Mitochondrial Calcium Uniporter Structure and Function in Different Types of Muscle Tissues in Health and Disease


 ,
, 
Abstract
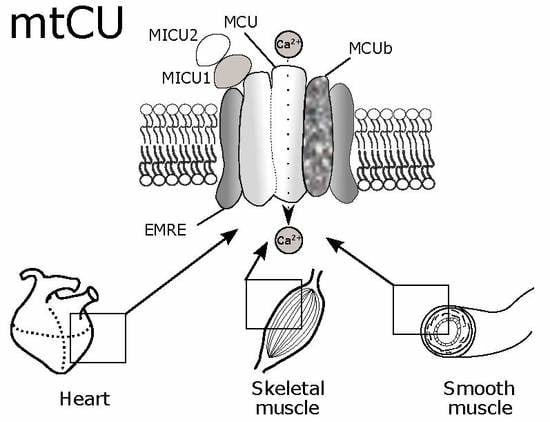
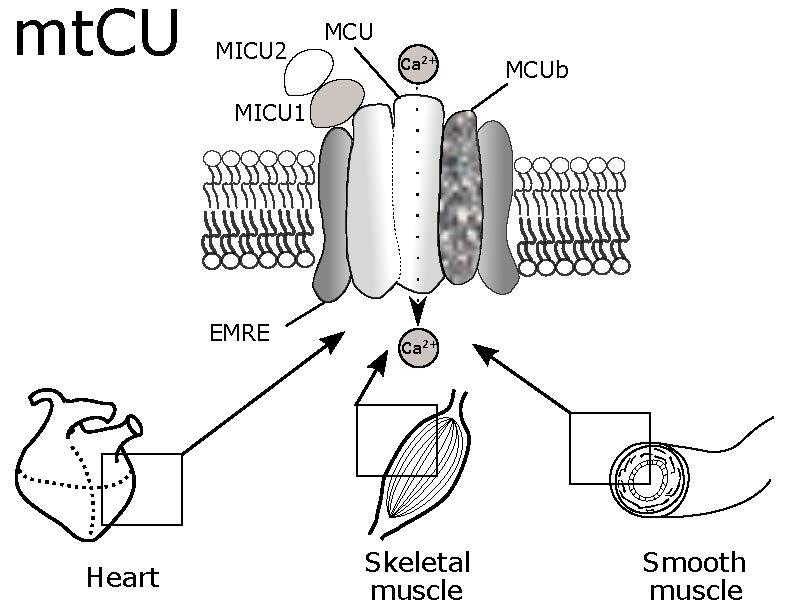
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
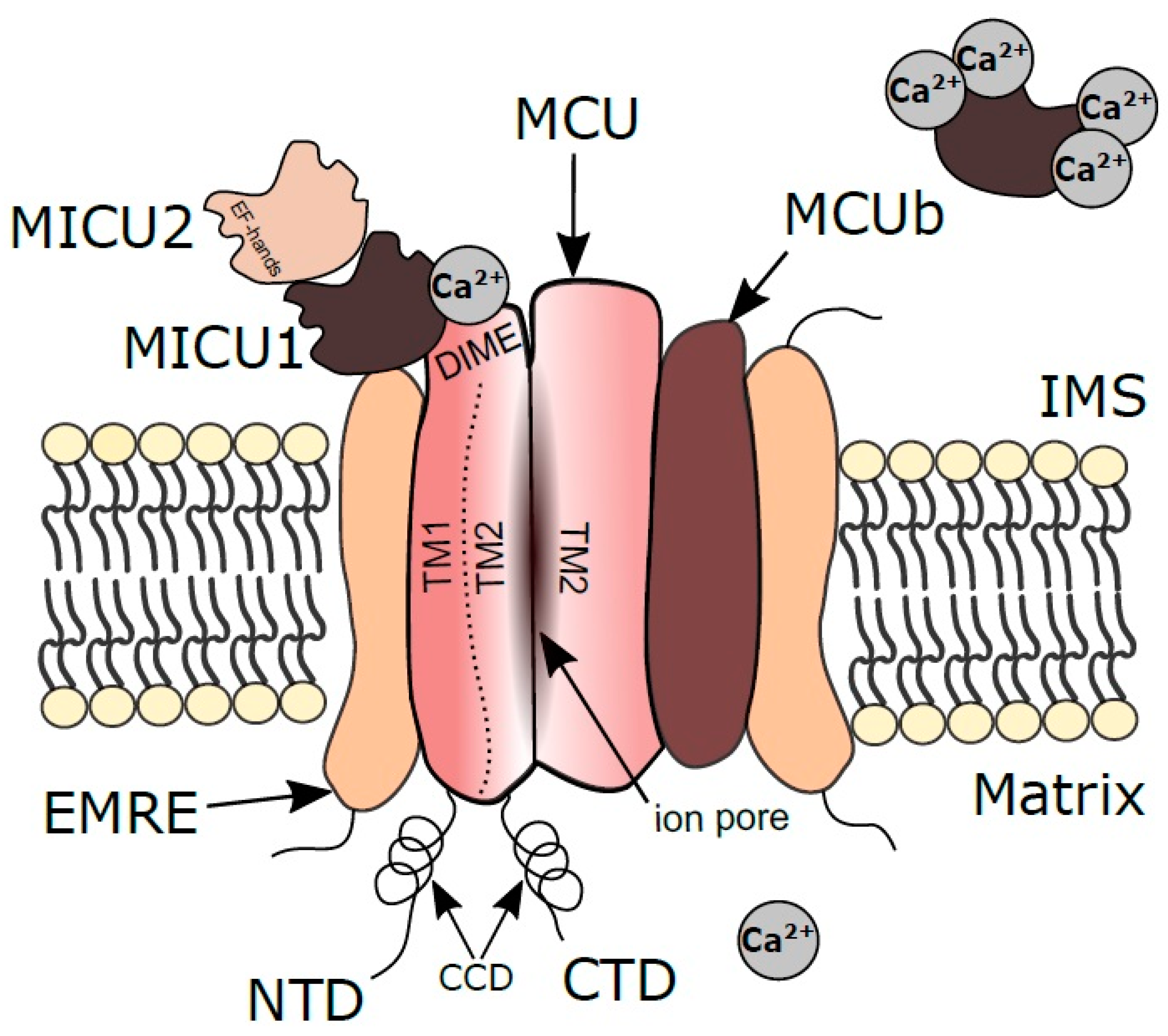
2. Structural and Electrophysiological Characteristics of MtCU
2.1. MCU
2.2. MCUb
2.3. MICU1
2.4. MICU2
2.5. MICU3
2.6. EMRE
2.7. MCUR1
3. MtCU Regulation by Intracellular Signaling Pathways
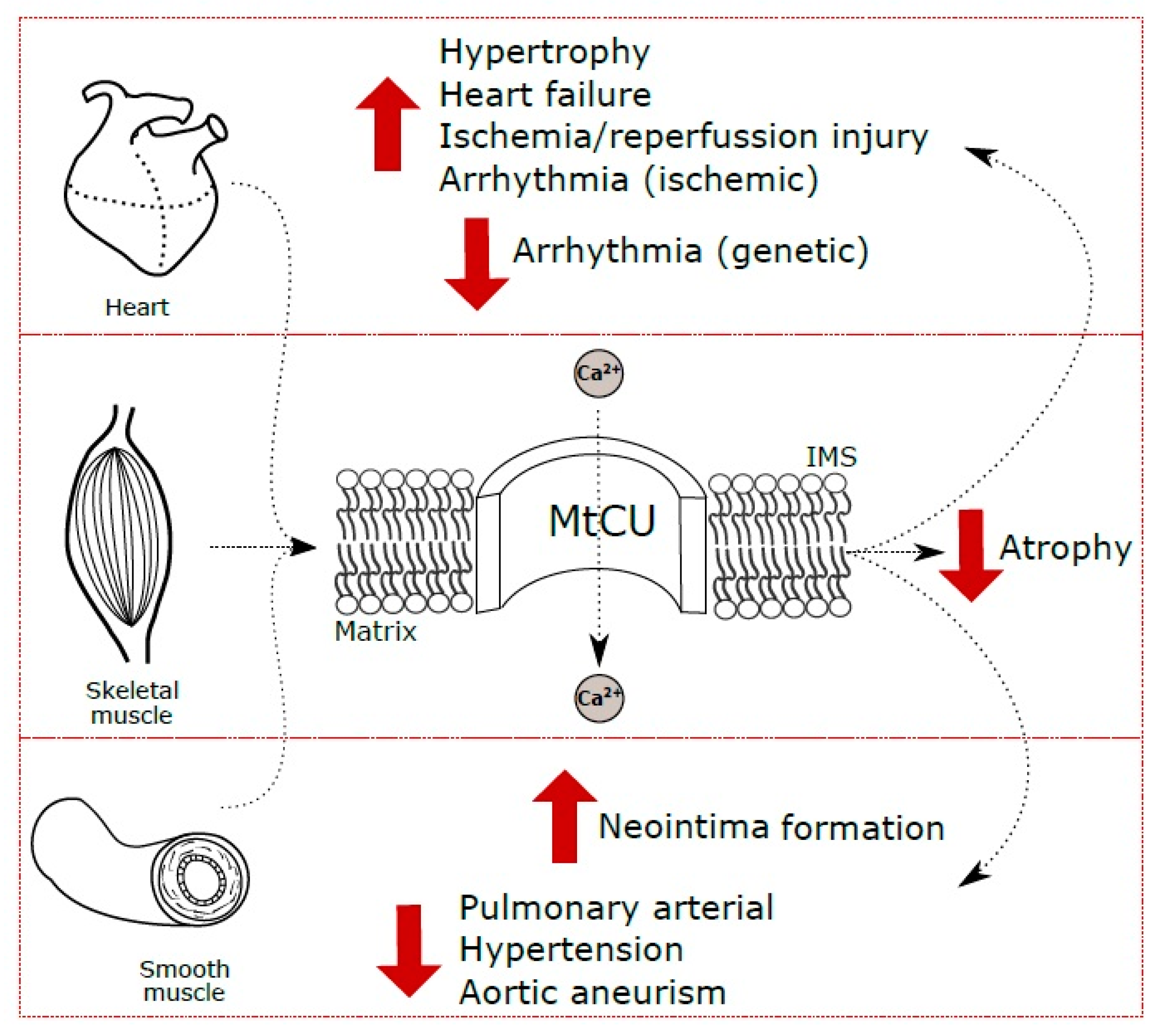
4. The Role of MtCU in Cardiac Muscle
4.1. MtCU in Normal Myocardium Function
4.2. MtCU in Cardiac Hypertrophy
4.3. MtCU in Arrhythmogenesis
4.4. MtCU in Myocardial Ischemia/Reperfusion Injury
4.5. MtCU in Heart Failure
5. The Role of MtCU in Skeletal Muscle
5.1. General Role of MtCU in Skeletal Muscle
5.2. Specific Features of MtCU in Skeletal Muscle
5.3. MtCU in Skeletal Muscle Dysfunction, Injury and Ageing
6. MtCU in Smooth Muscle Pathologies
7. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CaMKII | calmodulin-dependent protein kinase II |
| CCD | coiled-coil domain |
| CPVT | catecholaminergic polymorphic ventricular tachycardia |
| CREB | cyclic AMP response element binding protein |
| cryo-EM | cryo-electron microscopy |
| DRP1 | dynamin-related protein 1 |
| EMRE | essential MCU regulator |
| KO | knockout |
| MCU | mitochondrial calcium uniporter pore-forming subunit |
| MICU1 | mitochondrial calcium uptake 1 |
| MICU2 | mitochondrial calcium uptake 2 |
| miR | microRNA |
| mtCU | mitochondrial calcium uniporter complex |
| NCX | Na+/Ca2+ exchanger |
| NMR | nuclear magnetic resonance |
| NTD | N-terminal domain |
| PASMC | pulmonary artery smooth muscle cells |
| PRMT1 | protein arginine methyl transferase 1 |
| RIPK1 | receptor-interacting protein kinase 1 |
| ROS | reactive oxygen species |
| RyR2 | ryanodine receptor 2 |
| SERCA | sarcoplasmic/endoplasmic reticulum Ca2+ ATPase |
| SR | sarcoplasmic reticulum |
| TMD | transmembrane domain |
| VSMCs | vascular smooth muscle cells |
References
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q. The mitochondrial calcium uniporter in the heart: Energetics and beyond. J. Physiol. 2017, 595, 3743–3751. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Eshima, H.; Poole, D.C.; Kano, Y. Mitochondrial calcium regulation during and following contractions in skeletal muscle. J. Phys. Fit. Sports Med. 2018, 7, 205–211. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, M.; Meng, X.; Chu, X.; Cai, C.; Zou, F. Phosphorylation of dynamin-related protein 1 at Ser616 regulates mitochondrial fission and is involved in mitochondrial calcium uniporter-mediated neutrophil polarization and chemotaxis. Mol. Immunol. 2017, 87, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Paupe, V.; Prudent, J. New insights into the role of mitochondrial calcium homeostasis in cell migration. Biochem. Biophys. Res. Commun. 2018, 500, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef] [PubMed]
- Koval, O.M.; Nguyen, E.K.; Santhana, V.; Fidler, T.P.; Sebag, S.C.; Rasmussen, T.P.; Mittauer, D.J.; Strack, S.; Goswami, P.C.; Abel, E.D.; et al. Loss of MCU prevents mitochondrial fusion in G1-S phase and blocks cell cycle progression and proliferation. Sci. Signal. 2019, 12, eaav1439. [Google Scholar] [CrossRef]
- Penna, E.; Espino, J.; De Stefani, D.; Rizzuto, R. The MCU complex in cell death. Cell Calcium 2018, 69, 73–80. [Google Scholar] [CrossRef]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Yang, J.; Fu, L.; Wang, M.; Wang, X. Progress in understanding mitochondrial calcium uniporter complex-mediated calcium signalling: A potential target for cancer treatment. Br. J. Pharmacol. 2019, 176, 1190–1205. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.L.; Adaniya, S.M.; Cypress, M.W.; Suzuki, Y.; Kusakari, Y.; Jhun, B.S.; O-Uchi, J. Role of mitochondrial Ca2+ homeostasis in cardiac muscles. Arch. Biochem. Biophys. 2019, 663, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Patron, M.; Sprenger, H.G.; Langer, T. m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res. 2018, 28, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; De, S. Mitochondrial VDAC, the Na+/Ca2+ Exchanger, and the Ca2+ Uniporter in Ca2+ Dynamics and Signaling. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2017; Volume 981, pp. 323–347. [Google Scholar]
