Smad7 Binds Differently to Individual and Tandem WW3 and WW4 Domains of WWP2 Ubiquitin Ligase Isoforms

 ,
,
Abstract
1. Introduction
2. Results
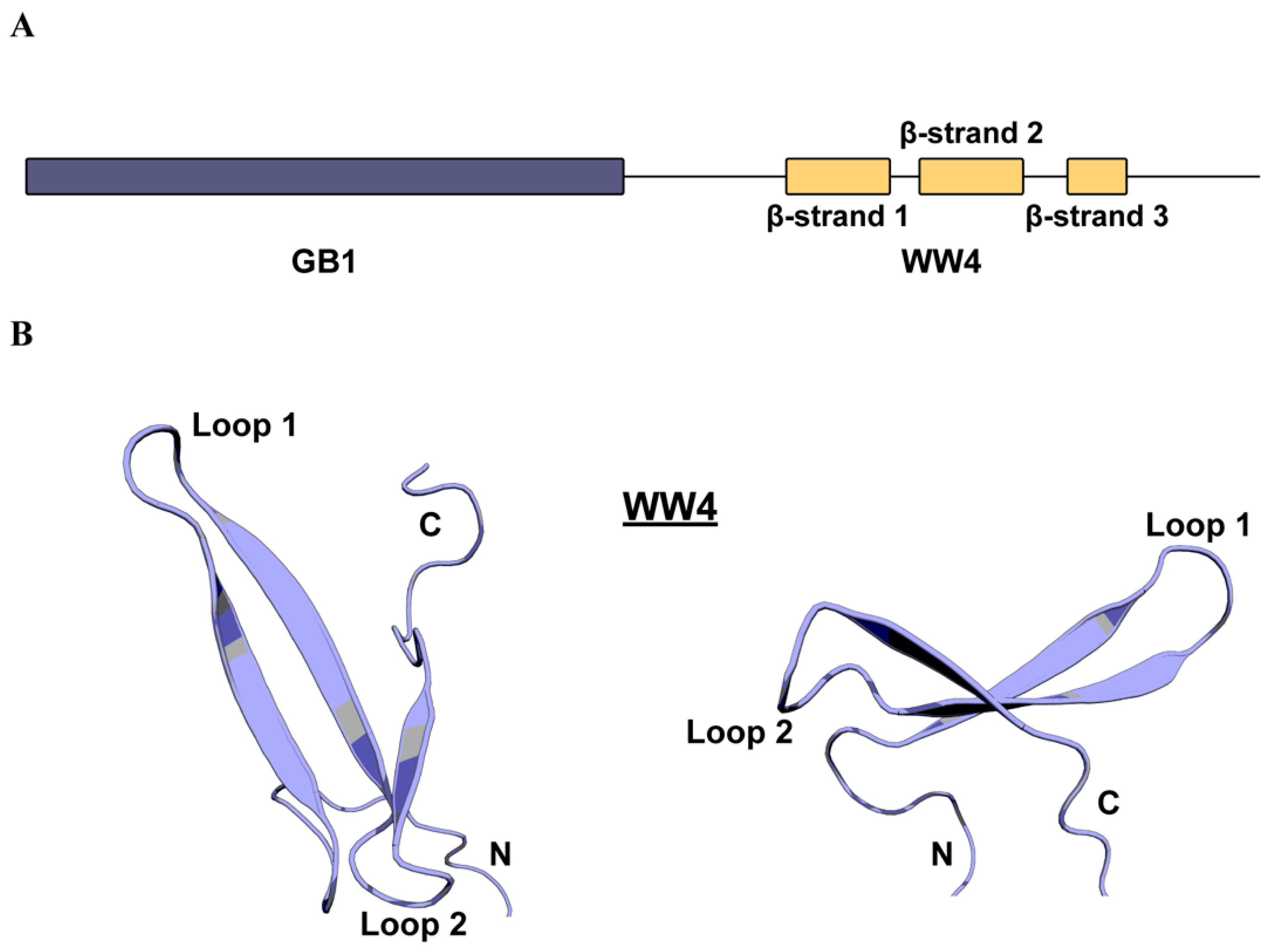
2.1. WWP2 WW4 Adopts The Canonical WW Domain Fold
2.2. WWP2 WW4 Binds to A Smad7 and Phospho-Smad7 PPxY Motif
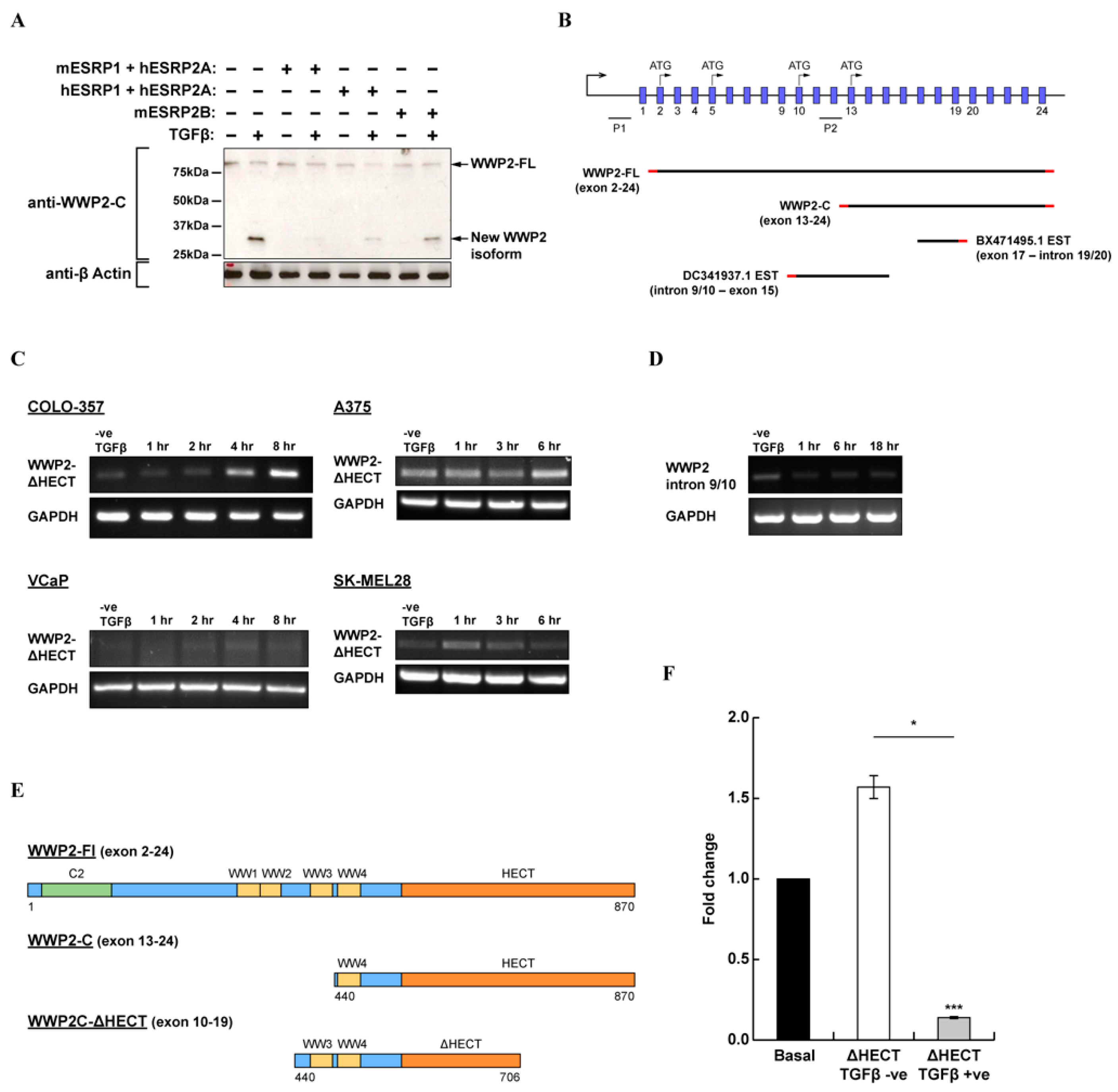
2.3. Evidence for A Novel WWP2 Isoform Which Contains Tandem WW3–WW4 Domains and A Truncated HECT Domain That Can Inhibit TGFβ-Dependent Signalling Activity
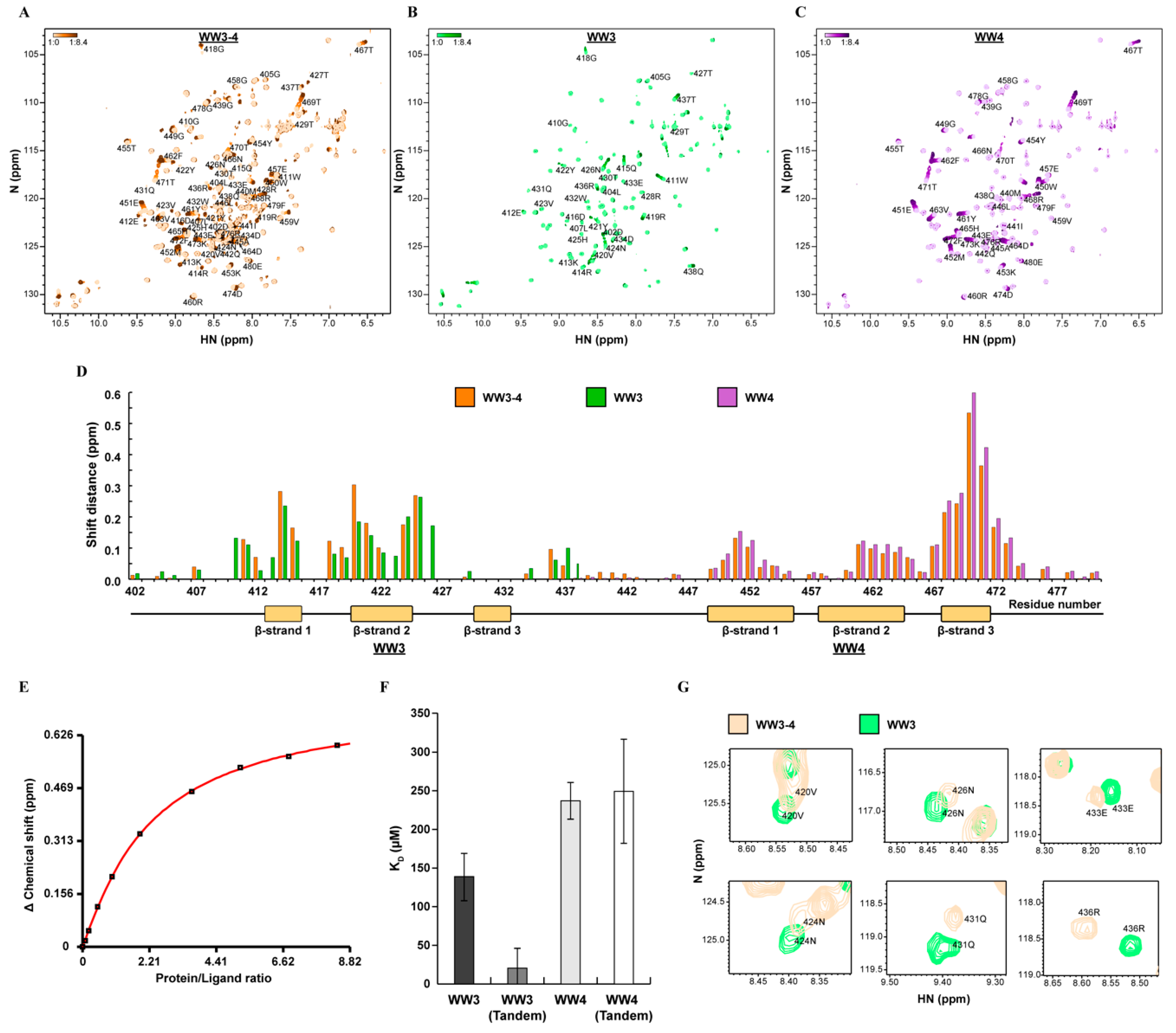
2.4. WWP2 WW Domain Affinity for Smad7 Is Switched On when Expressed in Tandem
3. Discussion
4. Materials and Methods
4.1. Protein and Peptide Expression and Purification
4.2. NMR Spectroscopy
4.3. NMR Ligand Titration
4.4. Dissociation Constants
4.5. Structure Determination
4.6. Cytokine Stimulation and Immunoblotting
4.7. Semi-Quantitative RT-PCR
4.8. Luciferase Reporter Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMRB | Biological Magnetic Resonance Data Bank |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| ESRPs | Epithelial splicing regulatory proteins |
| EST | Expressed sequence tag |
| GB1 | B1 domain of streptococcal protein G |
| HSQC | Heteronuclear single quantum coherence |
| KD | Dissociation constants |
| NMR | Nuclear magnetic resonance |
| NOE | Nuclear Overhauser effect |
| OCT4 | Octamer-binding transcription factor 4 |
| PDB | Protein Data Bank |
| TGFβ | Transforming growth factor-β |
| TβR-I | TGFβ receptor I |
| ULP-1 | Ubiquitin-like-specific protease-1 |
| UPS | Ubiquitin/proteasome system |
References
