rep. 2016, 6, 30892. [Google Scholar] [CrossRef]
- Wäldchen, S.; Lehmann, J.; Klein, T.; van de Linde, S.; Sauer, M. Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 2015, 5, 15348. [Google Scholar] [CrossRef]
- Oliinyk, O.S.; Shemetov, A.A.; Pletnev, S.; Shcherbakova, D.M.; Verkhusha, V.V. Smallest near-infrared fluorescent protein evolved from cyanobacteriochrome as versatile tag for spectral multiplexing. Nat. Commun. 2019, 10, 279. [Google Scholar] [CrossRef]
- Shcherbakova, D.M.; Baloban, M.; Emelyanov, A.V.; Brenowitz, M.; Guo, P.; Verkhusha, V.V. Bright monomeric near-infrared fluorescent proteins as tags and biosensors for multiscale imaging. Nat. Commun. 2016, 7, 12405. [Google Scholar] [CrossRef]
- Rodriguez, E.A.; Tran, G.N.; Gross, L.A.; Crisp, J.L.; Shu, X.; Lin, J.Y.; Tsien, R.Y. A far-red fluorescent protein evolved from a cyanobacterial phycobiliprotein. Nat. Methods 2016, 13, 763–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, J.M. Super-resolution through illumination by diffraction-born evanescent waves. Appl. Phys. Lett. 1995, 66, 3555–3557. [Google Scholar] [CrossRef]
- Hess, S.T.; Girirajan, T.P.K.; Mason, M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedecker, P.; Mo, G.C.H.; Dertinger, T.; Zhang, J. Widely accessible method for superresolution fluorescence imaging of living systems. Proc. Natl. Acad. Sci. USA 2012, 109, 10909–10914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, N.; Culley, S.; Ashdown, G.; Owen, D.M.; Pereira, P.M.; Henriques, R. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat. Commun. 2016, 7, 12471. [Google Scholar] [CrossRef] [Green Version]
- Cox, S.; Rosten, E.; Monypenny, J.; Jovanovic-Talisman, T.; Burnette, D.T.; Lippincott-Schwartz, J.; Jones, G.E.; Heintzmann, R. Bayesian localization microscopy reveals nanoscale podosome dynamics. Nat. Methods 2012, 9, 195–200. [Google Scholar] [CrossRef]
- Yahiatene, I.; Hennig, S.; Müller, M.; Huser, T. Entropy-based super-resolution imaging (ESI): From disorder to fine detail. ACS Photonics 2015, 2, 1049–1056. [Google Scholar] [CrossRef]
- Deng, Y.; Sun, M.; Lin, P.H.; Ma, J.; Shaevitz, J.W. Spatial covariance reconstructive (SCORE) super-resolution fluorescence microscopy. PLoS ONE 2014, 9, e94807. [Google Scholar] [CrossRef]
- Agarwal, K.; Macháň, R. Multiple signal classification algorithm for super-resolution fluorescence microscopy. Nat. Commun. 2016, 7, 13752. [Google Scholar] [CrossRef] [Green Version]
- Holden, S.J.; Uphoff, S.; Kapanidis, A.N. DAOSTORM: An algorithm for high-density super-resolution microscopy. Nat. Methods 2011, 8, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Ferrand, A.; Huser, T.; Eggeling, C.; Sauer, M.; Biehlmaier, O.; Drummen, G.P.C. Super-resolution microscopy demystified. Nat. Cell Biol. 2019, 21, 72. [Google Scholar] [CrossRef] [PubMed]
- Cloin, B.M.C.; Zitter, E.D.; Salas, D.; Gielen, V.; Folkers, G.E.; Mikhaylova, M.; Bergeler, M.; Krajnik, B.; Harvey, J.; Hoogenraad, C.C.; et al. Efficient switching of mCherry fluorescence using chemical caging. Proc. Natl. Acad. Sci. USA 2017, 114, 7013–7018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterflood, C.M.; Ewers, H. Single-molecule localization microscopy using mCherry. ChemPhysChem 2014, 15, 3447–3451. [Google Scholar] [CrossRef]
- Tiwari, D.K.; Nagai, T. Smart fluorescent proteins: Innovation for barrier-free superresolution imaging in living cells. Dev. Growth Differ. 2013, 55, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Takauchi, H.; Ogami, Y.; Fujiwara, S.; Nakano, M.; Matsuda, T.; Nagai, T. Spontaneously blinking fluorescent protein for simple single laser super-resolution live cell imaging. ACS Chem. Biol. 2018, 13, 1938–1943. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, H.; Lu, K.; Nakashima, R.; Wazawa, T.; Noguchi, K.; Matsuda, T.; Nagai, T. Acid-tolerant reversibly switchable green fluorescent protein for super-resolution imaging under acidic conditions. Cell Chem. Biol. 2019. [Google Scholar] [CrossRef]