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Nguyen, N.X.; She, J.; Zeng, W.; Yang, Y.; Bai, X.; Jiang, Y. Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter. Cell 2019, 177, 1252–1261. [Google Scholar] [CrossRef]
- Yoo, J.; Wu, M.; Yin, Y.; Herzik, M.A.; Lander, G.C.; Lee, S.Y. Cryo-EM structure of a mitochondrial calcium uniporter. Science 2018, 361, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baradaran, R.; Wang, C.; Siliciano, A.F.; Long, S.B. Cryo-EM structures of fungal and metazoan mitochondrial calcium uniporters. Nature 2018, 559, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Fan, M.; Orlando, B.J.; Fastman, N.M.; Zhang, J.; Xu, Y.; Chambers, M.G.; Xu, X.; Perry, K.; Liao, M.; et al. X-ray and cryo-EM structures of the mitochondrial calcium uniporter. Nature 2018, 559, 575–579. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.B.; Tsai, C.W.; Tsai, M.F. The conserved aspartate ring of MCU mediates MICU1 binding and regulation in the mitochondrial calcium uniporter complex. Elife 2019, 8, e41112. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Wang, S.; Cui, T.; Su, X.C.; Chou, J.J. Ion and inhibitor binding of the double-ring ion selectivity filter of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA 2017, 114, E2846–E2851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Kirichok, Y.Y. Patch-Clamp Analysis of the Mitochondrial Calcium Uniporter. In Calcium Signalling; Humana: New York, NY, USA, 2019; Volume 1925, pp. 75–86. [Google Scholar]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabò, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Li, S.; Zong, G.; Liu, X.; Fei, S.; Shen, L.; Guan, X.; Yang, X.; Shen, Y. Single channel recording of a mitochondrial calcium uniporter. Biochem. Biophys. Res. Commun. 2018, 496, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Joiner, M.A.; Koval, O.M.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Fieni, F.; Johnson, D.E.; Hudmon, A.; Kirichok, Y. Mitochondrial Ca2+ uniporter and CaMKII in heart. Nature 2014, 513, E1–E2. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.W.; Tsai, M.F. Electrical recordings of the mitochondrial calcium uniporter in Xenopus oocytes. J. Gen. Physiol. 2018, 150, 1035. [Google Scholar] [PubMed]
- Wacquier, B.; Romero Campos, H.E.; González-Vélez, V.; Combettes, L.; Dupont, G. Mitochondrial Ca2+ dynamics in cells and suspensions. FEBS J. 2017, 284, 4128–4142. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Shanmughapriya, S.; Mok, M.C.Y.; Dong, Z.; Tomar, D.; Carvalho, E.; Rajan, S.; Junop, M.S.; Madesh, M.; Stathopulos, P.B. Structural Insights into Mitochondrial Calcium Uniporter Regulation by Divalent Cations. Cell Chem. Biol. 2016, 23, 1157–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014–1028. [Google Scholar] [CrossRef] [PubMed]
- Niescier, R.F.; Hong, K.; Park, D.; Min, K.T. MCU Interacts with Miro1 to Modulate Mitochondrial Functions in Neurons. J. Neurosci. 2018, 38, 4666–4677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paillard, M.; Csordás, G.; Huang, K.T.; Várnai, P.; Joseph, S.K.; Hajnóczky, G. MICU1 Interacts with the D-Ring of the MCU Pore to Control Its Ca2+ Flux and Sensitivity to Ru360. Mol. Cell 2018, 72, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Csordás, G.; Szanda, G.; Golenár, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spät, A.; et al. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca 2+ Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep. 2017, 18, 2291–2300. [Google Scholar] [CrossRef]
- Checchetto, V.; Szabò, I. MCU Regulation in Lipid Bilayer and Electrophysiological Recording; Humana: New York, NY, USA, 2019; pp. 59–63. [Google Scholar]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High-affinity cooperative Ca2+ binding by MICU 1–MICU 2 serves as an on–off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411. [Google Scholar] [CrossRef]