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of Proteins by Ubiquitin and Ubiquitin-Like Proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef] [PubMed]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Wang, X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dixit, V.M. Drugging the undruggables: Exploring the ubiquitin system for drug development. Cell Res. 2016, 26, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Modell, A.E.; Blosser, S.L.; Arora, P.S. Systematic Targeting of Protein-Protein Interactions. Trends Pharmacol. Sci. 2016, 37, 702. [Google Scholar] [CrossRef]
- Corbi-Verge, C.; Kim, P.M. Motif mediated protein-protein interactions as drug targets. Cell Commun. Signal. 2016, 14, 8. [Google Scholar] [CrossRef]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1α Interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef]
- Ingham, R.J.; Gish, G.; Pawson, T. The Nedd4 family of E3 ubiquitin ligases: Functional diversity within a common modular architecture. Oncogene 2004, 23, 1972–1984. [Google Scholar] [CrossRef]
- Zou, X.; Levy-Cohen, G.; Blank, M. Molecular functions of NEDD4 E3 ubiquitin ligases in cancer. Biochim. Biophys. Acta Rev. Cancer 2015, 1856, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Kumar, S. Mammalian HECT ubiquitin-protein ligases: Biological and pathophysiological aspects. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Matesic, L.E. The Nedd4-like family of E3 ubiquitin ligases and cancer. Cancer Metastasis Rev. 2007, 26, 587–604. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Hill, C.S. TGF-β Superfamily Signaling in Embryonic Development and Homeostasis. Dev. Cell 2009, 16, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Massagué, J. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- MacFarlane, E.G.; Haupt, J.; Dietz, H.C.; Shore, E.M. TGF-β Family Signaling in Connective Tissue and Skeletal Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a022269. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Imamura, T. Regulation of TGF-β family signaling by E3 ubiquitin ligases. Cancer Sci. 2008, 99, 2107–2112. