The Human Ovary and Future of Fertility Assessment in the Post-Genome Era



Abstract
:
1. Introduction
2. Proteomics for Selection of Competent Embryos and Oocytes in IVF
2.1. Follicular Fluid: An Inexhaustible Biosource for Investigation of Competence Biomarkers
2.1.1. Role of Inflammation and Coagulation Cascades in IVF
2.1.2. Impact of Extracellular Matrix Turnover on Oocyte Quality
2.1.3. Involvement of Lipid Metabolism in the Success of IVF
2.2. A New Way of Predicting IVF Success: The Cumulus–Oocyte Complex
3. Ensuring Normal Pregnancy in Patients with Polycystic Ovary Syndrome through Proteomics
4. The Future of Human Ovary Proteomics
4.1. In Vivo Mass Spectrometry
4.2. Proteomics to Help Engineer a Transplantable Artificial Ovary
4.3. Challenges of Proteomic Studies of the Human Ovary
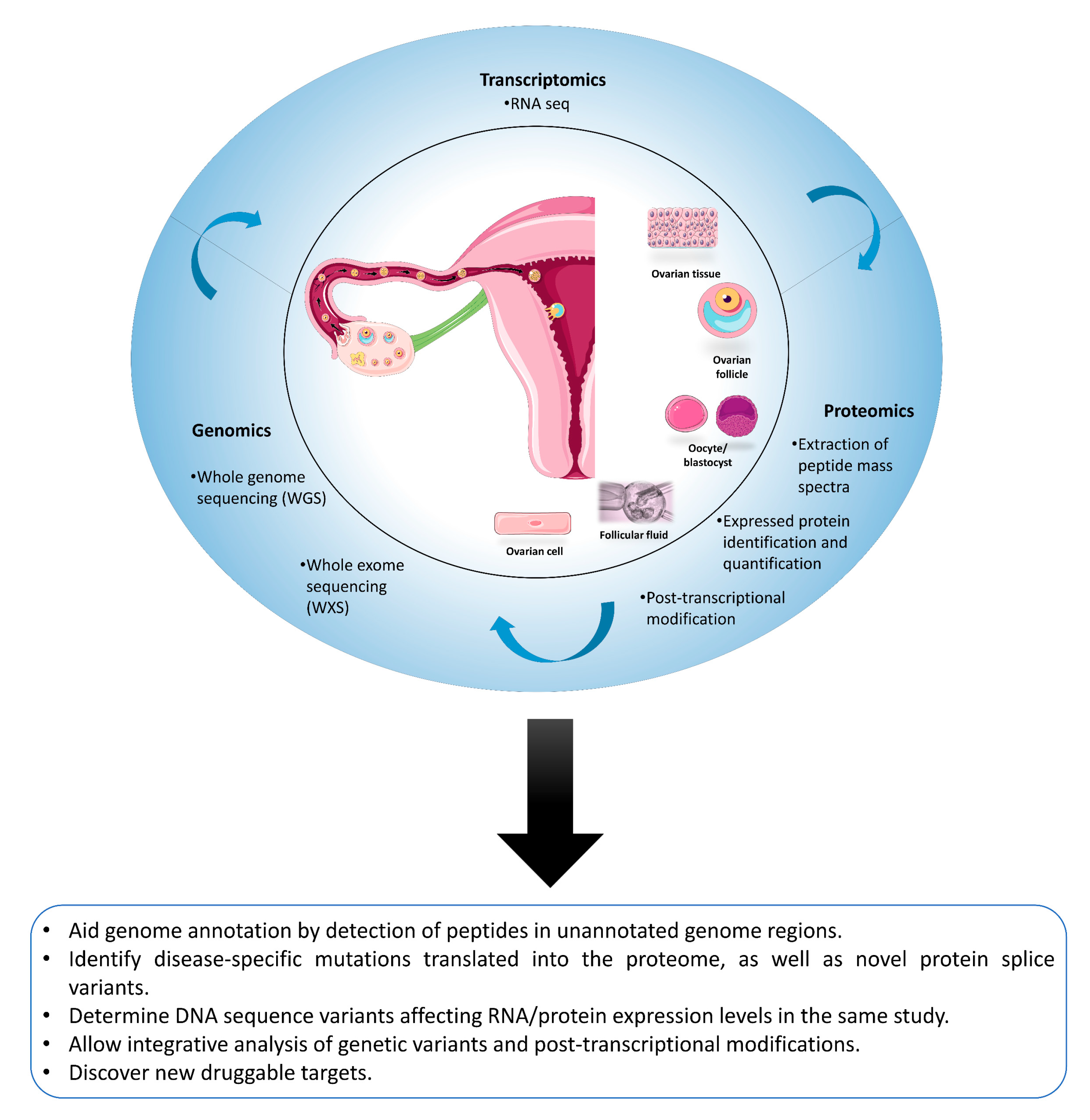
4.4. Proteomics Is Not an Island: The Added Value of Proteogenomics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Calderón-González, K.G.; Hernández-Monge, J.; Herrera-Aguirre, M.E.; Luna-Arias, J.P. Bioinformatics Tools for Proteomics Data Interpretation. In Modern Proteomics–Sample Preparation, Analysis and Practical Applications; Mirzaei, H., Carrasco, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 281–341. [Google Scholar]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N.; Yates, J.R., 3rd. Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13, 5293–5309. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.D.; Balasinor, N.H.; Kumar, A.V.; Sachdeva, G.; Parte, P.; Dumasia, K. Proteomics in reproductive biology: Beacon for unraveling the molecular complexities. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 8–15. [Google Scholar] [CrossRef]
- Braga, D.P.; Setti, A.S.; Lo Turco, E.G.; Cordeiro, F.B.; Cabral, E.C.; Cortezzi, S.S.; Ono, E.; Figueira, R.C.; Eberlin, M.N.; Borges, E., Jr. Protein expression in human cumulus cells as an indicator of blastocyst formation and pregnancy success. J. Assist. Reprod. Genet. 2016, 33, 1571–1583. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, L.; Gagliardi, A.; Campanella, G.; Landi, C.; Capaldo, A.; Carleo, A.; Armini, A.; De Leo, V.; Piomboni, P.; Focarelli, R.; et al. A methodological and functional proteomic approach of human follicular fluid en route for oocyte quality evaluation. J. Proteom. 2013, 90, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Liu, X.; Zhu, P.; Zhang, Y.; Wang, J.; Wang, Y.; Wang, W.; Liu, J.; Li, N.; Liu, F. Proteomic analysis of human follicular fluid associated with successful in vitro fertilization. Reprod. Biol. Endocrinol. 2017, 15, 58. [Google Scholar] [CrossRef]