- Kamer, K.J.; Sancak, Y.; Fomina, Y.; Meisel, J.D.; Chaudhuri, D.; Grabarek, Z.; Mootha, V.K. MICU1 imparts the mitochondrial uniporter with the ability to discriminate between Ca2+ and Mn2+. Proc. Natl. Acad. Sci. USA 2018, 115, E7960–E7969. [Google Scholar] [CrossRef]
- Wettmarshausen, J.; Goh, V.; Huang, K.T.; Arduino, D.M.; Tripathi, U.; Leimpek, A.; Cheng, Y.; Pittis, A.A.; Gabaldón, T.; Mokranjac, D.; et al. MICU1 Confers Protection from MCU-Dependent Manganese Toxicity. Cell Rep. 2018, 25, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Tufi, R.; Gleeson, T.P.; von Stockum, S.; Hewitt, V.L.; Lee, J.J.; Terriente-Felix, A.; Sanchez-Martinez, A.; Ziviani, E.; Whitworth, A.J. Comprehensive Genetic Characterization of Mitochondrial Ca2+ Uniporter Components Reveals Their Different Physiological Requirements In Vivo. Cell Rep. 2019, 27, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, A.; Patron, M.; Reane, D.V.; Gastaldello, S.; Amoroso, S.; Rizzuto, R.; Brini, M.; Raffaello, A.; Calì, T. Parkin-dependent regulation of the MCU complex component MICU1. Sci. Rep. 2018, 8, 14199. [Google Scholar] [CrossRef]
- Vecellio Reane, D.; Vallese, F.; Checchetto, V.; Acquasaliente, L.; Butera, G.; De Filippis, V.; Szabò, I.; Zanotti, G.; Rizzuto, R.; Raffaello, A. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca 2+ Uptake Machinery of Skeletal Muscle. Mol. Cell 2016, 64, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Jiang, W.; Kaushik, V.K.; Mootha, V.K.; Grabarek, Z. Crystal structure of MICU2 and comparison with MICU1 reveal insights into the uniporter gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 3546–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Wang, M.; Wang, J.; Nie, Z.; Wu, G.; Yang, X.; Shen, Y. Dimerization of MICU Proteins Controls Ca2+ Influx through the Mitochondrial Ca2+ Uniporter. Cell Rep. 2019, 26, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.; Hoff, H.; Roskowski, A.; Foskett, J.K. MICU2 Restricts Spatial Crosstalk between InsP3R and MCU Channels by Regulating Threshold and Gain of MICU1-Mediated Inhibition and Activation of MCU. Cell Rep. 2017, 21, 3141–3154. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Wakimoto, H.; Kamer, K.J.; Sancak, Y.; Goldberger, O.; Axelsson, A.; DeLaughter, D.M.; Gorham, J.M.; Mootha, V.K.; Seidman, J.G.; et al. Cardiovascular homeostasis dependence on MICU2, a regulatory subunit of the mitochondrial calcium uniporter. Proc. Natl. Acad. Sci. USA 2017, 114, E9096–E9104. [Google Scholar] [CrossRef] [Green Version]
- Shamseldin, H.E.; Alasmari, A.; Salih, M.A.; Samman, M.M.; Mian, S.A.; Alshidi, T.; Ibrahim, N.; Hashem, M.; Faqeih, E.; Al-Mohanna, F.; et al. A null mutation in MICU2 causes abnormal mitochondrial calcium homeostasis and a severe neurodevelopmental disorder. Brain 2017, 140, 2806–2813. [Google Scholar] [CrossRef] [Green Version]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yamagoshi, R.; Harada, K.; Kawano, M.; Minami, N.; Ido, Y.; Kuwahara, K.; Fujita, A.; Ozono, M.; Watanabe, A.; et al. Analysis of the structure and function of EMRE in a yeast expression system. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.W.; Wu, Y.; Williams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. Elife 2016, 5, e15545. [Google Scholar] [CrossRef] [PubMed]
- Vais, H.; Mallilankaraman, K.; Mak, D.O.D.; Hoff, H.; Payne, R.; Tanis, J.E.; Foskett, J.K. EMRE Is a Matrix Ca(2+) Sensor that Governs Gatekeeping of the Mitochondrial Ca(2+) Uniporter. Cell Rep. 2016, 14, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Pan, X.; Liu, J.C.; Menazza, S.; Liu, J.; Nguyen, T.T.; Pan, H.; Parks, R.J.; Anderson, S.; Noguchi, A.; et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell. Cardiol. 2015, 85, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Adlakha, J.; Karamichali, I.; Sangwallek, J.; Deiss, S.; Bär, K.; Coles, M.; Hartmann, M.D.; Lupas, A.N.; Hernandez Alvarez, B. Characterization of MCU-Binding Proteins MCUR1 and CCDC90B—Representatives of a Protein Family Conserved in Prokaryotes and Eukaryotic Organelles. Structure 2019, 27, 464–475. [Google Scholar] [CrossRef]