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Baek, K.-H. TGF-β signalling pathway mediated by deubiquitinating enzymes. Cell. Mol. Life Sci. 2019, 76, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Alarcón, C.; Sapkota, G.; Rahman, S.; Chen, P.-Y.; Goerner, N.; Macias, M.J.; Erdjument-Bromage, H.; Tempst, P.; Massagué, J. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell 2009, 36, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Aragón, E.; Goerner, N.; Zaromytidou, A.-I.; Xi, Q.; Escobedo, A.; Massagué, J.; Macias, M.J. A Smad action turnover switch operated by WW domain readers of a phosphoserine code. Genes Dev. 2011, 25, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Soond, S.M.; Chantry, A. Selective targeting of activating and inhibitory Smads by distinct WWP2 ubiquitin ligase isoforms differentially modulates TGFβ signalling and EMT. Oncogene 2011, 30, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef]
- Suzuki, C.; Murakami, G.; Fukuchi, M.; Shimanuki, T.; Shikauchi, Y.; Imamura, T.; Miyazono, K. Smurf1 regulates the inhibitory activity of Smad7 by targeting Smad7 to the plasma membrane. J. Biol. Chem. 2002, 277, 39919–39925. [Google Scholar] [CrossRef]
- Dodson, E.J.; Fishbain-Yoskovitz, V.; Rotem-Bamberger, S.; Schueler-Furman, O. Versatile communication strategies among tandem WW domain repeats. Exp. Biol. Med. (Maywood). 2015, 240, 351–360. [Google Scholar] [CrossRef]
- Chong, P.A.; Lin, H.; Wrana, J.L.; Forman-Kay, J.D. Coupling of tandem Smad ubiquitination regulatory factor (Smurf) WW domains modulates target specificity. Proc. Natl. Acad. Sci. USA 2010, 107, 18404–18409. [Google Scholar] [CrossRef] [PubMed]
