STAT3 Regulates the Type I IFN-Mediated Antiviral Response by Interfering with the Nuclear Entry of STAT1

Abstract
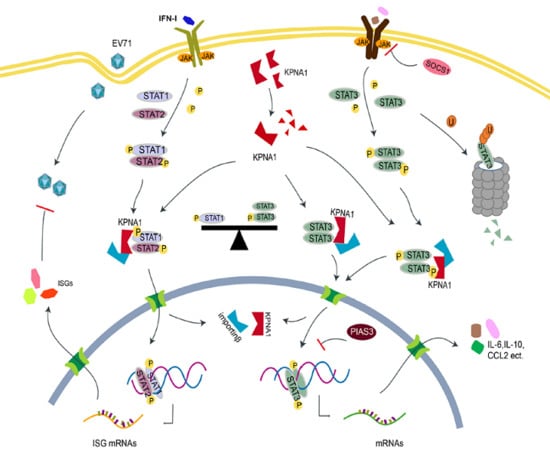
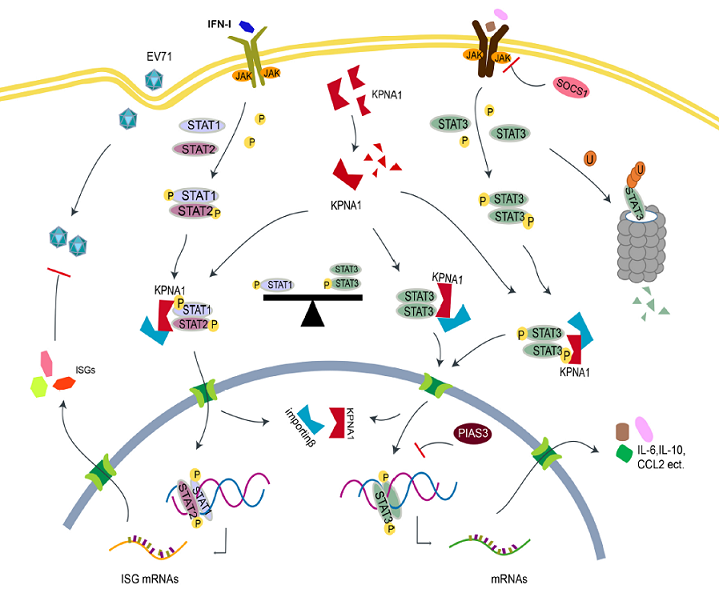{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. EV71 Infection Induces Activation of STAT3
2.2. STAT3 Targets are Upregulated Early and Downregulated Later during Infection
2.3. Cellular STAT3 Positively Regulates EV71 Infection
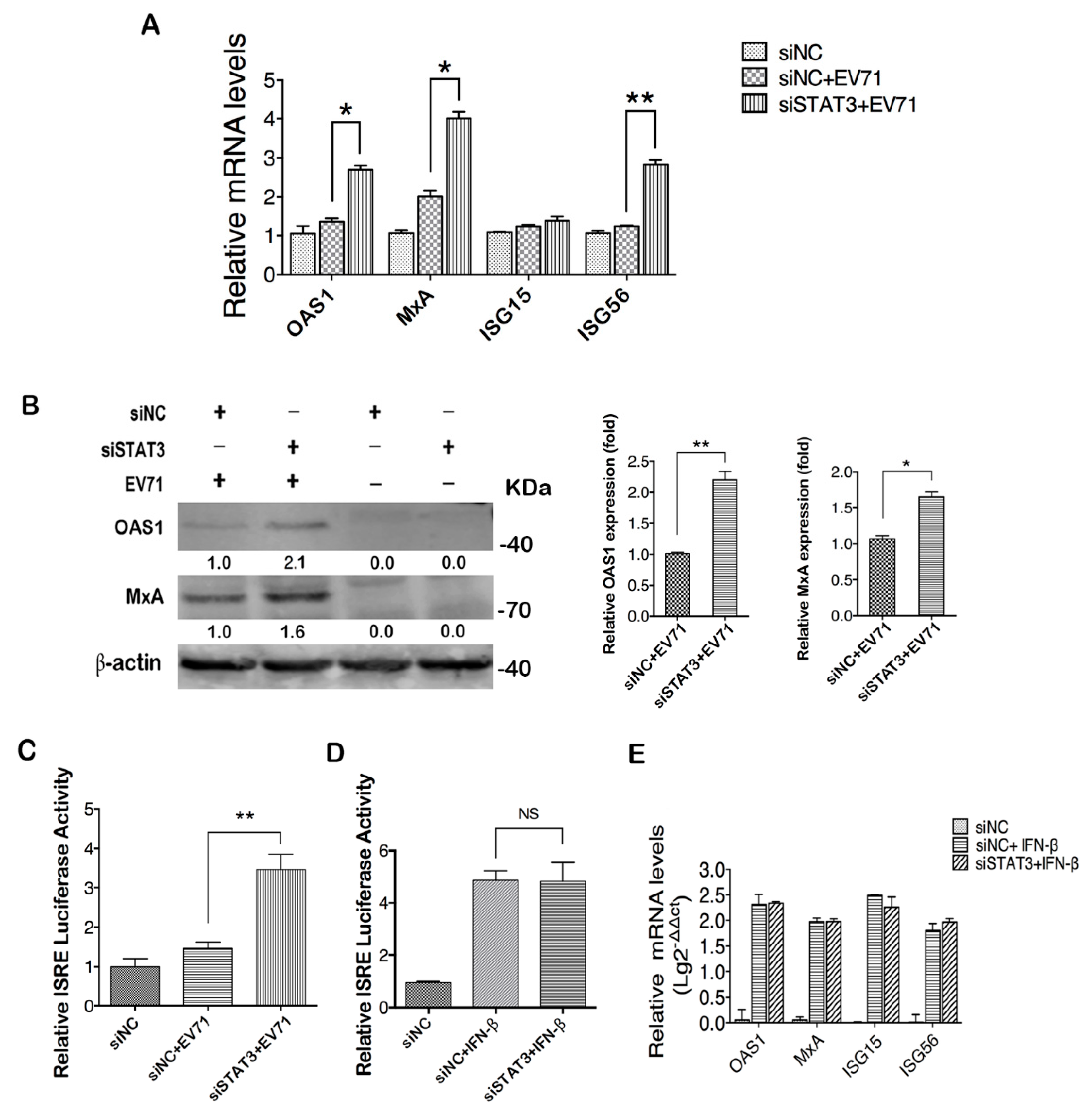
2.4. STAT3 Knockdown Elevates ISGs Production in EV71 Infected Cells
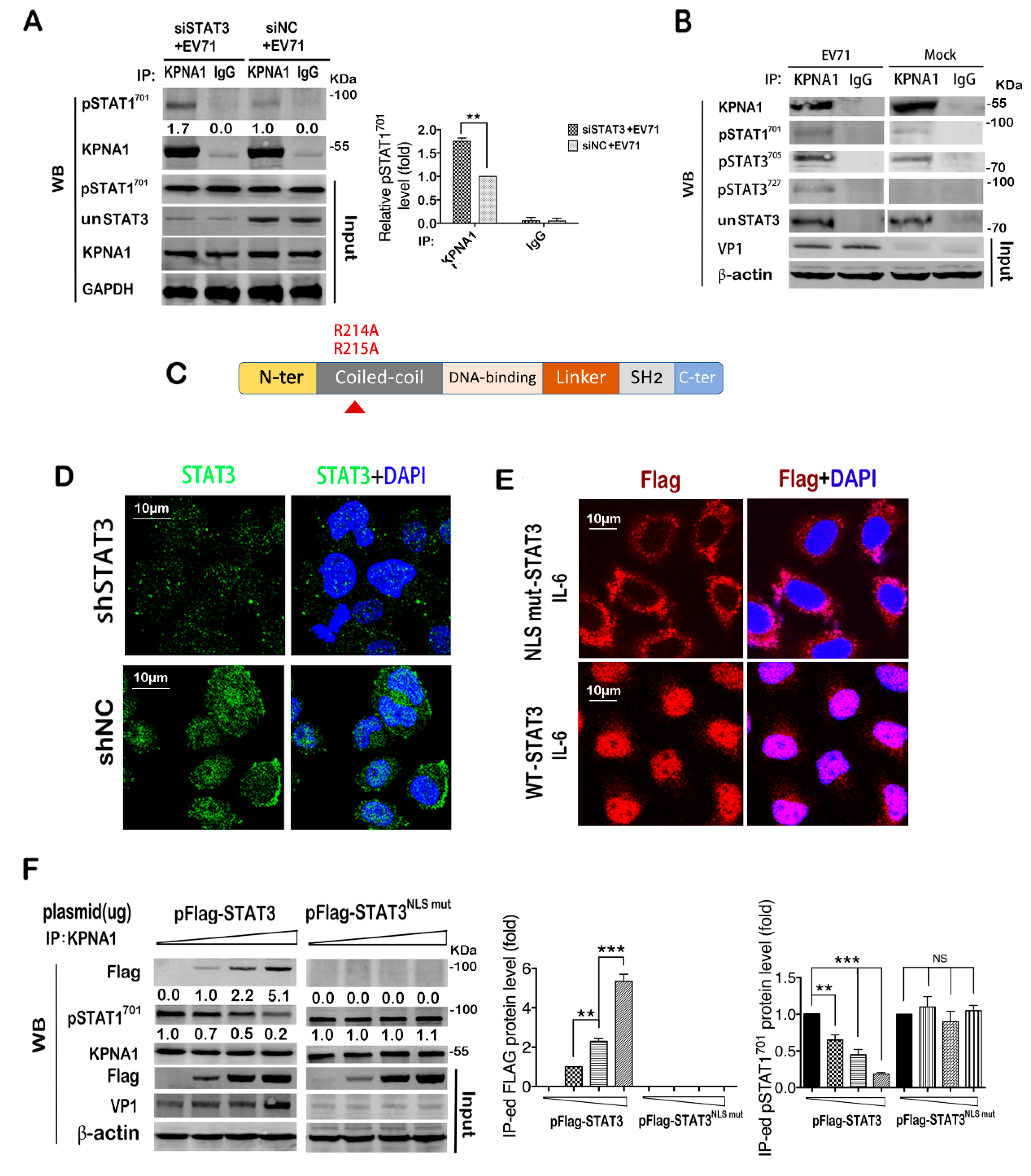
2.5. STAT3 Knockdown in Infected Cells Facilitates Nuclear Translocation of STAT1
2.6. STAT3 Competes with Activated STAT1 for Binding with KPNA1 during Infection
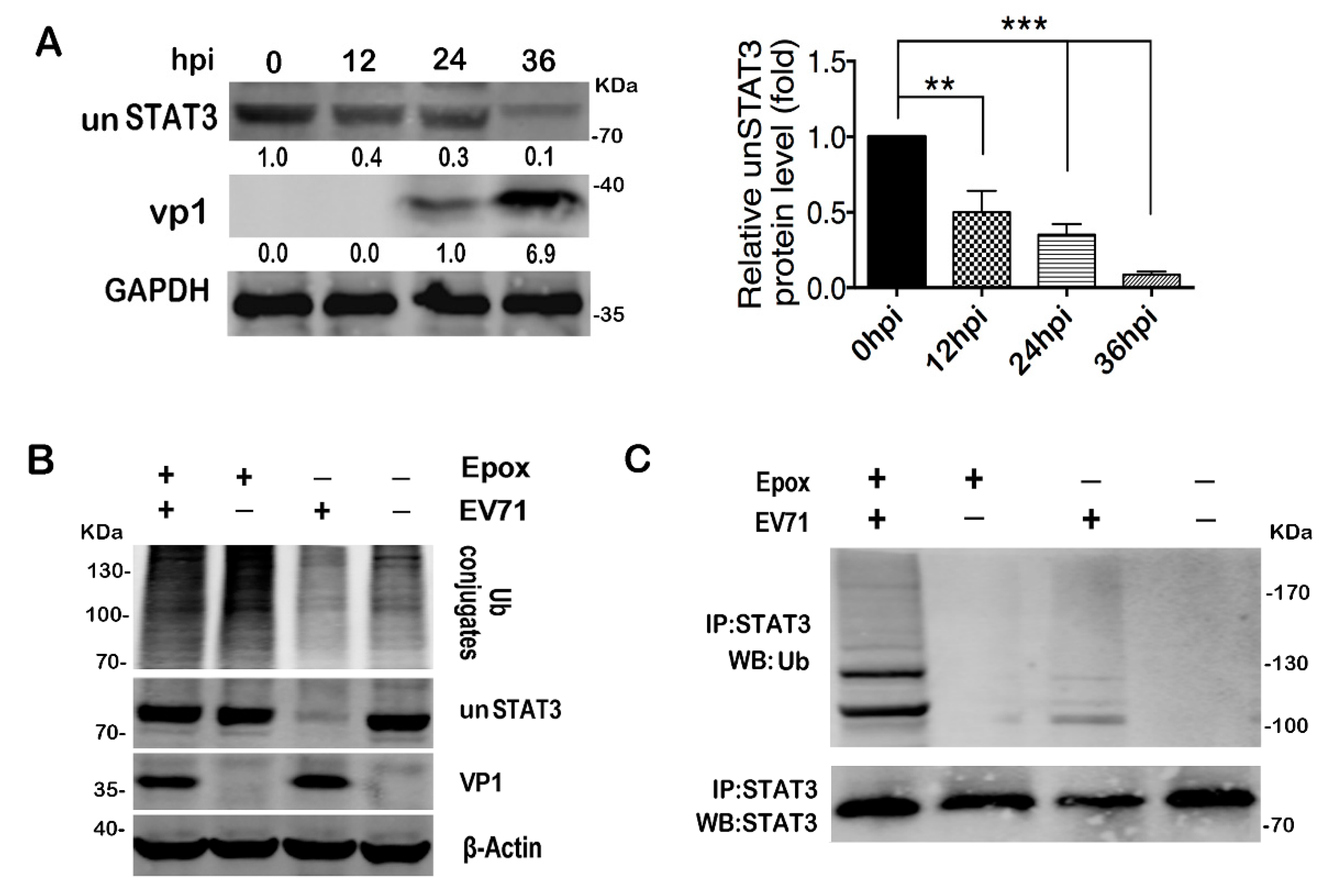
2.7. EV71 Infection Induces STAT3 Degradation via Proteasome Dependent Pathway
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cells and Virus
4.3. Plasmids, siRNAs, Antibodies and Inhibitors
4.4. Western Blot
4.5. PCR Array and qRT-PCR Analysis
4.6. ELISA Assay
4.7. Co-Immunoprecipitation Assay
4.8. Luciferase Assay
4.9. Immunofluorescence Staining and Confocal Microscopy
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EV71 | Enterovirus 71 |
| STAT | Signal transducer and activator of transcription |
| HFMD | Hand, foot and mouth disease |
| KPNA1 | Karyopherin-α1 |
| PAMPs | Pathogen-associated molecular patterns |
| CNS | Central nervous system |
| ISRE | IFN-stimulated response element |
| ISGs | IFN-stimulated genes |
| SOCS | Cytokine signaling proteins |
| unSTAT3 | un-phosphorylated STAT3 |
| un-phosphorylated STAT3 | Epoxomicin |
| ME | Meningoencephalitis |
| FC | Febrile convulsion |
| JAK | Janus-Activated Kinase |
| CSF | Cerebrospinal fluid |
| TCID50 | 50% tissue culture infectious dose |
References
- Wan, J.; Fu, A.K.; Ip, F.C.; Ng, H.K.; Hugon, J.; Page, G.; Wang, J.H.; Lai, K.O.; Wu, Z.; Ip, N.Y. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: Implications in Alzheimer’s disease. J. Neurosci. 2010, 30, 6873–6881. [Google Scholar] [CrossRef]