- Chaudhuri, D.; Artiga, D.J.; Abiria, S.A.; Clapham, D.E. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. USA 2016, 113, E1872–E1880. [Google Scholar] [CrossRef]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 136. [Google Scholar] [CrossRef]
- Nemani, N.; Shanmughapriya, S.; Madesh, M. Molecular regulation of MCU: Implications in physiology and disease. Cell Calcium 2018, 74, 86–93. [Google Scholar] [CrossRef]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Zhang, X.; Guo, S.; Kolesar, J.E.; Hines, K.J.; Ragheb, J.; Jog, N.R.; Caricchio, R.; et al. Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU. Sci. Signal. 2015, 8, ra23. [Google Scholar] [CrossRef]
- Shanmughapriya, S.; Tomar, D.; Dong, Z.; Slovik, K.J.; Nemani, N.; Natarajaseenivasan, K.; Carvalho, E.; Lu, C.; Corrigan, K.; Garikipati, V.N.S.; et al. FOXD1-dependent MICU1 expression regulates mitochondrial activity and cell differentiation. Nat. Commun. 2018, 9, 3449. [Google Scholar] [CrossRef] [PubMed]
- Jaquenod De Giusti, C.; Roman, B.; Das, S. The Influence of MicroRNAs on Mitochondrial Calcium. Front. Physiol. 2018, 9, 1291. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Wang, Y.; Peng, J.; Shen, Q.; Chen, M.; Tang, W.; Li, X.; Cai, C.; Wang, B.; Cai, S.; et al. Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 2017, 8, 83831–83844. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corrà, F.; Giorgi, C.; De Marchi, E.; et al. Downregulation of the Mitochondrial Calcium Uniporter by Cancer-Related miR-25. Curr. Biol. 2013, 23, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Z.; Chen, K.H.; DasGupta, A.; Potus, F.; Dunham-Snary, K.; Bonnet, S.; Tian, L.; Fu, J.; Breuils-Bonnet, S.; Provencher, S.; et al. MicroRNA-138 and MicroRNA-25 Down-regulate Mitochondrial Calcium Uniporter, Causing the Pulmonary Arterial Hypertension Cancer Phenotype. Am. J. Respir. Crit. Care Med. 2017, 195, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.; Huang, B.J.; Ma, X.E.; Wang, S.Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.M.; Liang, D.D.; et al. MiR-25 Protects Cardiomyocytes against Oxidative Damage by Targeting the Mitochondrial Calcium Uniporter. Int. J. Mol. Sci. 2015, 16, 5420–5433. [Google Scholar] [CrossRef] [Green Version]
- Zaglia, T.; Ceriotti, P.; Campo, A.; Borile, G.; Armani, A.; Carullo, P.; Prando, V.; Coppini, R.; Vida, V.; Stølen, T.O.; et al. Content of mitochondrial calcium uniporter (MCU) in cardiomyocytes is regulated by microRNA-1 in physiologic and pathologic hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E9006–E9015. [Google Scholar] [CrossRef] [Green Version]
- Paiva, S.; Agbulut, O. MiRroring the Multiple Potentials of MicroRNAs in Acute Myocardial Infarction. Front. Cardiovasc. Med. 2017, 4, 73. [Google Scholar] [CrossRef]
- Favaro, G.; Romanello, V.; Varanita, T.; Desbats, M.A.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Uchi, J.; Jhun, B.S.; Xu, S.; Hurst, S.; Raffaello, A.; Liu, X.; Yi, B.; Zhang, H.; Gross, P.; Mishra, J.; et al. Adrenergic Signaling Regulates Mitochondrial Ca2+ Uptake Through Pyk2-Dependent Tyrosine Phosphorylation of the Mitochondrial Ca2+ Uniporter. Antioxid. Redox Signal. 2014, 21, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yan, J.; Wang, L.; Tian, X.; Zhang, T.; Guo, L.; Li, B.; Wang, W.; Liu, X. The Pyk2/MCU pathway in the rat middle cerebral artery occlusion model of ischemic stroke. Neurosci. Res. 2018, 131, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.G.; Kohlhaas, M.; Bertero, E.; Wilhelm, D.; Wagner, M.; Sequeira, V.; Kreusser, M.M.; Dewenter, M.; Kappl, R.; Hoth, M.; et al. CaMKII does not control mitochondrial Ca2+ uptake in cardiac myocytes. J. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Min, C.K.; Kim, T.G.; Song, H.K.; Lim, Y.; Kim, D.; Shin, K.; Kang, M.; Kang, J.Y.; Youn, H.S.; et al. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep. 2015, 16, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, E.K.; Koval, O.M.; Noble, P.; Broadhurst, K.; Allamargot, C.; Wu, M.; Strack, S.; Thiel, W.H.; Grumbach, I.M. CaMKII (Ca2+/Calmodulin-Dependent Kinase II) in Mitochondria of Smooth Muscle Cells Controls Mitochondrial Mobility, Migration, and Neointima Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1333–1345. [Google Scholar] [CrossRef]