- Aragón, E.; Goerner, N.; Xi, Q.; Gomes, T.; Gao, S.; Massagué, J.; Macias, M.J. Structural basis for the versatile interactions of Smad7 with regulator WW domains in TGF-β Pathways. Structure 2012, 20, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616. [Google Scholar] [CrossRef] [PubMed]
- Chantry, A. WWP2 ubiquitin ligase and its isoforms: New biological insight and promising disease targets. Cell Cycle 2011, 10, 2437–2439. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Moreno-Moral, A.; Pesce, F.; Devapragash, N.; Mancini, M.; Heng, E.L.; Rotival, M.; Srivastava, P.K.; Harmston, N.; Shkura, K.; et al. WWP2 regulates pathological cardiac fibrosis by modulating SMAD2 signaling. Nat. Commun. 2019, 10, 3616. [Google Scholar] [CrossRef] [PubMed]
- Soond, S.M.; Smith, P.G.; Wahl, L.; Swingler, T.E.; Clark, I.M.; Hemmings, A.M.; Chantry, A. Novel WWP2 ubiquitin ligase isoforms as potential prognostic markers and molecular targets in cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2127–2135. [Google Scholar] [CrossRef]
- Gong, W.; Zhang, X.; Zhang, W.; Li, J.; Li, Z. Structure of the HECT domain of human WWP2. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jiang, H.; Xu, W.; Li, X.; Dempsey, D.R.; Zhang, X.; Devreotes, P.; Wolberger, C.; Amzel, L.M.; Gabelli, S.B.; et al. A Tunable Brake for HECT Ubiquitin Ligases. Mol. Cell 2017, 66, 345–357.e6. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, N.; Jiang, Y.; Tan, H.; Zheng, J.; Chen, G.; Jia, Z. Characterization of substrate binding of the WW domains in human WWP2 protein. FEBS Lett. 2015, 589, 1935–1942. [Google Scholar] [CrossRef]
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2007, 36, D402–D408. [Google Scholar] [CrossRef]
- Bouvignies, G.; Meier, S.; Grzesiek, S.; Blackledge, M. Ultrahigh-Resolution Backbone Structure of Perdeuterated Protein GB1 Using Residual Dipolar Couplings from Two Alignment Media. Angew. Chem. Int. Ed. 2006, 45, 8166–8169. [Google Scholar] [CrossRef] [PubMed]