- Skov, S.; Nielsen, M.; Bregenholt, S.; Odum, N.; Claesson, M.H. Activation of Stat-3 is involved in the induction of apoptosis after ligation of major histocompatibility complex class I molecules on human Jurkat T cells. Blood 1998, 91, 3566–3573. [Google Scholar] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Tsareva, S.A.; Moriggl, R.; Corvinus, F.M.; Wiederanders, B.; Schutz, A.; Kovacic, B.; Friedrich, K. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia 2007, 9, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Wang, H.-W.; Cuenca, A.; Huang, M.; Ghansah, T.; Brayer, J.; Kerr, W.G.; Takeda, K.; Akira, S.; Schoenberger, S.P.; et al. A Critical Role for Stat3 Signaling in Immune Tolerance. Immunity 2003, 19, 425–436. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Holst, J.; Coffre, M.; Mielke, L.; De Pauw, A.; Lhocine, N.; Smith, A.M.; Rutschman, R.; Kaushal, D.; Shen, Y.; et al. General nature of the STAT3-activated anti-inflammatory response. J. Immunol. 2006, 177, 7880–7888. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and Functions of the IL-10 Family of Cytokines in Inflammation and Disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.R.; Dambuza, I.M.; Lee, Y.J.; Frank, G.M.; Egwuagu, C.E. STAT3 regulates proliferation and survival of CD8+ T cells: Enhances effector responses to HSV-1 infection, and inhibits IL-10+ regulatory CD8+ T cells in autoimmune uveitis. Mediat. Inflamm. 2013, 2013, 359674. [Google Scholar] [CrossRef] [PubMed]
- Kuchipudi, S.V. The Complex Role of STAT3 in Viral Infections. J. Immunol. Res. 2015, 2015, 272359. [Google Scholar] [CrossRef] [PubMed]
- Suarez, A.A.R.; Van Renne, N.; Baumert, T.F.; Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. PLoS Pathog. 2018, 14, e1006839. [Google Scholar]
- Wang, W.-B.; Levy, D.E.; Lee, C.-K. STAT3 Negatively Regulates Type I IFN-Mediated Antiviral Response. J. Immunol. 2011, 187, 2578–2585. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-H.; Lee, C.-K. STAT3 Cooperates with Phospholipid Scramblase 2 to Suppress Type I Interferon Response. Front. Immunol. 2018, 9, 1886. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.H.; Ivashkiv, L.B. Role of STAT3 in Type I Interferon Responses: Negative Regulation of Stat1-Dependent Inflammatory Gene Activation. J. Biol. Chem. 2006, 281, 14111–14118. [Google Scholar] [CrossRef] [PubMed]