- Bianchi, L.; Gagliardi, A.; Landi, C.; Focarelli, R.; De Leo, V.; Luddi, A.; Bini, L.; Piomboni, P. Protein pathways working in human follicular fluid: The future for tailored IVF? Expert Rev. Mol. Med. 2016, 18, e9. [Google Scholar] [CrossRef]
- O’Brien, M. The reciprocal relationship between inflammation and coagulation. Top. Companion Anim. Med. 2012, 27, 46–52. [Google Scholar] [CrossRef]
- Jarkovska, K.; Martinkova, J.; Liskova, L.; Halada, P.; Moos, J.; Rezabek, K.; Gadher, S.J.; Kovarova, H. Proteome mining of human follicular fluid reveals a crucial role of complement cascade and key biological pathways in women undergoing in vitro fertilization. J. Proteome Res. 2010, 9, 1289–1301. [Google Scholar] [CrossRef]
- Hillmeister, P.; Persson, P.B. The Kallikrein-Kinin system. Acta Physiol. (Oxf.) 2012, 206, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.; Shalev, E. MMPS and TIMPS in ovarian physiology and pathophysiology. Front. Biosci. 2004, 9, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Horka, P.; Malickova, K.; Jarosova, R.; Janatkova, I.; Zima, T.; Kalousova, M. Matrix metalloproteinases in serum and the follicular fluid of women treated by in vitro fertilization. J. Assist. Reprod. Genet. 2012, 29, 1207–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlage, P.; auf dem Keller, U. Proteomic approaches to uncover MMP function. Matrix Biol. 2015, 44–46, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Sun, Y.; Wan, J.; Luan, T.; Cheng, Q.; Tan, Y. A proteomic analysis identifies candidate early biomarkers to predict ovarian hyperstimulation syndrome in polycystic ovarian syndrome patients. Mol. Med. Rep. 2017, 16, 272–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, G.H.; Galazis, N.; Docheva, N.; Layfield, R.; Atiomo, W. Overlap of proteomics biomarkers between women with pre-eclampsia and PCOS: A systematic review and biomarker database integration. Hum. Reprod. 2015, 30, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Galazis, N.; Docheva, N.; Nicolaides, K.H.; Atiomo, W. Proteomic biomarkers of preterm birth risk in women with polycystic ovary syndrome (PCOS): A systematic review and biomarker database integration. PLoS ONE 2013, 8, e53801. [Google Scholar] [CrossRef] [PubMed]
- Saudemont, P.; Quanico, J.; Robin, Y.M.; Baud, A.; Balog, J.; Fatou, B.; Tierny, D.; Pascal, Q.; Minier, K.; Pottier, M.; et al. Real-Time Molecular Diagnosis of Tumors Using Water-Assisted Laser Desorption/Ionization Mass Spectrometry Technology. Cancer Cell 2018, 34, 840–851. [Google Scholar] [CrossRef]