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877. [Google Scholar] [CrossRef]
- Subach, F.V.; Patterson, G.H.; Manley, S.; Gillette, J.M.; Lippincott-Schwartz, J.; Verkhusha, V.V. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat. Methods 2009, 6, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Wiedenmann, J.; Ivanchenko, S.; Oswald, F.; Schmitt, F.; Röcker, C.; Salih, A.; Spindler, K.D.; Nienhaus, G.U. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc. Natl. Acad. Sci. USA 2004, 101, 15905–15910. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Chang, H.; Zhang, Y.; Yu, J.; Wu, L.; Ji, W.; Chen, J.; Liu, B.; Lu, J.; Liu, Y.; et al. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat. Methods 2012, 9, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Brakemann, T.; Stiel, A.C.; Weber, G.; Andresen, M.; Testa, I.; Grotjohann, T.; Leutenegger, M.; Plessmann, U.; Urlaub, H.; Eggeling, C.; et al. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat. Biotechnol. 2011, 29, 942–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andresen, M.; Stiel, A.C.; Fölling, J.; Wenzel, D.; Schönle, A.; Egner, A.; Eggeling, C.; Hell, S.W.; Jakobs, S. Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy. Nat. Biotechnol. 2008, 26, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Ando, R.; Mizuno, H.; Miyawaki, A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 2004, 306, 1370–1373. [Google Scholar] [CrossRef]
- Acharya, A.; Bogdanov, A.M.; Grigorenko, B.L.; Bravaya, K.B.; Nemukhin, A.V.; Lukyanov, K.A.; Krylov, A.I. Photoinduced chemistry in fluorescent proteins: Curse or blessing? Chem. Rev. 2017, 117, 758–795. [Google Scholar] [CrossRef]
- Isaacoff, B.P.; Li, Y.; Lee, S.A.; Biteen, J.S. SMALL-LABS: Measuring single-molecule intensity and position in obscuring backgrounds. Biophys. J. 2019, 116, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Geissbuehler, S.; Bocchio, N.L.; Dellagiacoma, C.; Berclaz, C.; Leutenegger, M.; Lasser, T. Mapping molecular statistics with balanced super-resolution optical fluctuation imaging (bSOFI). Opt. Nanoscopy 2012, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Hertel, F.; Mo, G.C.H.; Duwé, S.; Dedecker, P.; Zhang, J. RefSOFI for mapping nanoscale organization of protein-protein interactions in living cells. Cell Rep. 2016, 14, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Nickerson, A.; Huang, T.; Lin, L.J.; Nan, X. Photoactivated localization microscopy with bimolecular fluorescence complementation (BiFC-PALM) for nanoscale imaging of protein-protein interactions in cells. PLoS ONE 2014, 9, e100589. [Google Scholar] [CrossRef]
- Liu, Z.; Xing, D.; Su, Q.P.; Zhu, Y.; Zhang, J.; Kong, X.; Xue, B.; Wang, S.; Sun, H.; Tao, Y.; et al. Super-resolution imaging and tracking of protein–protein interactions in sub-diffraction cellular space. Nat. Commun. 2014, 5, 4443. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Liu, X.; Wu, B.; Zhang, S.; Song, X.; Yao, P.Y.; Lippincott-Schwartz, J.; Yao, X.; Zheng, Y. Superresolution imaging reveals structural features of EB1 in microtubule plus-end tracking. Mol. Biol. Cell 2014, 25, 4166–4173. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, S.; Li, W.; Zhang, Z.; Zhang, X.; Zhang, X.-E.; Cui, Z. Three-fragment fluorescence complementation coupled with photoactivated localization microscopy for nanoscale imaging of ternary complexes. ACS Nano 2016, 10, 8482–8490. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ding, M.; Chen, X.; Chang, L.; Sun, Y. Development of bimolecular fluorescence complementation using rsEGFP2 for detection and super-resolution imaging of protein-protein interactions in live cells. Biomed. Opt. Express 2017, 8, 3119–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebo, A.G.; Gautier, A. A split fluorescent reporter with rapid and reversible complementation. Nat. Commun. 2019, 10, 2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharonov, A.; Hochstrasser, R.M. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. USA 2006, 103, 18911–18916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungmann, R.; Steinhauer, C.; Scheible, M.; Kuzyk, A.; Tinnefeld, P.; Simmel, F.C. Single-molecule kinetics and super-resolution microscopy by fluorescence imaging of transient binding on DNA origami. Nano Lett. 2010, 10, 4756–4761. [Google Scholar] [CrossRef]
- De Keersmaecker, H.; Camacho, R.; Rantasa, D.M.; Fron, E.; Uji-i, H.; Mizuno, H.; Rocha, S. Mapping transient protein interactions at the nanoscale in living mammalian cells. ACS Nano 2018, 12, 9842–9854. [Google Scholar] [CrossRef]
- Etheridge, T.J.; Boulineau, R.L.; Herbert, A.; Watson, A.T.; Daigaku, Y.; Tucker, J.; George, S.; Jönsson, P.; Palayret, M.; Lando, D.; et al. Quantification of DNA-associated proteins inside eukaryotic cells using single-molecule localization microscopy. Nucleic Acids Res. 2014, 42, e146. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, Z.; Li, L.; Chen, B.C.; Revyakin, A.; Hajj, B.; Legant, W.; Dahan, M.; Lionnet, T.; Betzig, E.; et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 2014, 156, 1274–1285. [Google Scholar] [CrossRef] [Green Version]
- Cisse, I.I.; Izeddin, I.; Causse, S.Z.; Boudarene, L.; Senecal, A.; Muresan, L.; Dugast-Darzacq, C.; Hajj, B.; Dahan, M.; Darzacq, X. Real-time dynamics of RNA polymerase II clustering in live human cells. Science 2013, 341, 664–667. [Google Scholar] [CrossRef]
- Cho, W.K.; Jayanth, N.; English, B.P.; Inoue, T.; Andrews, J.O.; Conway, W.; Grimm, J.B.; Spille, J.H.; Lavis, L.D.; Lionnet, T.; et al. RNA polymerase II cluster dynamics predict mRNA output in living cells. eLife 2016, 5, e13617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loveland, A.B.; Habuchi, S.; Walter, J.C.; van Oijen, A.M. A general approach to break the concentration barrier in single-molecule imaging. Nat. Methods 2012, 9, 987–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, S.; Sun, R.; Wang, W.; Chen, C. Single-molecule photoactivation FRET: A general and easy-to-implement approach to break the concentration barrier. Angew. Chem. Int. Ed. Engl. 2017, 56, 6882–6885. [Google Scholar] [CrossRef] [PubMed]
- Fox-Roberts, P.; Marsh, R.; Pfisterer, K.; Jayo, A.; Parsons, M.; Cox, S. Local dimensionality determines imaging speed in localization microscopy. Nat. Commun. 2017, 8, 13558. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.M.; Sauer, M.; Gaus, K. Fluorescence localization microscopy. Commun. Integr. Biol. 2012, 5, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Banterle, N.; Bui, K.H.; Lemke, E.A.; Beck, M. Fourier ring correlation as a resolution criterion for super-resolution microscopy. J. Struct. Biol. 2013, 183, 363–367. [Google Scholar] [CrossRef]
- Mo, G.C.H.; Ross, B.; Hertel, F.; Manna, P.; Yang, X.; Greenwald, E.; Booth, C.; Plummer, A.M.; Tenner, B.; Chen, Z.; et al. Genetically encoded biosensors for visualizing live-cell biochemical activity at super-resolution. Nat. Methods 2017, 14, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Dickson, R.M.; Cubitt, A.B.; Tsien, R.Y.; Moerner, W.E. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 1997, 388, 355–358. [Google Scholar] [CrossRef]
