Corticosteroids in Acute Lung Injury: The Dilemma Continues


Abstract
1. Introduction
2. Acute Lung Injury
2.1. Definitions and Incidence
2.2. Aetiology of ARDS
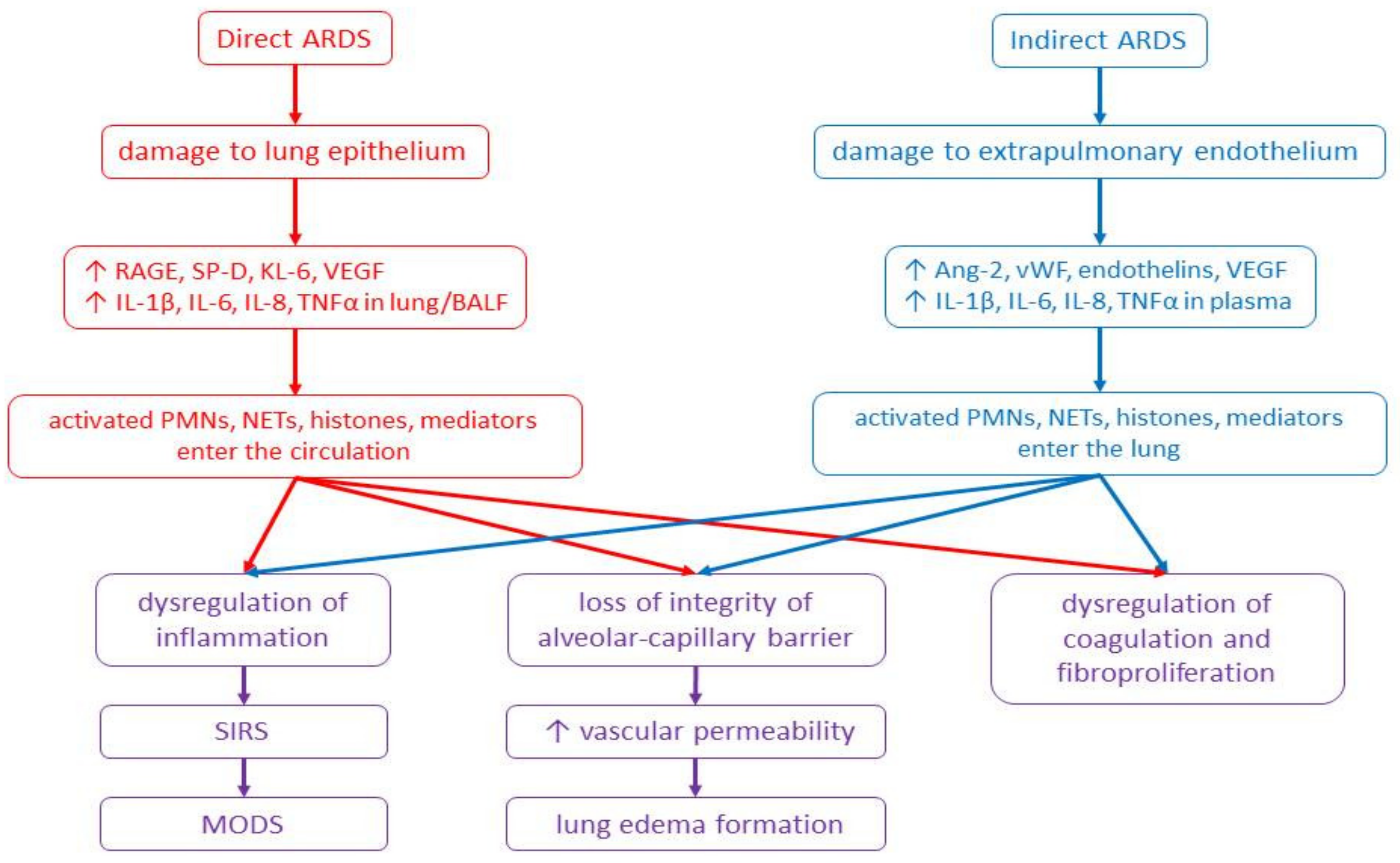
2.3. Pathophysiology of ARDS
2.4. Therapeutic Options in ARDS
3. Corticosteroids (CS)
3.1. Mechanisms of CS Action
3.2. Corticosteroids in Experimental Models of ALI
3.3. Corticosteroids in Patients with ARDS
3.4. Limitations
3.4.1. Animal vs. Human Studies
3.4.2. Human vs. Human Studies
3.4.3. Adverse Effects of CS
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| ALI | Acute lung injury |
| AP-1 | Activator protein-1 |
| ARDS | Acute respiratory distress syndrome |
| ATP | Adenosine triphosphate |
| BALF | Bronchoalveolar lavage fluid |
| CAP | Community-acquired pneumonia |
| cGCR | Cytosolic glucocorticoid receptor |
| CRP | C-reactive protein |
| CS | Corticosteroids |
| DAMPs | Danger-associated molecular patterns |
| DNA | Deoxyribonucleic acid |
| eNOS | Endothelial nitric oxide synthase |
| FiO2 | Fraction of inspired oxygen |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte macrophage colony-stimulating factor |
| GR | Glucocorticoid receptor |
| GRE | Glucocorticoid response element |
| HCAP | Healthcare-associated pneumonia |
| i.p. | Intraperitoneal |
| i.t. | Intratracheal |
| i.v. | Intravenous |
| ICAM | Intercellular adhesion molecule |
| ICU | Intensive care unit |
| IFN | Interferon |
| IL | Interleukin |
| LIS | Lung injury score |
| LPS | Lipopolysaccharide |
| MCP | Monocyte chemoattractant protein |
| MIP | Macrophage inflammatory protein |
| MMP | Matrix metalloproteinase |
| MODS | Multiple organ dysfunction syndrome |
| MPO | Myeloperoxidase |
| mRNA | Mitochondrial ribonucleic acid |
| NETs | Neutrophil extracellular traps |
| NF-κB | Nuclear factor-κB |
| NMBA | Neuromuscular blocking agents |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| PaCO2 | Arterial partial pressure of carbon dioxide |
| PaO2 | Arterial partial pressure of oxygen |
| PEEP | Positive end-expiratory pressure |
| RAGE | Receptor for advanced glycation end products |
| RCT | Randomized controlled trial |
| SIRS | Systemic inflammatory response syndrome |
| TGFβ | Transforming growth factor |
| TNF | Tumour necrosis factor |
| VILI | Ventilator-induced lung injury |
| VT | Tidal volume |
| vWf | Von Willebrand factor |
| WD | Wet-dry lung weight ratio |
References
- Matthay, M.A.; Zemans, R.L. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu. Rev. Pathol. 2011, 6, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Umbrello, M.; Formenti, P.; Bolgiaghi, L.; Chiumello, D. Current Concepts of ARDS: A Narrative Review. Int. J. Mol. Sci. 2016, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.R.; Artigas, A.; Brigham, K.L.; Carlet, J.; Falke, K.; Hudson, L.; Lamy, M.; LeGall, J.R.; Morris, A.; Spragg, R.; et al. Report of the American-European Consensus conference on acute respiratory distress syndrome: Definitions, mechanisms, relevant outcomes, and clinical trial coordination. J. Crit. Care 1994, 9, 72–81. [Google Scholar] [CrossRef]
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S.; ARDS Definition Task Force. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [PubMed]
- Fioretto, J.R.; de Carvalho, W.B. ARDS definitions in children: One step forward. J. Pediatr. 2014, 90, 211–212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Standiford, T.J.; Ward, P.A. Therapeutic targeting of acute lung injury and acute respiratory distress syndrome. Transl. Res. 2016, 167, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Mortelliti, M.P.; Manning, H.L. Acute respiratory distress syndrome. Am. Fam. Physician 2002, 65, 1823–1830. [Google Scholar]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef]
- Bhargava, M.; Wendt, C.H. Biomarkers in acute lung injury. Transl. Res. 2012, 159, 205–217. [Google Scholar] [CrossRef]
- Cross, L.J.; Matthay, M.A. Biomarkers in acute lung injury: Insights into the pathogenesis of acute lung injury. Crit. Care Clin. 2011, 27, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Sarma, J.V.; Ward, P.A. Oxidants and redox signaling in acute lung injury. Compr. Physiol. 2011, 1, 1365–1381. [Google Scholar] [PubMed]