- Mahony, R.; Gargan, S.; Roberts, K.L.; Bourke, N.; Keating, S.E.; Bowie, A.G.; O’Farrelly, C.; Stevenson, N.J. A novel anti-viral role for STAT3 in IFN-α signalling responses. Cell. Mol. Life Sci. 2016, 74, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- McCartney, E.M.; Helbig, K.J.; Narayana, S.K.; Eyre, N.S.; Aloia, A.L.; Beard, M.R. Signal transducer and activator of transcription 3 is a proviral host factor for hepatitis C virus. Hepatology 2013, 58, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Ulane, C.M.; Rodriguez, J.J.; Parisien, J.-P.; Horvath, C.M. STAT3 Ubiquitylation and Degradation by Mumps Virus Suppress Cytokine and Oncogene Signaling. J. Virol. 2003, 77, 6385–6393. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, R.; Ma, Z.; Xiao, Y.; Nan, Y.; Wang, Y.; Lin, S.; Zhang, Y.-J. Porcine Reproductive and Respiratory Syndrome Virus Antagonizes JAK/STAT3 Signaling via nsp5, Which Induces STAT3 Degradation. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Li, H.S.; Cheung, M.C.; Chan, R.W.Y.; Yuen, K.M.; Mok, C.K.P.; Nicholls, J.M.; Peiris, J.S.M.; Chan, M.C.W. Highly pathogenic avian influenza H5N1 virus delays apoptotic responses via activation of STAT3. Sci. Rep. 2016, 6, 28593. [Google Scholar] [CrossRef] [PubMed]
- Lieu, K.G.; Brice, A.; Wiltzer, L.; Hirst, B.; Jans, D.A.; Blondel, D.; Moseley, G.W. The Rabies Virus Interferon Antagonist P Protein Interacts with Activated STAT3 and Inhibits Gp130 Receptor Signaling. J. Virol. 2013, 87, 8261–8265. [Google Scholar] [CrossRef]
- Chandra, V.; Kar-Roy, A.; Kumari, S.; Mayor, S.; Jameel, S. The Hepatitis E Virus ORF3 Protein Modulates Epidermal Growth Factor Receptor Trafficking, STAT3 Translocation, and the Acute-Phase Response. J. Virol. 2008, 82, 7100–7110. [Google Scholar] [CrossRef][Green Version]
- Daugherty, M.D.; Malik, H.S. How a virus blocks a cellular emergency access lane to the nucleus, STAT! Cell Host Microbe 2014, 16, 150–152. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reitsma, J.M.; Sato, H.; Nevels, M.; Terhune, S.S.; Paulus, C. Human Cytomegalovirus IE1 Protein Disrupts Interleukin-6 Signaling by Sequestering STAT3 in the Nucleus. J. Virol. 2013, 87, 10763–10776. [Google Scholar] [CrossRef] [PubMed]