- Tiwari, D.K.; Arai, Y.; Yamanaka, M.; Matsuda, T.; Agetsuma, M.; Nakano, M.; Fujita, K.; Nagai, T. A fast- and positively photoswitchable fluorescent protein for ultralow-laser-power RESOLFT nanoscopy. Nat. Methods 2015, 12, 515–518. [Google Scholar] [CrossRef]
- Wazawa, T.; Arai, Y.; Kawahara, Y.; Takauchi, H.; Washio, T.; Nagai, T. Highly biocompatible super-resolution fluorescence imaging using the fast photoswitching fluorescent protein Kohinoor and SPoD-ExPAN with Lp-regularized image reconstruction. Microscopy 2018, 67, 89–98. [Google Scholar]
- Hafi, N.; Grunwald, M.; van den Heuvel, L.S.; Aspelmeier, T.; Chen, J.-H.; Zagrebelsky, M.; Schütte, O.M.; Steinem, C.; Korte, M.; Munk, A.; et al. Fluorescence nanoscopy by polarization modulation and polarization angle narrowing. Nat. Methods 2014, 11, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rainey, K.H.; Patterson, G.H. Photoswitching FRET to monitor protein–protein interactions. Proc. Natl. Acad. Sci. USA 2019, 116, 864–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fosque, B.F.; Sun, Y.; Dana, H.; Yang, C.-T.; Ohyama, T.; Tadross, M.R.; Patel, R.; Zlatic, M.; Kim, D.S.; Ahrens, M.B.; et al. Labeling of active neural circuits in vivo with designed calcium integrators. Science 2015, 347, 755–760. [Google Scholar] [CrossRef] [PubMed]
- McKinney, S.A.; Murphy, C.S.; Hazelwood, K.L.; Davidson, M.W.; Looger, L.L. A bright and photostable photoconvertible fluorescent protein. Nat. Methods 2009, 6, 131–133. [Google Scholar] [CrossRef]
- Zolnik, T.A.; Sha, F.; Johenning, F.W.; Schreiter, E.R.; Looger, L.L.; Larkum, M.E.; Sachdev, R.N.S. All-optical functional synaptic connectivity mapping in acute brain slices using the calcium integrator CaMPARI. J. Physiol. 2017, 595, 1465–1477. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Needham, L.-M.; Lando, D.; Taylor, E.J.R.; Wohlfahrt, K.J.; Shah, D.; Boucher, W.; Tan, Y.L.; Bates, L.E.; Tkachenko, O.; et al. FRET-enhanced photostability allows improved single-molecule tracking of proteins and protein complexes in live mammalian cells. Nat. Commun. 2018, 9, 2520. [Google Scholar] [CrossRef] [Green Version]
- Duwé, S.; De Zitter, E.; Gielen, V.; Moeyaert, B.; Vandenberg, W.; Grotjohann, T.; Clays, K.; Jakobs, S.; Van Meervelt, L.; Dedecker, P. Expression-enhanced fluorescent proteins based on enhanced green fluorescent protein for super-resolution microscopy. ACS Nano 2015, 9, 9528–9541. [Google Scholar] [CrossRef] [Green Version]
- Roebroek, T.; Duwé, S.; Vandenberg, W.; Dedecker, P. Reduced fluorescent protein switching fatigue by binding-induced emissive state stabilization. Int. J. Mol. Sci. 2017, 18, 2015. [Google Scholar] [CrossRef] [Green Version]
- Kirchhofer, A.; Helma, J.; Schmidthals, K.; Frauer, C.; Cui, S.; Karcher, A.; Pellis, M.; Muyldermans, S.; Casas-Delucchi, C.S.; Cardoso, M.C.; et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 2010, 17, 133–138. [Google Scholar] [CrossRef]
- Eshaghi, M.; Sun, G.; Grüter, A.; Lim, C.L.; Chee, Y.C.; Jung, G.; Jauch, R.; Wohland, T.; Chen, S.L. Rational structure-based design of bright GFP-based complexes with tunable dimerization. Angew. Chem. Int. Ed. Engl. 2015, 54, 13952–13956. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Application | Name | Target | Class | FP | FP Config. | Split Site(s) | Microscopy | Localization Precision 1 | FWHM 2 | Kinetics | Reversibility | Cell Type(s) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rapamycin-induced FKBP and FRB interaction | refSOFI | Protein-protein interaction | BiFC | DMVF | cp; split | a.a. 181 | SOFI | N/A | ~100 nm | Slow 3 | No | HeLa | [58] |