- Wilton, D.J.; Tunnicliffe, R.B.; Kamatari, Y.O.; Akasaka, K.; Williamson, M.P. Pressure-induced changes in the solution structure of the GB1 domain of protein G. Proteins Struct. Funct. Bioinformat. 2007, 71, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Ikeya, T.; Hanashima, T.; Hosoya, S.; Shimazaki, M.; Ikeda, S.; Mishima, M.; Güntert, P.; Ito, Y. Improved in-cell structure determination of proteins at near-physiological concentration. Sci. Rep. 2016, 6, 38312. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Sicheritz-Pontén, T.; Gupta, R.; Gammeltoft, S.; Brunak, S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 2004, 4, 1633–1649. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Ren, J.; Gao, X.; Jin, C.; Wen, L.; Yao, X. GPS 2.0, a Tool to Predict Kinase-specific Phosphorylation Sites in Hierarchy. Mol. Cell. Proteomics 2008, 7, 1598–1608. [Google Scholar] [CrossRef]
- Pulaski, L.; Landström, M.; Heldin, C.H.; Souchelnytskyi, S. Phosphorylation of Smad7 at Ser-249 does not interfere with its inhibitory role in transforming growth factor-beta-dependent signaling but affects Smad7-dependent transcriptional activation. J. Biol. Chem. 2001, 276, 14344–14349. [Google Scholar] [CrossRef]
- Zarrinpar, A.; Lim, W.A. Converging on proline: The mechanism of WW domain peptide recognition. Nat. Struct. Biol. 2000, 7, 611–613. [Google Scholar] [CrossRef]
- Kato, Y.; Ito, M.; Kawai, K.; Nagata, K.; Tanokura, M. Determinants of ligand specificity in groups I and IV WW domains as studied by surface plasmon resonance and model building. J. Biol. Chem. 2002, 277, 10173–10177. [Google Scholar] [CrossRef]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef]