- Mitzel, D.N.; Jaramillo, R.J.; Stout-Delgado, H.; Senft, A.P.; Harrod, K.S. Human metapneumovirus inhibits the IL-6-induced JAK/STAT3 signalling cascade in airway epithelium. J. Gen. Virol. 2014, 95, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Liao, Q.; Viboud, C.; Zhang, J.; Sun, J.; Wu, J.T.; Chang, Z.; Liu, F.; Fang, V.J.; Zheng, Y.; et al. Hand, foot, and mouth disease in China, 2008-12: An epidemiological study. Lancet Infect. Dis. 2014, 14, 308–318. [Google Scholar] [CrossRef]
- Li, H.; Li, S.; Zheng, J.; Cai, C.; Ye, B.; Yang, J.; Chen, Z. Cerebrospinal fluid Th1/Th2 cytokine profiles in children with enterovirus 71-associated meningoencephalitis. Microbiol. Immunol. 2015, 59, 152–159. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Cai, C.; Xu, Y.; Jiang, Y.; Chen, Z. Cerebrospinal fluid chemokine patterns in children with enterovirus 71-related encephalitis. Sci. Rep. 2018, 8, 1658. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Lei, H.-Y.; Huang, K.-J.; Wu, J.-M.; Yu, C.-K.; Su, I.-J.; Liu, C.-C. Pathogenesis of Enterovirus 71 Brainstem Encephalitis in Pediatric Patients: Roles of Cytokines and Cellular Immune Activation in Patients with Pulmonary Edema. J. Infect. Dis. 2003, 188, 564–570. [Google Scholar] [CrossRef]
- Das Sarma, J. Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J. Neurovirol. 2014, 20, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Guo, S.J.; Fan, S.T.; Zeng, X.F.; Zhang, Y.; Liao, Y.; Wang, J.B.; Zhao, T.; Wang, L.C.; Che, Y.C.; et al. The Preferential Infection of Astrocytes by Enterovirus 71 Plays a Key Role in the Viral Neurogenic Pathogenesis. Front. Cell. Infect. Microbiol. 2016, 6, 192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, Z.; Shu, B.; Liu, X.; Zhang, Z.; Liu, Y.; Bai, B.; Hu, Q.; Mao, P.; Wang, H. Human Astrocytic Cells Support Persistent Coxsackievirus B3 Infection. J. Virol. 2013, 87, 12407–12421. [Google Scholar] [CrossRef]