- Ward, P.A. Oxidative stress: Acute and progressive lung injury. Ann. N. Y. Acad. Sci. 2010, 1203, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Pierrakos, C.; Karanikolas, M.; Scolletta, S.; Karamouzos, V.; Velissaris, D. Acute respiratory distress syndrome: Pathophysiology and therapeutic options. J. Clin. Med. Res. 2012, 4, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huang, Y.; Mao, P.; Zhang, J.; Li, Y. Sepsis and ARDS: The Dark Side of Histones. Mediat. Inflamm. 2015, 2015, 205054. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Wen, T.; Song, J.; Xie, D.; Wu, L.; Jiang, X.; Jiang, P.; Wen, Z. Extracellular histones are clinically relevant mediators in the pathogenesis of acute respiratory distress syndrome. Respir. Res. 2017, 18, 165. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Moochhala, S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J. Pathol. 2004, 202, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, S. Pathophysiology and biomarkers of acute respiratory distress syndrome. J. Intensive Care 2014, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, P.; D’Onofrio, D.; Chiumello, D.; Paolo, S.; Chiara, G.; Capelozzi, V.L.; Barbas, C.S.; Chiaranda, M.; Gattinoni, L. Pulmonary and extrapulmonary acute respiratory distress syndrome are different. Eur. Respir. J. 2003, 42, 48s–56s. [Google Scholar] [CrossRef]
- Shaver, C.M.; Bastarache, J.A. Clinical and biological heterogeneity in acute respiratory distress syndrome: Direct versus indirect lung injury. Clin. Chest Med. 2014, 35, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Calfee, C.S.; Janz, D.R.; Bernard, G.R.; May, A.K.; Kangelaris, K.N.; Matthay, M.A.; Ware, L.B. Distinct molecular phenotypes of direct vs. indirect ARDS in single-center and multicenter studies. Chest 2015, 147, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Dreyfuss, D.; Saumon, G. Ventilator-induced lung injury: Lessons from experimental studies. Am. J. Respir. Crit. Care Med. 1998, 157, 294–323. [Google Scholar] [CrossRef] [PubMed]
- Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A.; Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar]
- Needham, D.M.; Colantuoni, E.; Mendez-Tellez, P.A.; Dinglas, V.D.; Sevransky, J.E.; Dennison Himmelfarb, C.R.; Desai, S.V.; Shanholtz, C.; Brower, R.G.; Pronovost, P.J. Lung protective mechanical ventilation and two year survival in patients with acute lung injury: Prospective cohort study. BMJ 2012, 344, e2124. [Google Scholar] [CrossRef] [PubMed]
- Needham, D.M.; Yang, T.; Dinglas, V.D.; Mendez-Tellez, P.A.; Shanholtz, C.; Sevransky, J.E.; Brower, R.G.; Pronovost, P.J.; Colantuoni, E. Timing of low tidal volume ventilation and intensive care unit mortality in acute respiratory distress syndrome. A prospective cohort study. Am. J. Respir. Crit. Care Med. 2015, 191, 177–185. [Google Scholar] [CrossRef]
- Haberthür, C.; Seeberger, M.D. Acute respiratory distress syndrome and mechanical ventilation: Ups and downs of an ongoing relationship trap. J. Thorac. Dis. 2016, 8, E1608–E1609. [Google Scholar] [CrossRef]
- Umbrello, M.; Marino, A.; Chiumello, D. Tidal volume in acute respiratory distress syndrome: How best to select it. Ann. Transl. Med. 2017, 5, 287. [Google Scholar] [CrossRef]
- Chiumello, D.; Algieri, I.; Grasso, S.; Terragni, P.; Pelosi, P. Recruitment maneuvers in acute respiratory distress syndrome and during general anesthesia. Minerva Anestesiol. 2016, 82, 210–220. [Google Scholar]
- Lichtenstein, D.A.; Mezière, G.A. Relevance of lung ultrasound in the diagnosis of acute respiratory failure: The BLUE protocol. Chest 2008, 134, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Taccone, P.; Carlesso, E.; Marini, J.J. Prone position in acute respiratory distress syndrome. Rationale, indications, and limits. Am. J. Respir. Crit. Care Med. 2013, 188, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Guérin, C.; Mancebo, J. Prone positioning and neuromuscular blocking agents are part of standard care in severe ARDS patients: Yes. Intensive Care Med. 2015, 41, 2195–2197. [Google Scholar] [CrossRef] [PubMed]
- Dushianthan, A.; Grocott, M.P.; Postle, A.D.; Cusack, R. Acute respiratory distress syndrome and acute lung injury. Postgrad. Med. J. 2011, 87, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.J.; Mac Sweeney, R.; McAuley, D.F. Pharmacological treatments in ARDS; A state-of-the-art update. BMC Med. 2013, 11, 166. [Google Scholar] [CrossRef] [PubMed]
- Neto, A.S.; Pereira, V.G.; Espósito, D.C.; Damasceno, M.C.; Schultz, M.J. Neuromuscular blocking agents in patients with acute respiratory distress syndrome: A summary of the current evidence from three randomized controlled trials. Ann. Intensive Care 2012, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Hraiech, S.; Yoshida, T.; Papazian, L. Balancing neuromuscular blockade versus preserved muscle activity. Curr. Opin. Crit. Care 2015, 21, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef]
- Stahn, C.; Buttgereit, F. Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pract. Rheumatol. 2008, 4, 525–533. [Google Scholar] [CrossRef]
- Rogatsky, I.; Ivashkiv, L.B. Glucocorticoid modulation of cytokine signaling. Tissue Antigens 2006, 68, 1–12. [Google Scholar] [CrossRef]