- Hafsa, N.E.; Arndt, D.; Wishart, D.S. CSI 3.0: A web server for identifying secondary and super-secondary structure in proteins using NMR chemical shifts. Nucleic Acids Res. 2015, 43, W370–W377. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.A.; Lin, H.; Wrana, J.L.; Forman-Kay, J.D. An expanded WW domain recognition motif revealed by the interaction between Smad7 and the E3 ubiquitin ligase Smurf2. J. Biol. Chem. 2006, 281, 17069–17075. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.A.; Bouchard, J.J.; Peng, J.W. Interdomain interactions support interdomain communication in human Pin1. Biochemistry 2013, 52, 6968–6981. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Shan, Z.; Chen, X.; Cai, Y.; Cui, L.; Yao, W.; Wang, Z.; Shi, P.; Tian, C.; Lou, J.; et al. Allosteric auto-inhibition and activation of the Nedd4 family E3 ligase Itch. EMBO Rep. 2017, 18, 1618–1630. [Google Scholar] [CrossRef] [PubMed]
- Debonneville, C.; Flores, S.Y.; Kamynina, E.; Plant, P.J.; Tauxe, C.; Thomas, M.A.; Münster, C.; Chraïbi, A.; Pratt, J.H.; Horisberger, J.D.; et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 2001, 20, 7052–7059. [Google Scholar] [CrossRef]
- Liu, Y.; Lau, J.; Li, W.; Tempel, W.; Li, L.; Dong, A.; Narula, A.; Qin, S.; Min, J. Structural basis for the regulatory role of the PPxY motifs in the thioredoxin-interacting protein TXNIP. Biochem. J. 2016, 473, 179–187. [Google Scholar] [CrossRef]
- Iglesias-Bexiga, M.; Luque, I.; Macias, M. Human NEDD4 3RD WW Domain Complex with Human T-cell Leukemia virus GAP-Pro Poliprotein Derived Peptide. 2010. Available online: http://www.rcsb.org/pdb/explore/litView.do?structureId=2KPZ (accessed on 1 September 2018).
- Iglesias-Bexiga, M. Human NEDD4 3rd WW Domain Complex with Ebola Zaire Virus Matrix Protein VP40 Derived Peptide. 2005. Available online: https://www.ncbi.nlm.nih.gov/Structure/pdb/2KQ0 (accessed on 1 September 2018).