- Zhao, H.; Li, T.; Wang, K.; Zhao, F.; Chen, J.; Xu, G.; Zhao, J.; Li, T.; Chen, L.; Li, L.; et al. AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nat. Cell Biol. 2019, 21, 476–486. [Google Scholar] [CrossRef]
- Zeng, F.; Chen, X.; Cui, W.; Wen, W.; Lu, F.; Sun, X.; Ma, D.; Yuan, Y.; Li, Z.; Hou, N.; et al. RIPK1 Binds MCU to Mediate Induction of Mitochondrial Ca2+ Uptake and Promotes Colorectal Oncogenesis. Cancer Res. 2018, 78, 2876–2885. [Google Scholar] [CrossRef]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J. 2019, 38, e99435. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Klec, C.; Parichatikanond, W.; Stryeck, S.; Gottschalk, B.; Pulido, S.; Rost, R.; Eroglu, E.; Hofmann, N.A.; Bondarenko, A.I.; et al. PRMT1-mediated methylation of MICU1 determines the UCP2/3 dependency of mitochondrial Ca2+ uptake in immortalized cells. Nat. Commun. 2016, 7, 12897. [Google Scholar] [CrossRef]
- Trenker, M.; Malli, R.; Fertschai, I.; Levak-Frank, S.; Graier, W.F. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 2007, 9, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Parker, N.; Buckingham, J.A.; Vidal-Puig, A.; Halestrap, A.P.; Gunter, T.E.; Nicholls, D.G.; Bernardi, P.; Lemasters, J.J.; Brand, M.D. UCPs—Unlikely calcium porters. Nat. Cell Biol. 2008, 10, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Motloch, L.J.; Larbig, R.; Gebing, T.; Reda, S.; Schwaiger, A.; Leitner, J.; Wolny, M.; Eckardt, L.; Hoppe, U.C. By Regulating Mitochondrial Ca2+-Uptake UCP2 Modulates Intracellular Ca2+. PLoS ONE 2016, 11, e0148359. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, A.I.; Parichatikanond, W.; Madreiter, C.T.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. UCP2 modulates single-channel properties of a MCU-dependent Ca2+ inward current in mitochondria. Pflügers Arch. Eur. J. Physiol. 2015, 467, 2509–2518. [Google Scholar] [CrossRef] [PubMed]
- Opalińska, M.; Jańska, H.; Opalińska, M.; Jańska, H. AAA Proteases: Guardians of Mitochondrial Function and Homeostasis. Cells 2018, 7, 163. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.W.; Wu, Y.; Pao, P.C.; Phillips, C.B.; Williams, C.; Miller, C.; Ranaghan, M.; Tsai, M.F. Proteolytic control of the mitochondrial calcium uniporter complex. Proc. Natl. Acad. Sci. USA 2017, 114, 4388–4393. [Google Scholar] [CrossRef] [Green Version]
- König, T.; Tröder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Mühlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef]
- Hurst, S.; Baggett, A.; Csordas, G.; Sheu, S.S. SPG7 targets the m-AAA protease complex to process MCU for uniporter assembly, Ca2+ influx, and regulation of mPTP opening. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Pareek, G.; Thomas, R.E.; Pallanck, L.J. Loss of the Drosophila m-AAA mitochondrial protease paraplegin results in mitochondrial dysfunction, shortened lifespan, and neuronal and muscular degeneration. Cell Death Dis. 2018, 9, 304. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Fieni, F.; Bae Lee, S.; Nung Jan, Y.; Kirichok, Y. ARTICLE Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun. 2012, 3, 1317. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Lu, X.; Correll, R.N.; Schwanekamp, J.A.; Vagnozzi, R.J.; Sargent, M.A.; York, A.J.; Zhang, J.; Bers, D.M.; Molkentin, J.D. The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 2015, 12, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Fuente, S.; Sheu, S.S. SR-mitochondria communication in adult cardiomyocytes: A close relationship where the Ca2+ has a lot to say. Arch. Biochem. Biophys. 2019, 663, 259–268. [Google Scholar] [CrossRef]
- Lu, X.; Ginsburg, K.S.; Kettlewell, S.; Bossuyt, J.; Smith, G.L.; Bers, D.M. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ. Res. 2013, 112, 424–431. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente, S.; Fernandez-Sanz, C.; Vail, C.; Agra, E.J.; Holmstrom, K.; Sun, J.; Mishra, J.; Williams, D.; Finkel, T.; Murphy, E.; et al. Strategic Positioning and Biased Activity of the Mitochondrial Calcium Uniporter in Cardiac Muscle. J. Biol. Chem. 2016, 291, 23343–23362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Fuente, S.; Lambert, J.P.; Nichtova, Z.; Fernandez Sanz, C.; Elrod, J.W.; Sheu, S.S.; Csordás, G. Spatial Separation of Mitochondrial Calcium Uptake and Extrusion for Energy-Efficient Mitochondrial Calcium Signaling in the Heart. Cell Rep. 2018, 24, 3099–3107. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227. [Google Scholar] [CrossRef]