- Kleinert, H.; Schwarz, P.M.; Förstermann, U. Regulation of the expression of inducible nitric oxide synthase. Biol. Chem. 2003, 384, 1343–1364. [Google Scholar] [CrossRef] [PubMed]
- Song, C.Z.; Tian, X.; Gelehrter, T.D. Glucocorticoid receptor inhibits transforming growth factor-beta signaling by directly targeting the transcriptional activation function of Smad3. Proc. Natl. Acad. Sci. USA 1999, 96, 11776–11781. [Google Scholar] [CrossRef] [PubMed]
- Lösel, R.; Wehling, M. Nongenomic actions of steroid hormones. Nat. Rev. Mol. Cell Biol. 2003, 4, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Buttgereit, F.; Scheffold, A. Rapid glucocorticoid effects on immune cells. Steroids 2002, 67, 529–534. [Google Scholar] [CrossRef]
- Bartholome, B.; Spies, C.M.; Gaber, T.; Schuchmann, S.; Berki, T.; Kunkel, D.; Bienert, M.; Radbruch, A.; Burmester, G.R.; Lauster, R.; et al. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J. 2004, 18, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Croxtall, J.D.; Choudhury, Q.; Flower, R.J. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br. J. Pharmacol. 2000, 130, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, K.; Matsuda, N.; Yamamoto, S.; Takano, K.; Takano, Y.; Yamazaki, H.; Kageyama, S.; Yokoo, H.; Nagata, T.; Hatakeyama, N.; et al. Modulation of glucocorticoid receptor expression, inflammation, and cell apoptosis in septic guinea pig lungs using methylprednisolone. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L998–L1006. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Annane, D.; Chrousos, G.P.; Marik, P.E.; Sinclair, S.E. Activation and regulation of systemic inflammation in ARDS: Rationale for prolonged glucocorticoid therapy. Chest 2009, 136, 1631–1643. [Google Scholar] [CrossRef]
- Incerpi, E.K.; Oliveira, L.M.; Pereira, E.M.; Soncini, R. Inhibition of endogenous glucocorticoid synthesis aggravates lung injury triggered by septic shock in rats. Int. J. Exp. Pathol. 2015, 96, 133–139. [Google Scholar] [CrossRef]
- Meduri, G.U.; Bell, W.; Sinclair, S.; Annane, D. Pathophysiology of acute respiratory distress syndrome. Glucocorticoid receptor-mediated regulation of inflammation and response to prolonged glucocorticoid treatment. Presse Med. 2011, 40, e543–e560. [Google Scholar] [CrossRef]
- Schwingshackl, A.; Meduri, G.U. Rationale for Prolonged Glucocorticoid Use in Pediatric ARDS: What the Adults Can Teach Us. Front. Pediatr. 2016, 4, 58. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Lange, D.W.; Meulenbelt, J. Do corticosteroids have a role in preventing or reducing acute toxic lung injury caused by inhalation of chemical agents? Clin. Toxicol. 2011, 49, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Brown, R.; Jugg, B.; Platt, J.; Mann, T.; Masey, C.; Jenner, J.; Rice, P. The effect of steroid treatment with inhaled budesonide or intravenous methylprednisolone on phosgene-induced acute lung injury in a porcine model. Mil. Med. 2009, 174, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Pauluhn, J.; Trübel, H.; Wang, C. Corticosteroids found ineffective for phosgene-induced acute lung injury in rats. Toxicol. Lett. 2014, 229, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Pauluhn, J.; Trübel, H.; Wang, C. Single high-dose dexamethasone and sodium salicylate failed to attenuate phosgene-induced acute lung injury in rats. Toxicology 2014, 315, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Sjöblom, E.; Höjer, J.; Kulling, P.E.; Stauffer, K.; Suneson, A.; Ludwigs, U. A placebo-controlled experimental study of steroid inhalation therapy in ammonia-induced lung injury. J. Toxicol. Clin. Toxicol. 1999, 37, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Rocco, P.R.; Souza, A.B.; Faffe, D.S.; Pássaro, C.P.; Santos, F.B.; Negri, E.M.; Lima, J.G.; Contador, R.S.; Capelozzi, V.L.; Zin, W.A. Effect of corticosteroid on lung parenchyma remodeling at an early phase of acute lung injury. Am. J. Respir. Crit. Care Med. 2003, 168, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; Wang, L.F.; Su, B.; Hsu, H.H. Methylprednisolone effects on oxygenation and histology in a rat model of acute lung injury. Pulm. Pharmacol. Ther. 2003, 16, 215–220. [Google Scholar] [CrossRef]
- Chen, J.; Mo, Y.; Schlueter, C.F.; Hoyle, G.W. Inhibition of chlorine-induced pulmonary inflammation and edema by mometasone and budesonide. Toxicol. Appl. Pharmacol. 2013, 272, 408–413. [Google Scholar] [CrossRef]
- Musah, S.; Chen, J.; Schlueter, C.; Humphrey, D.M., Jr.; Stocke, K.; Hoyle, M.I.; Hoyle, G.W. Inhibition of chlorine-induced airway fibrosis by budesonide. Toxicol. Appl. Pharmacol. 2019, 363, 11–21. [Google Scholar] [CrossRef]