- Othman, Z.; Cillero Pastor, B.; van Rijt, S.; Habibovic, P. Understanding interactions between biomaterials and biological systems using proteomics. Biomaterials 2018, 167, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Groen, N.; Guvendiren, M.; Rabitz, H.; Welsh, W.J.; Kohn, J.; de Boer, J. Stepping into the omics era: Opportunities and challenges for biomaterials science and engineering. Acta Biomater. 2016, 34, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Ouni, E.; Vertommen, D.; Chiti, M.C.; Dolmans, M.M.; Amorim, C.A. A draft map of the human ovarian proteome for tissue engineering and clinical applications. Mol. Cell. Proteom. 2018, 18 (Suppl. 1), S159–S173. [Google Scholar] [CrossRef] [PubMed]
- Insenser, M.; Escobar-Morreale, H.F. Proteomics and polycystic ovary syndrome. Expert Rev. Proteom. 2013, 10, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Spiessens, C.; D’Hooghe, T.; Peeraer, K.; Carpentier, S. Follicular fluid biomarkers for human in vitro fertilization outcome: Proof of principle. Proteome Sci. 2016, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, J.; Nagai, Y.; Matsuyama, Y.; Terada, T.; Era, S. The influence of the redox state of follicular fluid albumin on the viability of aspirated human oocytes. Syst. Biol. Reprod. Med. 2012, 58, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Amorim, C.A. Special Issue Devoted to a New Field of Regenerative Medicine: Reproductive Tissue Engineering. Ann. Biomed. Eng. 2017, 45, 1589–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Clauser, K.R.; Hynes, R.O. Enrichment of Extracellular Matrix Proteins from Tissues and Digestion into Peptides for Mass Spectrometry Analysis. J. Vis. Exp. 2015, e53057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.C.; Yates, J.R., 3rd. The application of mass spectrometry to membrane proteomics. Nat. Biotechnol. 2003, 21, 262–267. [Google Scholar] [CrossRef]
- Tabb, D.L. Quality assessment for clinical proteomics. Clin. Biochem. 2013, 46, 411–420. [Google Scholar] [CrossRef]
- Mischak, H.; Apweiler, R.; Banks, R.E.; Conaway, M.; Coon, J.; Dominiczak, A.; Ehrich, J.H.; Fliser, D.; Girolami, M.; Hermjakob, H.; et al. Clinical proteomics: A need to define the field and to begin to set adequate standards. Proteom. Clin. Appl. 2007, 1, 148–156. [Google Scholar] [CrossRef]
- Dimitrakopoulos, L.; Prassas, I.; Diamandis, E.P.; Nesvizhskii, A.; Kislinger, T.; Jaffe, J.; Drabovich, A. Proteogenomics: Opportunities and Caveats. Clin. Chem. 2016, 62, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Whiteaker, J.R.; Hoofnagle, A.N.; Baird, G.S.; Rodland, K.D.; Paulovich, A.G. Clinical potential of mass spectrometry-based proteogenomics. Nat. Rev. Clin. Oncol. 