- Au-Yeung, N.; Mandhana, R.; Horvath, C.M. Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway. JAK-STAT 2013, 2, e23931. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E. STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Kochs, G. Interferon-induced mx proteins: Dynamin-like GTPases with antiviral activity. Traffic 2002, 3, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Liu, X.; Ma, Y.; Sun, Z.; Yang, Y.; Jin, Q.; He, B.; Wang, J. The 3C Protein of Enterovirus 71 Inhibits Retinoid Acid-Inducible Gene I-Mediated Interferon Regulatory Factor 3 Activation and Type I Interferon Responses. J. Virol. 2010, 84, 8051–8061. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 Protease 2Apro Targets MAVS to Inhibit Anti-Viral Type I Interferon Responses. PLoS Pathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sun, M.; Yuan, X.; Ji, L.; Jin, Y.; Cardona, C.J.; Xing, Z. Enterovirus 71 suppresses interferon responses by blocking Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling through inducing karyopherin-alpha1 degradation. J. Biol. Chem. 2017, 292, 10262–10274. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.M.; Reich, N.C. The Ins and Outs of STAT1 Nuclear Transport. Sci. Signal. 2003, 2003, re13. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.; Hoyer, D. Astrocytes: Adhesion molecules and immunomodulation. Curr. Drug Targets 2016, 17, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, H.; Xu, F.; Huang, Y.; Liu, Z.; Liu, T. Enterovirus 71 induces apoptosis of SH-SY5Y human neuroblastoma cells through stimulation of endogenous microRNA let-7b expression. Mol. Med. Rep. 2015, 12, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Too, I.H.K.; Bonne, I.; Tan, E.L.; Chu, J.J.H.; Alonso, S. Prohibitin plays a critical role in Enterovirus 71 neuropathogenesis. PLoS Pathog. 2018, 14, e1006778. [Google Scholar] [CrossRef] [PubMed]
- Haolong, C.; Du, N.; Hongchao, T.; Yang, Y.; Wei, Z.; Hua, Z.; Wenliang, Z.; Lei, S.; Po, T. Enterovirus 71 VP1 Activates Calmodulin-Dependent Protein Kinase II and Results in the Rearrangement of Vimentin in Human Astrocyte Cells. PLoS ONE 2013, 8, e73900. [Google Scholar] [CrossRef] [PubMed]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A Small-Molecule Inhibitor of STAT3 Activation and Dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef]