- Gunnarsson, M.; Walther, S.M.; Seidal, T.; Lennquist, S. Effects of inhalation of corticosteroids immediately after experimental chlorine gas lung injury. J. Trauma Acute Care Surg. 2000, 48, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Walther, S.M. Inhaled budesonide in experimental chlorine gas lung injury: Influence of time interval between injury and treatment. Intensive Care Med. 2002, 28, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Winskog, C.; Edston, E.; Walther, S.M. Inhaled and intravenous corticosteroids both attenuate chlorine gas-induced lung injury in pigs. Acta Anaesthesiol. Scand. 2005, 49, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Xinmin, D.; Yunyou, D.; Chaosheng, P.; Huasong, F.; Pingkun, Z.; Jiguang, M.; Zhiqian, X.; Qinzhi, X. Dexamethasone treatment attenuates early seawater instillation-induced acute lung injury in rabbits. Pharmacol. Res. 2006, 53, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Kosutova, P.; Mikolka, P.; Balentova, S.; Adamkov, M.; Kolomaznik, M.; Calkovska, A.; Mokra, D. Intravenous dexamethasone attenuated inflammation and influenced apoptosis of lung cells in an experimental model of acute lung injury. Physiol. Res. 2016, 65, S663–S672. [Google Scholar] [PubMed]
- Mokra, D.; Kosutova, P.; Balentova, S.; Adamkov, M.; Mikolka, P.; Mokry, J.; Antosova, M.; Calkovska, A. Effects of budesonide on the lung functions, inflammation and apoptosis in a saline-lavage model of acute lung injury. J. Physiol. Pharmacol. 2016, 67, 919–932. [Google Scholar]
- Nin, N.; Peñuelas, O.; de Paula, M.; Lorente, J.A.; Fernández-Segoviano, P.; Esteban, A. Ventilation-induced lung injury in rats is associated with organ injury and systemic inflammation that is attenuated by dexamethasone. Crit. Care Med. 2006, 34, 1093–1098. [Google Scholar] [CrossRef]
- Ju, Y.N.; Yu, K.J.; Wang, G.N. Budesonide ameliorates lung injury induced by large volume ventilation. BMC. Pulm. Med. 2016, 16, 90. [Google Scholar] [CrossRef]
- Held, H.D.; Boettcher, S.; Hamann, L.; Uhlig, S. Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am. J. Respir. Crit. Care Med. 2001, 163, 711–716. [Google Scholar] [CrossRef]
- Jansson, A.H.; Eriksson, C.; Wang, X. Effects of budesonide and N-acetylcysteine on acute lung hyperinflation, inflammation and injury in rats. Vasc. Pharmacol. 2005, 43, 101–111. [Google Scholar] [CrossRef]
- Rocksén, D.; Lilliehöök, B.; Larsson, R.; Johansson, T.; Bucht, A. Differential anti-inflammatory and anti-oxidative effects of dexamethasone and N-acetylcysteine in endotoxin-induced lung inflammation. Clin. Exp. Immunol. 2000, 122, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.L.; Garcia, C.S.; Maronas, P.A.; Cagido, V.R.; Negri, E.M.; Damaceno-Rodrigues, N.R.; Ventura, G.M.; Bozza, P.T.; Zin, W.A.; Capelozzi, V.L.; et al. Early short-term versus prolonged low-dose methylprednisolone therapy in acute lung injury. Eur. Respir. J. 2009, 33, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Q.; Zhou, X.; Zhou, Y.; Rong, L.; Gao, L.; Xu, W. Low-dose dexamethasone alleviates lipopolysaccharide-induced acute lung injury in rats and upregulates pulmonary glucocorticoid receptors. Respirology 2008, 13, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Ju, Y.N. Budesonide Attenuates Ventilator-induced Lung Injury in a Rat Model of Inflammatory Acute Respiratory Distress Syndrome. Arch. Med. Res. 2016, 47, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Borg, T.; Gerdin, B.; Modig, J. Prophylactic and delayed treatment with high-dose methylprednisolone in a porcine model of early ARDS induced by endotoxaemia. Acta Anaesthesiol. Scand. 1985, 29, 831–845. [Google Scholar] [CrossRef]
- Kuwabara, K.; Furue, S.; Tomita, Y.; Ueno, M.; Ono, T.; Matsukawa, A.; Yoshinaga, M.; Mikawa, K.; Nishina, K.; Shiga, M.; et al. Effect of methylprednisolone on phospholipase A(2) activity and lung surfactant degradation in acute lung injury in rabbits. Eur. J. Pharmacol. 2001, 433, 209–216. [Google Scholar] [CrossRef]
- Ottosson, J.; Dawidson, I.J.; Svensjö, E.; Brattsand, R.; Dahlbäck, M. Intravenous versus intrapulmonary administration of corticosteroids in combination with fluid infusion in experimental septic shock. Acta Chir. Scand. 1987, 153, 507–512. [Google Scholar]
- Walther, S.; Jansson, I.; Gunnarsson, M.; Lennquist, S. Nebulized corticosteroid improves pulmonary function and outcome in experimental porcine septicemia. Acta Anaesthesiol. Scand. 1991, 35, 635–641. [Google Scholar] [CrossRef]
- Gao, W.; Ju, N. Budesonide inhalation ameliorates endotoxin-induced lung injury in rabbits. Exp. Biol. Med. 2015, 240, 1708–1716. [Google Scholar] [CrossRef]
- Hicks, C.W.; Sweeney, D.A.; Danner, R.L.; Eichacker, P.Q.; Suffredini, A.F.; Feng, J.; Sun, J.; Behrend, E.N.; Solomon, S.B.; Natanson, C. Efficacy of selective mineralocorticoid and glucocorticoid agonists in canine septic shock. Crit. Care Med. 2012, 40, 199–207. [Google Scholar] [CrossRef]