- Piquereau, J.; Ventura-Clapier, R. Maturation of Cardiac Energy Metabolism during Perinatal Development. Front. Physiol. 2018, 9, 959. [Google Scholar] [CrossRef]
- Yu, Z.; Chen, R.; Li, M.; Yu, Y.; Liang, Y.; Han, F.; Qin, S.; Chen, X.; Su, Y.; Ge, J. Mitochondrial calcium uniporter inhibition provides cardioprotection in pressure overload-induced heart failure through autophagy enhancement. Int. J. Cardiol. 2018, 271, 161–168. [Google Scholar] [CrossRef]
- Wong, C.X.; Brown, A.; Lau, D.H.; Chugh, S.S.; Albert, C.M.; Kalman, J.M.; Sanders, P. Epidemiology of Sudden Cardiac Death: Global and Regional Perspectives. Heart Lung Circ. 2019, 28, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Di Diego, J.M.; Antzelevitch, C. Ischemic ventricular arrhythmias: Experimental models and their clinical relevance. Heart Rhythm 2011, 8, 1963–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, P.; Quinn, T.A. Altered Calcium Handling and Ventricular Arrhythmias in Acute Ischemia. Clin. Med. Insights Cardiol. 2016, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Zhou, A.; Liu, H.; Shi, G.; Liu, M.; Boheler, K.R.; Dudley, S.C. Mitochondrial Ca2+ flux modulates spontaneous electrical activity in ventricular cardiomyocytes. PLoS ONE 2018, 13, e0200448. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Terentyeva, R.; Kim, T.Y.; Bronk, P.; Clements, R.T.; O-Uchi, J.; Csordás, G.; Choi, B.R.; Terentyev, D. Pharmacological Modulation of Mitochondrial Ca2+ Content Regulates Sarcoplasmic Reticulum Ca2+ Release via Oxidation of the Ryanodine Receptor by Mitochondria-Derived Reactive Oxygen Species. Front. Physiol. 2018, 9, 1831. [Google Scholar] [CrossRef]
- Xie, A.; Song, Z.; Liu, H.; Zhou, A.; Shi, G.; Wang, Q.; Gu, L.; Liu, M.; Xie, L.H.; Qu, Z.; et al. Mitochondrial Ca2+ Influx Contributes to Arrhythmic Risk in Nonischemic Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e007805. [Google Scholar] [CrossRef]
- Schweitzer, M.K.; Wilting, F.; Sedej, S.; Dreizehnter, L.; Dupper, N.J.; Tian, Q.; Moretti, A.; My, I.; Kwon, O.; Priori, S.G.; et al. Suppression of Arrhythmia by Enhancing Mitochondrial Ca2+ Uptake in Catecholaminergic Ventricular Tachycardia Models. JACC Basic Transl. Sci. 2017, 2, 737–747. [Google Scholar] [CrossRef]
- Larbig, R.; Reda, S.; Paar, V.; Trost, A.; Leitner, J.; Weichselbaumer, S.; Motloch, K.A.; Wernly, B.; Arrer, A.; Strauss, B.; et al. Through modulation of cardiac Ca2+ handling, UCP2 affects cardiac electrophysiology and influences the susceptibility for Ca2+-mediated arrhythmias. Exp. Physiol. 2017, 102, 650–662. [Google Scholar] [CrossRef]
- Matsunaga, T.; Gu, N.; Yamazaki, H.; Tsuda, M.; Adachi, T.; Yasuda, K.; Moritani, T.; Tsuda, K.; Nonaka, M.; Nishiyama, T. Association of UCP2 and UCP3 polymorphisms with heart rate variability in Japanese men. J. Hypertens. 2009, 27, 305–313. [Google Scholar] [CrossRef]
- Lang, D.; Glukhov, A.V.; Efimova, T.; Efimov, I.R. Role of Pyk2 in cardiac arrhythmogenesis. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H975–H983. [Google Scholar] [CrossRef]
- Ezeani, M.; Elom, S. Necessity to evaluate PI3K/Akt signalling pathway in proarrhythmia. Open Heart 2017, 4, e000596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinaglia, T.; Jerosch-Herold, M.; Coelho-Filho, O.R. State-of-the-Art Quantitative Assessment of Myocardial Ischemia by Stress Perfusion Cardiac Magnetic Resonance. Magn. Reson. Imaging Clin. 2019, 27, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Boyman, L.; Williams, G.S.B.; Lederer, W.J. Mitochondrial Calcium and Ischemia: Reperfusion Injury in Heart. In Mitochondria and Cell Death; Springer: New York, NY, USA, 2016; pp. 17–43. [Google Scholar]