- Sen, N.; Che, X.B.; Rajamani, J.; Zerboni, L.; Sung, P.; Ptacek, J.; Arvin, A.M. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Matsubara, K.; Qian, G.-S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific Inhibition of Stat3 Signal Transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef]
- Endo, T.A.; Masuhara, M.; Yokouchi, M.; Suzuki, R.; Sakamoto, H.; Mitsui, K.; Matsumoto, A.; Tanimura, S.; Ohtsubo, M.; Misawa, H.; et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature 1997, 387, 921–924. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A Road Map for Those Who Don’t Know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef] [PubMed]
- Nardozzi, J.; Wenta, N.; Yasuhara, N.; Vinkemeier, U.; Cingolani, G. Molecular basis for the recognition of phosphorylated STAT1 by importin alpha5. J. Mol. Biol. 2010, 402, 83–100. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.M.; Banninger, G.; McDonald, C.; Reich, N.C. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-alpha. EMBO J. 2002, 21, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, T.; Imamoto, N.; Nakajima, K.; Hirano, T.; Yoneda, Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J. 1997, 16, 7067–7077. [Google Scholar] [CrossRef]
- Ma, J.; Cao, X. Regulation of Stat3 nuclear import by importin alpha5 and importin alpha7 via two different functional sequence elements. Cell Signal. 2006, 18, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Cimica, V.; Chen, H.C.; Iyer, J.K.; Reich, N.C. Dynamics of the STAT3 transcription factor: Nuclear import dependent on Ran and importin-beta1. PLoS ONE 2011, 6, e20188. [Google Scholar] [CrossRef]
- Pumroy, R.A.; Cingolani, G. Diversification of importin-alpha isoforms in cellular trafficking and disease states. Biochem. J. 2015, 466, 13–28. [Google Scholar] [CrossRef]
- Wang, L.-C.; Chen, S.-O.; Chang, S.-P.; Lee, Y.-P.; Yu, C.-K.; Chen, C.-L.; Tseng, P.-C.; Hsieh, C.-Y.; Chen, S.-H.; Lin, C.-F. Enterovirus 71 Proteins 2A and 3D Antagonize the Antiviral Activity of Gamma Interferon via Signaling Attenuation. J. Virol. 2015, 89, 7028–7037. [Google Scholar] [CrossRef]
- Lei, X.; Sun, Z.; Liu, X.; Jin, Q.; He, B.; Wang, J. Cleavage of the Adaptor Protein TRIF by Enterovirus 71 3C Inhibits Antiviral Responses Mediated by Toll-Like Receptor 3. J. Virol. 2011, 85, 8811–8818. [Google Scholar] [CrossRef]