- Wang, Z.; Kang, J.S.; Li, Y.; Yuan, Z.X.; Liu, S.S.; Sun, L.K. The effects of dexamethasone on rat brain cortical nuclear factor kappa B (NF-kappaB) in endotoxic shock. Toxicol. Appl. Pharmacol. 2006, 214, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.R.; Luce, J.M.; Sprung, C.L.; Rinaldo, J.E.; Tate, R.M.; Sibbald, W.J.; Kariman, K.; Higgins, S.; Bradley, R.; Metz, C.A.; et al. High-dose corticosteroids in patients with the adult respiratory distress syndrome. N. Engl. J. Med. 1987, 317, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Headley, A.S.; Golden, E.; Carson, S.J.; Umberger, R.A.; Kelso, T.; Tolley, E.A. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: A randomized controlled trial. JAMA 1998, 280, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Confalonieri, M.; Urbino, R.; Potena, A.; Piattella, M.; Parigi, P.; Puccio, G.; Della Porta, R.; Giorgio, C.; Blasi, F.; Umberger, R.; et al. Hydrocortisone infusion for severe community-acquired pneumonia: A preliminary randomized study. Am. J. Respir. Crit. Care Med. 2005, 171, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, K.P.; Hudson, L.D.; Goodman, R.B.; Hough, C.L.; Lanken, P.N.; Hyzy, R.; Thompson, B.T.; Ancukiewicz, M. National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N. Engl. J. Med. 2006, 354, 1671–1684. [Google Scholar] [PubMed]
- Oppert, M.; Schindler, R.; Husung, C.; Offermann, K.; Gräf, K.J.; Boenisch, O.; Barckow, D.; Frei, U.; Eckardt, K.U. Low-dose hydrocortisone improves shock reversal and reduces cytokine levels in early hyperdynamic septic shock. Crit. Care Med. 2005, 33, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Golden, E.; Freire, A.X.; Taylor, E.; Zaman, M.; Carson, S.J.; Gibson, M.; Umberger, R. Methylprednisolone infusion in early severe ARDS: Results of a randomized controlled trial. Chest 2007, 131, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Meijvis, S.C.; Hardeman, H.; Remmelts, H.H.; Heijligenberg, R.; Rijkers, G.T.; van Velzen-Blad, H.; Voorn, G.P.; van de Garde, E.M.; Endeman, H.; Grutters, J.C.; et al. Dexamethasone and length of hospital stay in patients with community-acquired pneumonia: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 377, 2023–2030. [Google Scholar] [CrossRef]
- Tongyoo, S.; Permpikul, C.; Mongkolpun, W.; Vattanavanit, V.; Udompanturak, S.; Kocak, M.; Meduri, G.U. Hydrocortisone treatment in early sepsis-associated acute respiratory distress syndrome: Results of a randomized controlled trial. Crit. Care 2016, 20, 329. [Google Scholar] [CrossRef]
- Annane, D.; Renault, A.; Brun-Buisson, C.; Megarbane, B.; Quenot, J.P.; Siami, S.; Cariou, A.; Forceville, X.; Schwebel, C.; Martin, C.; et al. Hydrocortisone plus Fludrocortisone for Adults with Septic Shock. N. Engl. J. Med. 2018, 378, 809–818. [Google Scholar] [CrossRef]
- Keh, D.; Trips, E.; Marx, G.; Wirtz, S.P.; Abduljawwad, E.; Bercker, S.; Bogatsch, H.; Briegel, J.; Engel, C.; Gerlach, H.; et al. Effect of Hydrocortisone on Development of Shock Among Patients with Severe Sepsis: The HYPRESS Randomized Clinical Trial. JAMA 2016, 316, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, B.; Finfer, S.; Cohen, J.; Rajbhandari, D.; Arabi, Y.; Bellomo, R.; Billot, L.; Correa, M.; Glass, P.; Harward, M.; et al. Adjunctive Glucocorticoid Therapy in Patients with Septic Shock. N. Engl. J. Med. 2018, 378, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Headley, S.; Tolley, E.; Shelby, M.; Stentz, F.; Postlethwaite, A. Plasma and BAL cytokine response to corticosteroid rescue treatment in late ARDS. Chest 1995, 108, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Tolley, E.A.; Chrousos, G.P.; Stentz, F. Prolonged methylprednisolone treatment suppresses systemic inflammation in patients with unresolving acute respiratory distress syndrome: Evidence for inadequate endogenous glucocorticoid secretion and inflammation-induced immune cell resistance to glucocorticoids. Am. J. Respir. Crit. Care Med. 2002, 165, 983–991. [Google Scholar]
- Peter, J.V.; John, P.; Graham, P.L.; Moran, J.L.; George, I.A.; Bersten, A. Corticosteroids in the prevention and treatment of acute respiratory distress syndrome (ARDS) in adults: Meta-analysis. BMJ 2008, 336, 1006–1009. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.M.; Craig, J.C.; Eslick, G.D.; Seppelt, I.; McLean, A.S. Use of corticosteroids in acute lung injury and acute respiratory distress syndrome: A systematic review and meta-analysis. Crit. Care Med. 2009, 37, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Sessler, C.N.; Gay, P.C. Are corticosteroids useful in late-stage acute respiratory distress syndrome? Respir. Care 2010, 55, 43–55. [Google Scholar]