- Spagnol, G.; Kieken, F.; Kopanic, J.L.; Li, H.; Zach, S.; Stauch, K.L.; Grosely, R.; Sorgen, P.L. Structural Studies of the Nedd4 WW Domains and Their Selectivity for the Connexin43 (Cx43) Carboxyl Terminus. J. Biol. Chem. 2016, 291, 7637–7650. [Google Scholar] [CrossRef]
- Kowalski, K.; Merkel, A.L.; Booker, G.W. Solution structures of WW domains of Nedd4-2. 2018. Available online: https://www.rcsb.org/structure/1WR3 (accessed on 1 September 2018).
- Qi, S.; O’Hayre, M.; Gutkind, J.S.; Hurley, J.H. Structural and biochemical basis for ubiquitin ligase recruitment by arrestin-related domain-containing protein-3 (ARRDC3). J. Biol. Chem. 2014, 289, 4743–4752. [Google Scholar] [CrossRef]
- Carter, P.; Wells, J.A. Dissecting the catalytic triad of a serine protease. Nature 1988, 332, 564–568. [Google Scholar] [CrossRef]
- Li, S.; Hong, M. Protonation, tautomerization, and rotameric structure of histidine: A comprehensive study by magic-angle-spinning solid-state NMR. J. Am. Chem. Soc. 2011, 133, 1534–1544. [Google Scholar] [CrossRef]
- Ferrigno, O.; Lallemand, F.; Verrecchia, F.; L’Hoste, S.; Camonis, J.; Atfi, A.; Mauviel, A. Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-β/Smad signaling. Oncogene 2002, 21, 4879–4884. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.J.; Martin-Malpartida, P.; Massagué, J. Structural determinants of Smad function in TGF-β signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.; Zaromytidou, A.-I.; Xi, Q.; Gao, S.; Yu, J.; Fujisawa, S.; Barlas, A.; Miller, A.N.; Manova-Todorova, K.; Macias, M.J.; et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 2009, 139, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Strasen, J.; Sarma, U.; Jentsch, M.; Bohn, S.; Sheng, C.; Horbelt, D.; Knaus, P.; Legewie, S.; Loewer, A. Cell-specific responses to the cytokine TGFβ are determined by variability in protein levels. Mol. Syst. Biol. 2018, 14, e7733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Mechanistic insight into contextual TGF-β signaling. Curr. Opin. Cell Biol. 2018, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Vranken, W.F.; Boucher, W.; Stevens, T.J.; Fogh, R.H.; Pajon, A.; Llinas, M.; Ulrich, E.L.; Markley, J.L.; Ionides, J.; Laue, E.D. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins Struct. Funct. Bioinformat. 2005, 59, 687–696. [Google Scholar] [CrossRef]
- Boyko, R.; Sykes, B. Xcrvfit: A Graphical X-Windows Program for Binding Curve Studies and NMR Spectroscopic Analysis. Available online: http://www.bionmr.ualberta.ca/bds/software/xcrvfit (accessed on 5 August 2019).