- Li, J.; Yao, Y.; Chen, Y.; Xu, X.; Lin, Y.; Yang, Z.; Qiao, W.; Tan, J. Enterovirus 71 3C Promotes Apoptosis through Cleavage of PinX1, a Telomere Binding Protein. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Lei, X.; Xiao, X.; Xue, Q.; Jin, Q.; He, B.; Wang, J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 2013, 87, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y. Enterovirus 71 infection and neurological complications. Korean J. Pediatr. 2016, 59, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Hong, J.-S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.E. Immune responses to RNA-virus infections of the CNS. Nat. Rev. Immunol. 2003, 3, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Type I interferon: Friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.; Chang, C.W.; Rona, G.; Smith, K.M.; Stewart, A.G.; Takeda, A.A.S.; Fontes, M.R.M.; Stewart, M.; Vertessy, B.G.; Forwood, J.K.; et al. Structural Biology and Regulation of Protein Import into the Nucleus. J. Mol. Biol. 2016, 428, 2060–2090. [Google Scholar] [CrossRef] [PubMed]
- Kohler, M.; Speck, C.; Christiansen, M.; Bischoff, F.R.; Prehn, S.; Haller, H.; Gorlich, D.; Hartmann, E. Evidence for distinct substrate specificities of importin alpha family members in nuclear protein import. Mol. Cell. Biol. 1999, 19, 7782–7791. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Lopes, M.H.; Bleggi-Torres, L.F.; Suzuki, S.; Cunha, I.W.; Sanematsu, P.; Martins, V.R. Nuclear unphosphorylated STAT3 correlates with a worse prognosis in human glioblastoma. Pathol. Res. Pract. 2016, 212, 517–523. [Google Scholar] [CrossRef]
- Liu, L.; McBride, K.M.; Reich, N.C. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha 3. Proc. Natl. Acad. Sci. USA 2005, 102, 8150–8155. [Google Scholar] [CrossRef]
- Meyer, T.; Gavenis, K.; Vinkemeier, U. Cell Type-Specific and Tyrosine Phosphorylation-Independent Nuclear Presence of STAT1 and STAT3. Exp. Cell Res. 2002, 272, 45–55. [Google Scholar] [CrossRef]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAK-STAT 2012, 1, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Nivarthi, H.; Gordziel, C.; Themanns, M.; Kramer, N.; Eberl, M.; Rabe, B.; Schlederer, M.; Rose-John, S.; Knösel, T.; Kenner, L.; et al. The ratio of STAT1 to STAT3 expression is a determinant of colorectal cancer growth. Oncotarget 2016, 7, 51096–51106. [Google Scholar] [CrossRef] [PubMed]