- Seam, N.; Meduri, G.U.; Wang, H.; Nylen, E.S.; Sun, J.; Schultz, M.J.; Tropea, M.; Suffredini, A.F. Effects of methylprednisolone infusion on markers of inflammation, coagulation, and angiogenesis in early acute respiratory distress syndrome. Crit. Care Med. 2012, 40, 495–501. [Google Scholar] [CrossRef]
- Meduri, G.U.; Siemieniuk, R.A.C.; Ness, R.A.; Seyler, S.J. Prolonged low-dose methylprednisolone treatment is highly effective in reducing duration of mechanical ventilation and mortality in patients with ARDS. J. Intensive Care 2018, 6, 53. [Google Scholar] [CrossRef]
- Kido, T.; Muramatsu, K.; Asakawa, T.; Otsubo, H.; Ogoshi, T.; Oda, K.; Kubo, T.; Fujino, Y.; Matsuda, S.; Mayumi, T.; et al. The relationship between high-dose corticosteroid treatment and mortality in acute respiratory distress syndrome: A retrospective and observational study using a nationwide administrative database in Japan. BMC Pulm. Med. 2018, 18, 28. [Google Scholar] [CrossRef]
- Ariani, F.; Liu, K.; Jing, Z.; Qu, J. Glucocorticosteroid in treatment of severe pneumonia. Mediat. Inflamm. 2013, 2013, 865635. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vidal, C.; Calbo, E.; Pascual, V.; Ferrer, C.; Quintana, S.; Garau, J. Effects of systemic steroids in patients with severe community-acquired pneumonia. Eur. Respir. J. 2007, 30, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Snijders, D.; Daniels, J.M.; de Graaff, C.S.; van der Werf, T.S.; Boersma, W.G. Efficacy of corticosteroids in community-acquired pneumonia: A randomized double-blinded clinical trial. Am. J. Respir. Crit. Care Med. 2010, 181, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Polverino, E.; Cillóniz, C.; Dambrava, P.; Gabarrús, A.; Ferrer, M.; Agustí, C.; Prina, E.; Montull, B.; Menendez, R.; Niederman, M.S.; et al. Systemic corticosteroids for community-acquired pneumonia: Reasons for use and lack of benefit on outcome. Respirology 2013, 18, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Skalsky, K.; Avni, T.; Carrara, E.; Leibovici, L.; Paul, M. Corticosteroids for pneumonia. Cochrane Database Syst. Rev. 2017, 12, CD007720. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.N.; Chen, G.; Sun, J.; Liang, B.M.; Liang, Z.A. The effect of corticosteroids on mortality of patients with influenza pneumonia: A systematic review and meta-analysis. Crit. Care 2019, 23, 99. [Google Scholar] [CrossRef] [PubMed]
- Drago, B.B.; Kimura, D.; Rovnaghi, C.R.; Schwingshackl, A.; Rayburn, M.; Meduri, G.U.; Anand, K.J. Double-blind, placebo-controlled pilot randomized trial of methylprednisolone infusion in pediatric acute respiratory distress syndrome. Pediatr. Crit. Care Med. 2015, 16, e74–e81. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, A.; Kimura, D.; Rovnaghi, C.R.; Saravia, J.S.; Cormier, S.A.; Teng, B.; West, A.N.; Meduri, U.G.; Anand, K.J. Regulation of inflammatory biomarkers by intravenous methylprednisolone in pediatric ARDS patients: Results from a double-blind, placebo-controlled randomized pilot trial. Cytokine 2016, 77, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Kimura, D.; Saravia, J.; Rovnaghi, C.R.; Meduri, G.U.; Schwingshackl, A.; Cormier, S.A.; Anand, K.J. Plasma Biomarker Analysis in Pediatric ARDS: Generating Future Framework from a Pilot Randomized Control Trial of Methylprednisolone: A Framework for Identifying Plasma Biomarkers Related to Clinical Outcomes in Pediatric ARDS. Front. Pediatr. 2016, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, F.; Rochwerg, B.; Lytvyn, L.; Guyatt, G.H.; Møller, M.H.; Annane, D.; Kho, M.E.; Adhikari, N.K.J.; Machado, F.; Vandvik, P.O.; et al. Corticosteroid therapy for sepsis: A clinical practice guideline. BMJ 2018, 362, k3284. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Sébille, V.; Bellissant, E. Ger-Inf-05 Study Group. Effect of low doses of corticosteroids in septic shock patients with or without early acute respiratory distress syndrome. Crit. Care Med. 2006, 34, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Bellissant, E.; Bollaert, P.E.; Briegel, J.; Confalonieri, M.; De Gaudio, R.; Keh, D.; Kupfer, Y.; Oppert, M.; Meduri, G.U. Corticosteroids in the treatment of severe sepsis and septic shock in adults: A systematic review. JAMA 2009, 301, 2362–2375. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Bellissant, E.; Bollaert, P.E.; Briegel, J.; Keh, D.; Kupfer, Y. Corticosteroids for treating sepsis. Cochrane Database Syst. Rev. 2015, 12, CD002243. [Google Scholar] [CrossRef] [PubMed]