| Interaction of receptor tyrosine kinases HER2 & 3 | refSOFI | Protein-protein interaction | BiFC | DMVF | cp; split | a.a. 181 | SOFI | N/A | ~100 nm | Slow 3 | No | HeLa | [58] |
| Interaction of ER Ca2+ sensor STIM1 and Ca2+ channel protein ORAI1 | refSOFI | Protein-protein interaction | BiFC | DMVF | cp; split | a.a. 181 | SOFI | N/A | ~100 nm | Slow 3 | No | HeLa | [58] |
| Interactions of small GTPase Ras and its effector Raf | BiFC-PALM | Protein-protein interaction | BiFC | PA-mCherry1 | split | a.a. 159 | PALM | 18 nm | N/A | Slow 3 | No | U2OS | [59] |
| Interaction of MreB and EF-Tu | BiFC-PALM | Protein-protein interaction | BiFC | mEos3.2 | split | a.a. 164 | PALM | 12 nm | N/A | Slow 3 | No | E. coli | [60] |
| Homodimerization of microtubule plus-end hub protein EB1 | BiFC-PALM | Protein-protein interaction | BiFC | PA-GFP | split | not specified | PALM | 23 nm | N/A | Slow 3 | No | HeLa; MCF7 | [61] |
| Formation of bJun/bFos complexes | BiFC-PALM | Protein-protein interaction | BiFC | mIrisFP | split | a.a. 150; a.a. 165 | PALM | 18 nm | N/A | Slow 3 | No | Vero cells | [62] |
| Interaction among αs, β1, and γ2 subunits of Gs ternary complex | TFFC-PALM | Protein-protein interaction | TFFC 4 | mIrisFP | split | a.a. 150 & a.a. 165 | PALM | 18 nm | N/A | Slow 3 | No | Vero cells | [62] |
| Interaction of Bcl-xL and Bak | BiFC-RESOLFT | Protein-protein interaction | BiFC | rsEGFP2 | split | a.a. 158 | RESOLFT | N/A | 113 nm | Slow 3 | No | Hela | [63] |
| Membrane-binding of proteins in EGF signaling pathway | PAINT-PALM | Proteinprotein interaction | PAINT | mEos3.2 | default | N/A | PALM | 35 nm | N/A | fast | Yes 5 | HeLa; CHO | [67] |
| Binding of PCNA and Mcm4 proteins to genomic DNA | PAINT-PALM | Protein-DNA interaction | PAINT | mEos3.1 | default | N/A | PALM | 11 nm | N/A | fast | Yes 5 | fission yeast | [68] |
| Dynamics of RNA Pol II clustering at β-actin gene locus | PAINT-PALM | Protein-RNA interaction | PAINT | Dendra2 | default | N/A | PALM | 31 nm | N/A | fast | Yes 5 | MEF | [71] |
| Protein Kinase A (PKA) activity | FLINC-AKAR1 | Biochemical activity | FLINC | TagRFP-T | default | N/A | SOFI | N/A | 107–179 nm | fast | Yes | HeLa; α4CHO | [77] |
| Extracellular signal-regulated kinase (ERK) activity | FLINC-EKAR1 | Biochemical activity | FLINC | TagRFP-T | default | N/A | SOFI | N/A | 160 nm | fast | Yes | HEK293 | [77] |
| Rapamycin-induced FKBP and FRB interaction | bimolecular FLINC | Protein-protein interaction | FLINC | TagRFP-T | default | N/A | SOFI | N/A | ~107–160 nm | fast | Yes | HeLa | [77] |
| Interaction of FHA1 and PKA phosphosubstrate | bimolecular FLINC-AKAR1 | Protein–protein interaction | FLINC | TagRFP-T | default | N/A | SOFI | N/A | ~107–160 nm | fast | Yes | HeLa | [77] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, K.; Vu, C.Q.; Matsuda, T.; Nagai, T. Fluorescent Protein-Based Indicators for Functional Super-Resolution Imaging of Biomolecular Activities in Living Cells. Int. J. Mol. Sci. 2019, 20, 5784. https://doi.org/10.3390/ijms20225784
Lu K, Vu CQ, Matsuda T, Nagai T. Fluorescent Protein-Based Indicators for Functional Super-Resolution Imaging of Biomolecular Activities in Living Cells. International Journal of Molecular Sciences. 2019; 20(22):5784. https://doi.org/10.3390/ijms20225784
Chicago/Turabian StyleLu, Kai, Cong Quang Vu, Tomoki Matsuda, and Takeharu Nagai. 2019. "Fluorescent Protein-Based Indicators for Functional Super-Resolution Imaging of Biomolecular Activities in Living Cells" International Journal of Molecular Sciences 20, no. 22: 5784. https://doi.org/10.3390/ijms20225784