- Sarafian, T.A.; Montes, C.; Imura, T.; Qi, J.; Coppola, G.; Geschwind, D.H.; Sofroniew, M.V. Disruption of Astrocyte STAT3 Signaling Decreases Mitochondrial Function and Increases Oxidative Stress In Vitro. PLoS ONE 2010, 5, e9532. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Qiu, M.; Chen, D.; Zheng, N.; Jin, Y.; Wu, Z. Apigenin inhibits enterovirus 71 replication through suppressing viral IRES activity and modulating cellular JNK pathway. Antivir. Res. 2014, 109, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Dengue Virus and Autophagy. Viruses 2011, 3, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Liu, L.; Lei, X.; Zhou, Z.; He, B.; Wang, J. 3C Protease of Enterovirus D68 Inhibits Cellular Defense Mediated by Interferon Regulatory Factor 7. J. Virol. 2016, 90, 1613–1621. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Yuan, M.; Wang, S.; Zhang, L.; Zhang, R.; Zou, X.; Wang, X.; Chen, D.; Wu, Z. STAT3 Regulates the Type I IFN-Mediated Antiviral Response by Interfering with the Nuclear Entry of STAT1. Int. J. Mol. Sci. 2019, 20, 4870. https://doi.org/10.3390/ijms20194870
Wang H, Yuan M, Wang S, Zhang L, Zhang R, Zou X, Wang X, Chen D, Wu Z. STAT3 Regulates the Type I IFN-Mediated Antiviral Response by Interfering with the Nuclear Entry of STAT1. International Journal of Molecular Sciences. 2019; 20(19):4870. https://doi.org/10.3390/ijms20194870
Chicago/Turabian StyleWang, Huanru, Meng Yuan, Shuaibo Wang, Li Zhang, Rui Zhang, Xue Zou, Xiaohui Wang, Deyan Chen, and Zhiwei Wu. 2019. "STAT3 Regulates the Type I IFN-Mediated Antiviral Response by Interfering with the Nuclear Entry of STAT1" International Journal of Molecular Sciences 20, no. 19: 4870. https://doi.org/10.3390/ijms20194870
APA StyleWang, H., Yuan, M., Wang, S., Zhang, L., Zhang, R., Zou, X., Wang, X., Chen, D., & Wu, Z. (2019). STAT3 Regulates the Type I IFN-Mediated Antiviral Response by Interfering with the Nuclear Entry of STAT1. International Journal of Molecular Sciences, 20(19), 4870. https://doi.org/10.3390/ijms20194870