- Rygård, S.L.; Butler, E.; Granholm, A.; Møller, M.H.; Cohen, J.; Finfer, S.; Perner, A.; Myburgh, J.; Venkatesh, B.; Delaney, A. Low-dose corticosteroids for adult patients with septic shock: A systematic review with meta-analysis and trial sequential analysis. Intensive Care Med. 2018, 44, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Zhang, Y.; Tang, J.; Lunsford, L.D.; Li, T.; Tang, R.; He, J.; Xu, P.; Faramand, A.; Xu, J.; et al. Association of Corticosteroid Treatment with Outcomes in Adult Patients With Sepsis: A Systematic Review and Meta-analysis. JAMA Intern. Med. 2019, 179, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef] [PubMed]
- Jantz, M.A.; Sahn, S.A. Corticosteroids in acute respiratory failure. Am. J. Respir. Crit. Care Med. 1999, 160, 1079–1100. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.R. Glucocorticoids and cardiovascular disease. Eur. J. Endocrinol. 2007, 157, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Schäcke, H.; Döcke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Czock, D.; Keller, F.; Rasche, F.M.; Häussler, U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin. Pharmacokinet. 2005, 44, 61–98. [Google Scholar] [CrossRef]
- Hanson, P.; Dive, A.; Brucher, J.M.; Bisteau, M.; Dangoisse, M.; Deltombe, T. Acute corticosteroid myopathy in intensive care patients. Muscle Nerve 1997, 20, 1371–1380. [Google Scholar] [CrossRef]
- Eddelien, H.S.; Hoffmeyer, H.W.; Lund, E.L.; Lauritsen, A.Ø. Glucocorticoid-induced myopathy in the intensive care unit. BMJ Case Rep. 2015, 2015, bcr2015209793. [Google Scholar] [CrossRef] [PubMed]
- Haran, M.; Schattner, A.; Kozak, N.; Mate, A.; Berrebi, A.; Shvidel, L. Acute steroid myopathy: A highly overlooked entity. QJM 2018, 111, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Vatti, R.R.; Ali, F.; Teuber, S.; Chang, C.; Gershwin, M.E. Hypersensitivity reactions to corticosteroids. Clin. Rev. Allergy Immunol. 2014, 47, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Bahna, S.L. Immediate hypersensitivity reactions to corticosteroids. Ann. Allergy Asthma Immunol. 2015, 115, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, A.N.; Arvikar, S.; Stern, T.A.; Axelrod, L. The neuropsychiatric complications of glucocorticoid use: Steroid psychosis revisited. Psychosomatics 2012, 53, 103–115. [Google Scholar] [CrossRef] [PubMed]
- West, S.; Kenedi, C. Strategies to prevent the neuropsychiatric side-effects of corticosteroids: A case report and review of the literature. Curr. Opin. Organ Transplant. 2014, 19, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M. Clinical Pharmacology of Corticosteroids. Respir. Care 2018, 63, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Weber-Carstens, S.; Deja, M.; Bercker, S.; Dimroth, A.; Ahlers, O.; Kaisers, U.; Keh, D. Impact of bolus application of low-dose hydrocortisone on glycemic control in septic shock patients. Intensive Care Med. 2007, 33, 730–733. [Google Scholar] [CrossRef]
- Loisa, P.; Parviainen, I.; Tenhunen, J.; Hovilehto, S.; Ruokonen, E. Effect of mode of hydrocortisone administration on glycemic control in patients with septic shock: A prospective randomized trial. Crit. Care 2007, 11, R21. [Google Scholar] [CrossRef]
- Fernandes, A.B.; Zin, W.A.; Rocco, P.R. Corticosteroids in acute respiratory distress syndrome. Braz. J. Med. Biol. Res. 2005, 38, 147–159. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mok, Y.H.; Lee, J.H.; Rehder, K.J.; Turner, D.A. Adjunctive treatments in pediatric acute respiratory distress syndrome. Expert Rev. Respir. Med. 2014, 8, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Tamburro, R.F.; Kneyber, M.C. Pediatric Acute Lung Injury Consensus Conference Group. Pulmonary specific ancillary treatment for pediatric acute respiratory distress syndrome: Proceedings from the Pediatric Acute Lung Injury Consensus Conference. Pediatr. Crit. Care Med. 2015, 16, S61–S72. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author (Year) | Diagnosis | Total No. of Patients (CS/Placebo) | Treatment/Dose | Duration of Therapy (Days) | Outcomes in CS Groups |
|---|---|---|---|---|---|
| Bernard et al. (1987) [82] | Early ARDS | 99 (50 CS/49 placebo) | Methylprednisolone (30 mg/kg 6-hourly) | 1 | No differences in mortality, infectious complications or ventilatory characteristics 5 days after entry |
| Meduri et al. (1998) [83] | Severe persistent ARDS | 24 (16 CS/8 placebo) | Methylprednisolone (2 mg/kg loading dose, then 2 mg/kg/day for days 1–14, 1 mg/kg/day for days 15–21, 0.5 mg/kg/day for days 22–28, 0.25 mg/kg/day for days 29–30, and 0.125 mg/kg/day for days 31–32) | 14 | Improvements in LIS and PaO2/FiO2 Reduced ICU and hospital mortality No increase in infection complications |
| Confalonieri et al. (2005) [84] | Severe community-acquired pneumonia | 46 (23 CS/23 placebo) | Hydrocortisone (bolus 200 mg, then infusion 10 mg/hour) | 7 | Improvement in PaO2/FiO2 and chest X-ray score Reduction in CRP levels, MODS score, and delayed septic shock Reduction in length of hospital stay and mortality |