- Williams, T.C.; Shelling, J.G.; Sykes, B.D. NMR Approaches to the Characterization of the Interaction of Metal Ions with Proteins. In NMR in the Life Sciences; Springer US: Boston, MA, USA, 1986; pp. 93–103. [Google Scholar]
- Cheung, M.-S.; Maguire, M.L.; Stevens, T.J.; Broadhurst, R.W. DANGLE: A Bayesian inferential method for predicting protein backbone dihedral angles and secondary structure. J. Magn. Reson. 2010, 202, 223–233. [Google Scholar] [CrossRef]
- Rieping, W.; Habeck, M.; Bardiaux, B.; Bernard, A.; Malliavin, T.E.; Nilges, M. ARIA2: Automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 2007, 23, 381–382. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; van Dijk, M.; Loureiro-Ferreira, N.; van der Schot, G.; de Vries, S.J.; Schmitz, C.; van der Zwan, J.; Boelens, R.; Giachetti, A.; Ferella, L.; et al. WeNMR: Structural Biology on the Grid. J. Grid Comput. 2012, 10, 743–767. [Google Scholar] [CrossRef]
- Nilges, M.; Bernard, A.; Bardiaux, B.; Malliavin, T.; Habeck, M.; Rieping, W. Accurate NMR Structures Through Minimization of an Extended Hybrid Energy. Structure 2008, 16, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Fogh, R.H.; Boucher, W.; Vranken, W.F.; Pajon, A.; Stevens, T.J.; Bhat, T.N.; Westbrook, J.; Ionides, J.M.C.; Laue, E.D. A framework for scientific data modeling and automated software development. Bioinformatics 2005, 21, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Tejero, R.; Montelione, G.T. Evaluating protein structures determined by structural genomics consortia. Proteins Struct. Funct. Bioinformat. 2006, 66, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, C.C.; Jiang, P.; Amirikian, K.; Dittmar, K.A.; Lu, H.; Shen, S.; Guo, W.; Xing, Y.; Carstens, R.P. An ESRP-regulated splicing programme is abrogated during the epithelial–mesenchymal transition. EMBO J. 2010, 29, 3286–3300. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NMR Distance and Dihedral Constraints | GB1–WW4 | |
| Distance constraints | ||
| Total NOE | 1803 | |
| Intra-residue | 873 | |
| Inter-residue | ||
| Sequential (|i-j| = 1) | 400 | |
| Medium-range (|i-j| < 4) | 118 | |
| Long-range (|i-j| > 5) | 412 | |
| Total dihedral angle restraints | ||
| φ | 96 | |
| ψ | 96 | |
| Structure statistics | GB1 | WW4 |
| Violations (mean and s.d.) | ||
| Distance constraints (±0.3Å tolerance) | 2 | 0 |
| Dihedral angle constraints (±5˚ tolerance) | 2 | 1 |
| Max. dihedral angle violation (˚) | 1.75 | 2.24 |
| Max. distance constraint violation (Å) | 0.043 | - |
| Deviations from idealised geometry | ||
| Bond lengths (Å) | 0.008 | 0.009 |
| Bond angles (˚) | 0.9 | 0.8 |
| Impropers (˚) | - | - |
| Average pairwise RMSD 1 (20 refined structures) (Å) | ||
| Heavy | 0.8 | 0.9 |
| Backbone | 0.4 | 0.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wahl, L.C.; Watt, J.E.; Yim, H.T.T.; De Bourcier, D.; Tolchard, J.; Soond, S.M.; Blumenschein, T.M.A.; Chantry, A. Smad7 Binds Differently to Individual and Tandem WW3 and WW4 Domains of WWP2 Ubiquitin Ligase Isoforms. Int. J. Mol. Sci. 2019, 20, 4682. https://doi.org/10.3390/ijms20194682
Wahl LC, Watt JE, Yim HTT, De Bourcier D, Tolchard J, Soond SM, Blumenschein TMA, Chantry A. Smad7 Binds Differently to Individual and Tandem WW3 and WW4 Domains of WWP2 Ubiquitin Ligase Isoforms. International Journal of Molecular Sciences. 2019; 20(19):4682. https://doi.org/10.3390/ijms20194682
Chicago/Turabian StyleWahl, Lloyd C., Jessica E. Watt, Hiu T. T. Yim, Danielle De Bourcier, James Tolchard, Surinder M. Soond, Tharin M. A. Blumenschein, and Andrew Chantry. 2019. "Smad7 Binds Differently to Individual and Tandem WW3 and WW4 Domains of WWP2 Ubiquitin Ligase Isoforms" International Journal of Molecular Sciences 20, no. 19: 4682. https://doi.org/10.3390/ijms20194682
APA StyleWahl, L. C., Watt, J. E., Yim, H. T. T., De Bourcier, D., Tolchard, J., Soond, S. M., Blumenschein, T. M. A., & Chantry, A. (2019). Smad7 Binds Differently to Individual and Tandem WW3 and WW4 Domains of WWP2 Ubiquitin Ligase Isoforms. International Journal of Molecular Sciences, 20(19), 4682. https://doi.org/10.3390/ijms20194682