- Altamimi, T.R.; Karwi, Q.G.; Uddin, G.M.; Fukushima, A.; Kwong, J.Q.; Molkentin, J.D.; Lopaschuk, G.D. Cardiac-specific deficiency of the mitochondrial calcium uniporter augments fatty acid oxidation and functional reserve. J. Mol. Cell. Cardiol. 2019, 127, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Oropeza-Almazán, Y.; Vázquez-Garza, E.; Chapoy-Villanueva, H.; Torre-Amione, G.; García-Rivas, G. Small Interfering RNA Targeting Mitochondrial Calcium Uniporter Improves Cardiomyocyte Cell Viability in Hypoxia/Reoxygenation Injury by Reducing Calcium Overload. Oxidative Med. Cell. Longev. 2017, 2017, 5750897. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.P.; Wu, Y.; Joiner, M.A.; Koval, O.M.; Wilson, N.R.; Luczak, E.D.; Wang, Q.; Chen, B.; Gao, Z.; Zhu, Z.; et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. USA 2015, 112, 9129–9134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, R.J.; Menazza, S.; Holmström, K.M.; Amanakis, G.; Fergusson, M.; Ma, H.; Aponte, A.M.; Bernardi, P.; Finkel, T.; Murphy, E. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc. Res. 2019, 115, 385–394. [Google Scholar] [CrossRef]
- Xue, Q.; Pei, H.; Liu, Q.; Zhao, M.; Sun, J.; Gao, E.; Ma, X.; Tao, L. MICU1 protects against myocardial ischemia/reperfusion injury and its control by the importer receptor Tom70. Cell Death Dis. 2017, 8, e2923. [Google Scholar] [CrossRef]
- Kohlhaas, M.; Nickel, A.G.; Maack, C. Mitochondrial energetics and calcium coupling in the heart. J. Physiol. 2017, 595, 3753–3763. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Gong, X.; Yu, Y.; Li, M.; Liang, Y.; Qin, S.; Fulati, Z.; Zhou, N.; Shu, X.; Nie, Z.; et al. The mechanical effects of CRT promoting autophagy via mitochondrial calcium uniporter down-regulation and mitochondrial dynamics alteration. J. Cell. Mol. Med. 2019, 23, 3833–3842. [Google Scholar] [CrossRef]
- Cho, H.M.; Ryu, J.R.; Jo, Y.; Seo, T.W.; Choi, Y.N.; Kim, J.H.; Chung, J.M.; Cho, B.; Kang, H.C.; Yu, S.W.; et al. Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol. Cell 2019, 73, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Chen, Y.; He, R.; Shi, Y.; Su, L. Rescuing infusion of miRNA-1 prevents cardiac remodeling in a heart-selective miRNA deficient mouse. Biochem. Biophys. Res. Commun. 2018, 495, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Sygitowicz, G.; Polska, K.; Tomaniak, M.; Błaszczyk, O.; Kołtowski, Ł.; Puchta, D.; Malesa, K.; Kochanowski, J.; Sitkiewicz, D.; Filipiak, K.J. miR-1, miR-21, and galectin-3 in hypertensive patients with symptomatic heart failure and left ventricular hypertrophy. Kardiol. Pol. 2018, 76, 1009–1011. [Google Scholar]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Frontera, W.R.; Ochala, J. Skeletal Muscle: A Brief Review of Structure and Function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflugers Arch. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Mammucari, C.; Gherardi, G.; Zamparo, I.; Raffaello, A.; Boncompagni, S.; Chemello, F.; Cagnin, S.; Braga, A.; Zanin, S.; Pallafacchina, G.; et al. The Mitochondrial Calcium Uniporter Controls Skeletal Muscle Trophism In Vivo. Cell Rep. 2015, 10, 1269–1279. [Google Scholar] [CrossRef]
- Gherardi, G.; Nogara, L.; Ciciliot, S.; Fadini, G.P.; Blaauw, B.; Braghetta, P.; Bonaldo, P.; De Stefani, D.; Rizzuto, R.; Mammucari, C. Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ. 2019, 26, 362–381. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.Q.; Huo, J.; Bround, M.J.; Boyer, J.G.; Schwanekamp, J.A.; Ghazal, N.; Maxwell, J.T.; Jang, Y.C.; Khuchua, Z.; Shi, K.; et al. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, G.; Sharpe, J.A.; Koh, A.; Kouli, A.; Szabadkai, G.; Duchen, M.R. Pathological consequences of MICU1 mutations on mitochondrial calcium signalling and bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Smith, D.; Kamer, K.J.; Griffin, H.; Childs, A.M.; Pysden, K.; Titov, D.; Duff, J.; Pyle, A.; Taylor, R.W.; Yu-Wai-Man, P.; et al. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol. Genet. 2016, 2, e59. [Google Scholar] [CrossRef] [PubMed]