| Steinberg et al. (2006) [85] | Persistent ARDS | 180 (89 CS/91 placebo) | Methylprednisolone (2 mg/kg loading dose, then 0.5 mg/kg 6-hourly for 14 days, 0.5 mg/kg 12-hourly for 7 days) | 14 | Starting CS therapy later than 2 weeks after the onset of ARDS associated with increased mortality |
| Annane et al. (2006) [86] | Septic patients with ARDS | 177 (85 CS, including 23 responders/92 placebo, including 25 responders) | Hydrocortisone (50 mg 6-hourly) and 9-α- fludrocortisone (50 mg once a day) | 7 | In nonresponders to short corticotrophin test: decreased mortality and more ventilator days off, no difference in responders |
| Meduri et al. (2007) [87] | Early severe ARDS | 91 (63 CS/28 placebo) | Methylprednisolone (1 mg/kg loading dose, then 1 mg/kg/day for days 1–14, 0.5 mg/kg/day for days 15–21, 0.25 mg/kg/day for days 22–25, 0.125 mg/kg/day for days 26–28) | Shorter duration of mechanical ventilation Reduced ICU stay and ICU mortality No increase in infectious complications | |
| Meijvis et al. (2011) [88] | Community-acquired pneumonia | 304 (151 CS/153 placebo) | Dexamethasone (5 mg once a day) | 4 | Shorter length of stay No differences in hospital mortality or severe adverse events More common hyperglycaemia |
| Tongyoo et al. (2016) [89] | Severe sepsis or septic shock | 197 (98 CS, 99 placebo) | Hydrocortisone (50 mg 6-hourly) | 7 | Improvement in PaO2/FiO2 and LIS No survival benefit More frequent hyperglycaemia |
| Keh et al. (2016) [90] | Severe sepsis | 380 190 CS/190 placebo) | Hydrocortisone (200 mg continuous infusion for 5 days, then dose tapering until day 11) | 5 | No reduction of risk of septic shockNo differences in mortality in ICU or in the hospital Higher occurrence of secondary infections, muscle weakness, and hyperglycemia |
| Annane et al. (2018) [91] | Septic shock | 1241 (614 CS/627 placebo) | Hydrocortisone (50 mg 6-hourly) and 9-α- fludrocortisone (50 mg once a day) | 7 | Lower 90-day mortality More vasopressor-free days and organ-failure-free days to day 28 No difference in ventilator-free days and rate of serious adverse events More common hyperglycemia |
| Venkatesh et al. (2018) [92] | Septic shock | 3658 (1832 CS/1826 placebo) | Hydrocortisone (200 mg per day) | 7 | No improvement in 90-day mortality Faster resolution of shock Fewer blood transfusions Shorter duration of initial mechanical ventilation No difference in ventilation-free days |
| Author (Year) | Diagnosis | Total No. of Patients (CS/Placebo) | Treatment/Dose | Duration of Treatment (Days) | Outcomes in CS Groups |
|---|---|---|---|---|---|
| Drago et al. (2015) [107] | Paediatric ARDS | 35 (17 CS, 18 placebo) | Methylprednisolone (loading dose 2 mg/kg, then 1 mg/kg/day infusion) | 7 | No differences in length of mechanical ventilation, ICU stay, hospital stay, or mortality Lower PaCO2 on days 2 and 3, higher pH on day 2, and higher PaO2/FiO2 on days 8 and 9 Lower requirement for treatment of postextubation stridor or supplemental oxygen at ICU transfer No adverse effects |
| Schwingshackl et al. (2016) [108] | Paediatric ARDS | 35 (17 CS, 18 placebo) | Methylprednisolone (loading dose 2 mg/kg, then 1 mg/kg/day infusion) | 7 | On day 7, increased WBC and platelets counts, lower IFN-α, IL-6, IL-10, MCP-1, G-CSF and GM-CSF levels, and higher IL-17α levels in comparison to study entry |
| Kimura et al. (2016) [109] | Paediatric ARDS | 35 (17 CS, 18 placebo) | Methylprednisolone (loading dose 2 mg/kg, then 1 mg/kg/day infusion) | 7 | On day 7, reduction in MMP-8 levels, no increases in sICAM-1, on day 8 positive correlation of sRAGE levels with PaO2/FiO2, negative correlation of O2 requirements at ICU transfer with day 7 sICAM-1 levels |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokra, D.; Mikolka, P.; Kosutova, P.; Mokry, J. Corticosteroids in Acute Lung Injury: The Dilemma Continues. Int. J. Mol. Sci. 2019, 20, 4765. https://doi.org/10.3390/ijms20194765
Mokra D, Mikolka P, Kosutova P, Mokry J. Corticosteroids in Acute Lung Injury: The Dilemma Continues. International Journal of Molecular Sciences. 2019; 20(19):4765. https://doi.org/10.3390/ijms20194765
Chicago/Turabian StyleMokra, Daniela, Pavol Mikolka, Petra Kosutova, and Juraj Mokry. 2019. "Corticosteroids in Acute Lung Injury: The Dilemma Continues" International Journal of Molecular Sciences 20, no. 19: 4765. https://doi.org/10.3390/ijms20194765
APA StyleMokra, D., Mikolka, P., Kosutova, P., & Mokry, J. (2019). Corticosteroids in Acute Lung Injury: The Dilemma Continues. International Journal of Molecular Sciences, 20(19), 4765. https://doi.org/10.3390/ijms20194765