2019, 16, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Lobas, A.A.; Pyatnitskiy, M.A.; Chernobrovkin, A.L.; Ilina, I.Y.; Karpov, D.S.; Solovyeva, E.M.; Kuznetsova, K.G.; Ivanov, M.V.; Lyssuk, E.Y.; Kliuchnikova, A.A.; et al. Proteogenomics of Malignant Melanoma Cell Lines: The Effect of Stringency of Exome Data Filtering on Variant Peptide Identification in Shotgun Proteomics. J. Proteome Res. 2018, 17, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ma, Z.; Carr, S.A.; Mertins, P.; Zhang, H.; Zhang, Z.; Chan, D.W.; Ellis, M.J.; Townsend, R.R.; Smith, R.D.; et al. Proteome Profiling Outperforms Transcriptome Profiling for Coexpression Based Gene Function Prediction. Mol. Cell. Proteom. 2017, 16, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggles, K.V.; Krug, K.; Wang, X.; Clauser, K.R.; Wang, J.; Payne, S.H.; Fenyö, D.; Zhang, B.; Mani, D.R. Methods, Tools and Current Perspectives in Proteogenomics. Mol. Cell. Proteom. 2017, 16, 959–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Huang, Y.; Lei, J.; Luo, H.; Zhu, X. The single-cell sequencing: New developments and medical applications. Cell Biosci. 2019, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guo, F.; Gao, Y.; Ren, Y.; Yuan, P.; Yan, L.; Li, R.; Lian, Y.; Li, J.; Hu, B.; et al. Publisher Correction: Single-cell multi-omics sequencing of human early embryos. Nat. Cell Biol. 2018, 20, 1227. [Google Scholar] [CrossRef]
- Crinier, A.; Milpied, P.; Escaliere, B.; Piperoglou, C.; Galluso, J.; Balsamo, A.; Spinelli, L.; Cervera-Marzal, I.; Ebbo, M.; Girard-Madoux, M.; et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity 2018, 49, 971–986. [Google Scholar] [CrossRef]
- Couvillion, S.P.; Zhu, Y.; Nagy, G.; Adkins, J.N.; Ansong, C.; Renslow, R.S.; Piehowski, P.D.; Ibrahim, Y.M.; Kelly, R.T.; Metz, T.O. New mass spectrometry technologies contributing towards comprehensive and high throughput omics analyses of single cells. Analyst 2019, 144, 794–807. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouni, E.; Vertommen, D.; Amorim, C.A. The Human Ovary and Future of Fertility Assessment in the Post-Genome Era. Int. J. Mol. Sci. 2019, 20, 4209. https://doi.org/10.3390/ijms20174209
Ouni E, Vertommen D, Amorim CA. The Human Ovary and Future of Fertility Assessment in the Post-Genome Era. International Journal of Molecular Sciences. 2019; 20(17):4209. https://doi.org/10.3390/ijms20174209
Chicago/Turabian StyleOuni, Emna, Didier Vertommen, and Christiani A. Amorim. 2019. "The Human Ovary and Future of Fertility Assessment in the Post-Genome Era" International Journal of Molecular Sciences 20, no. 17: 4209. https://doi.org/10.3390/ijms20174209
APA StyleOuni, E., Vertommen, D., & Amorim, C. A. (2019). The Human Ovary and Future of Fertility Assessment in the Post-Genome Era. International Journal of Molecular Sciences, 20(17), 4209. https://doi.org/10.3390/ijms20174209