- Musa, S.; Eyaid, W.; Kamer, K.; Ali, R.; Al-Mureikhi, M.; Shahbeck, N.; Al Mesaifri, F.; Makhseed, N.; Mohamed, Z.; AlShehhi, W.A.; et al. A Middle Eastern Founder Mutation Expands the Genotypic and Phenotypic Spectrum of Mitochondrial MICU1 Deficiency: A Report of 13 Patients. JIMD Rep. 2019, 43, 79–83. [Google Scholar] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, A.; Van der Meulen, J.H.; Defour, A.; Hogarth, M.; Sreetama, S.C.; Reed, A.; Scheffer, L.; Chandel, N.S.; Jaiswal, J.K. Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci. Signal. 2017, 10, eaaj1978. [Google Scholar] [CrossRef]
- Zampieri, S.; Mammucari, C.; Romanello, V.; Barberi, L.; Pietrangelo, L.; Fusella, A.; Mosole, S.; Gherardi, G.; Hӧfer, C.; Lӧfler, S.; et al. Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiol. Rep. 2016, 4, e13005. [Google Scholar] [CrossRef]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Kannurpatti, S.S.; Biswal, B.B. Mitochondrial Ca2+ uniporter blockers influence activation-induced CBF response in the rat somatosensory cortex. J. Cereb. Blood Flow Metab. 2008, 28, 772–785. [Google Scholar] [CrossRef]
- Grossi, M.; Bhattachariya, A.; Nordström, I.; Turczyńska, K.M.; Svensson, D.; Albinsson, S.; Nilsson, B.O.; Hellstrand, P. Pyk2 inhibition promotes contractile differentiation in arterial smooth muscle. J. Cell. Physiol. 2017, 232, 3088–3102. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.; Xu, B.; Cai, Z.; Wang, L.; Tian, J.; Liu, Y.; Li, Y. The mitochondrial calcium uniporter is involved in mitochondrial calcium cycle dysfunction: Underlying mechanism of hypertension associated with mitochondrial tRNA Ile A4263G mutation. Int. J. Biochem. Cell Biol. 2016, 78, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Conaway, S.; Deshpande, D.A. Mitochondrial regulation of airway smooth muscle functions in health and pulmonary diseases. Arch. Biochem. Biophys. 2019, 663, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, P.; Yang, B.; Thompson, M.A.; Pabelick, C.M.; Prakash, Y.S.; Sieck, G.C. Inflammation alters regional mitochondrial Ca 2+ in human airway smooth muscle cells. Am. J. Physiol. Physiol. 2012, 303, C244–C256. [Google Scholar] [CrossRef] [PubMed]
- Vishnyakova, P.A.; Tarasova, N.V.; Volodina, M.A.; Tsvirkun, D.V.; Sukhanova, I.A.; Kurchakova, T.A.; Kan, N.E.; Medzidova, M.K.; Sukhikh, G.T.; Vysokikh, M.Y. Gestation age-associated dynamics of mitochondrial calcium uniporter subunits expression in feto-maternal complex at term and preterm delivery. Sci. Rep. 2019, 9, 5501. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.J.; Nemani, N.; Shanmughapriya, S.; Kumar, A.; Zhang, M.; Nathan, S.R.; Thomas, M.; Carvalho, E.; Ramachandran, K.; Srikantan, S.; et al. A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 2019, 5, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic Identification of MCU Modulators by Orthogonal Interspecies Chemical Screening. Mol. Cell 2017, 67, 711–723. [Google Scholar] [CrossRef]
- Imran, M.; Salehi, B.; Sharifi-Rad, J.; Aslam Gondal, T.; Saeed, F.; Imran, A.; Shahbaz, M.; Tsouh Fokou, P.V.; Umair Arshad, M.; Khan, H.; et al. Kaempferol: A Key Emphasis to Its Anticancer Potential. Molecules 2019, 24, 2277. [Google Scholar] [CrossRef]
- Imran, M.; Rauf, A.; Shah, Z.A.; Saeed, F.; Imran, A.; Arshad, M.U.; Ahmad, B.; Bawazeer, S.; Atif, M.; Peters, D.G.; et al. Chemo-preventive and therapeutic effect of the dietary flavonoid kaempferol: A comprehensive review. Phytother. Res. 2019, 33, 263–275. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarasova, N.V.; Vishnyakova, P.A.; Logashina, Y.A.; Elchaninov, A.V. Mitochondrial Calcium Uniporter Structure and Function in Different Types of Muscle Tissues in Health and Disease. Int. J. Mol. Sci. 2019, 20, 4823. https://doi.org/10.3390/ijms20194823
Tarasova NV, Vishnyakova PA, Logashina YA, Elchaninov AV. Mitochondrial Calcium Uniporter Structure and Function in Different Types of Muscle Tissues in Health and Disease. International Journal of Molecular Sciences. 2019; 20(19):4823. https://doi.org/10.3390/ijms20194823
Chicago/Turabian StyleTarasova, Nadezhda V., Polina A. Vishnyakova, Yulia A. Logashina, and Andrey V. Elchaninov. 2019. "Mitochondrial Calcium Uniporter Structure and Function in Different Types of Muscle Tissues in Health and Disease" International Journal of Molecular Sciences 20, no. 19: 4823. https://doi.org/10.3390/ijms20194823