A Cyclic Pentamethinium Salt Induces Cancer Cell Cytotoxicity through Mitochondrial Disintegration and Metabolic Collapse

 , ,
, ,
Abstract
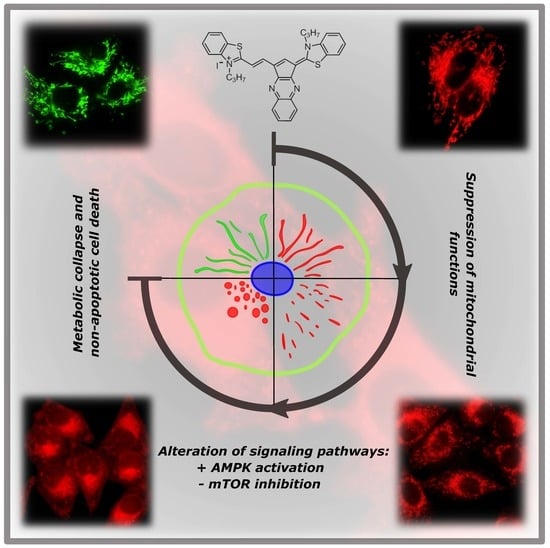
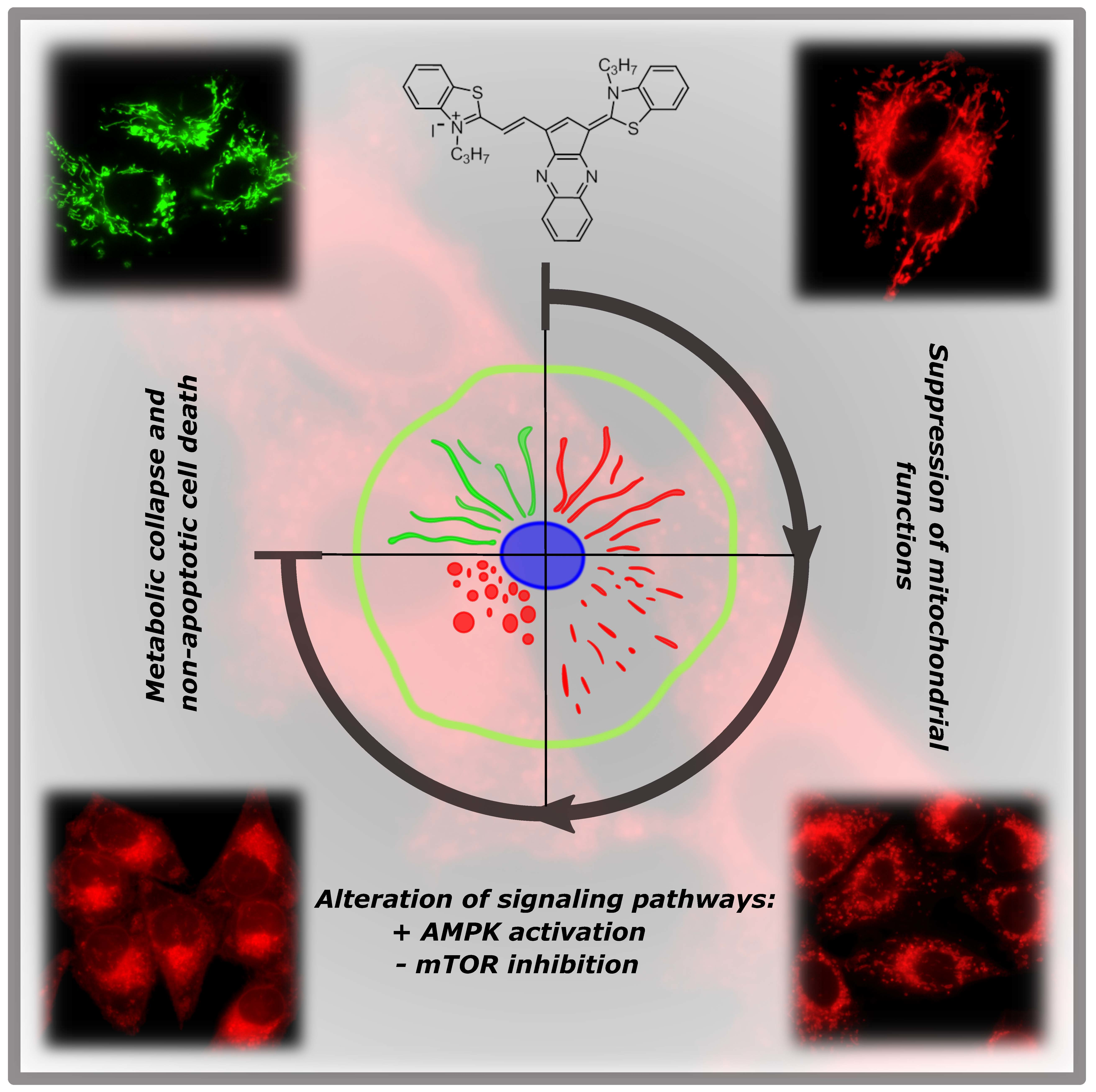{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
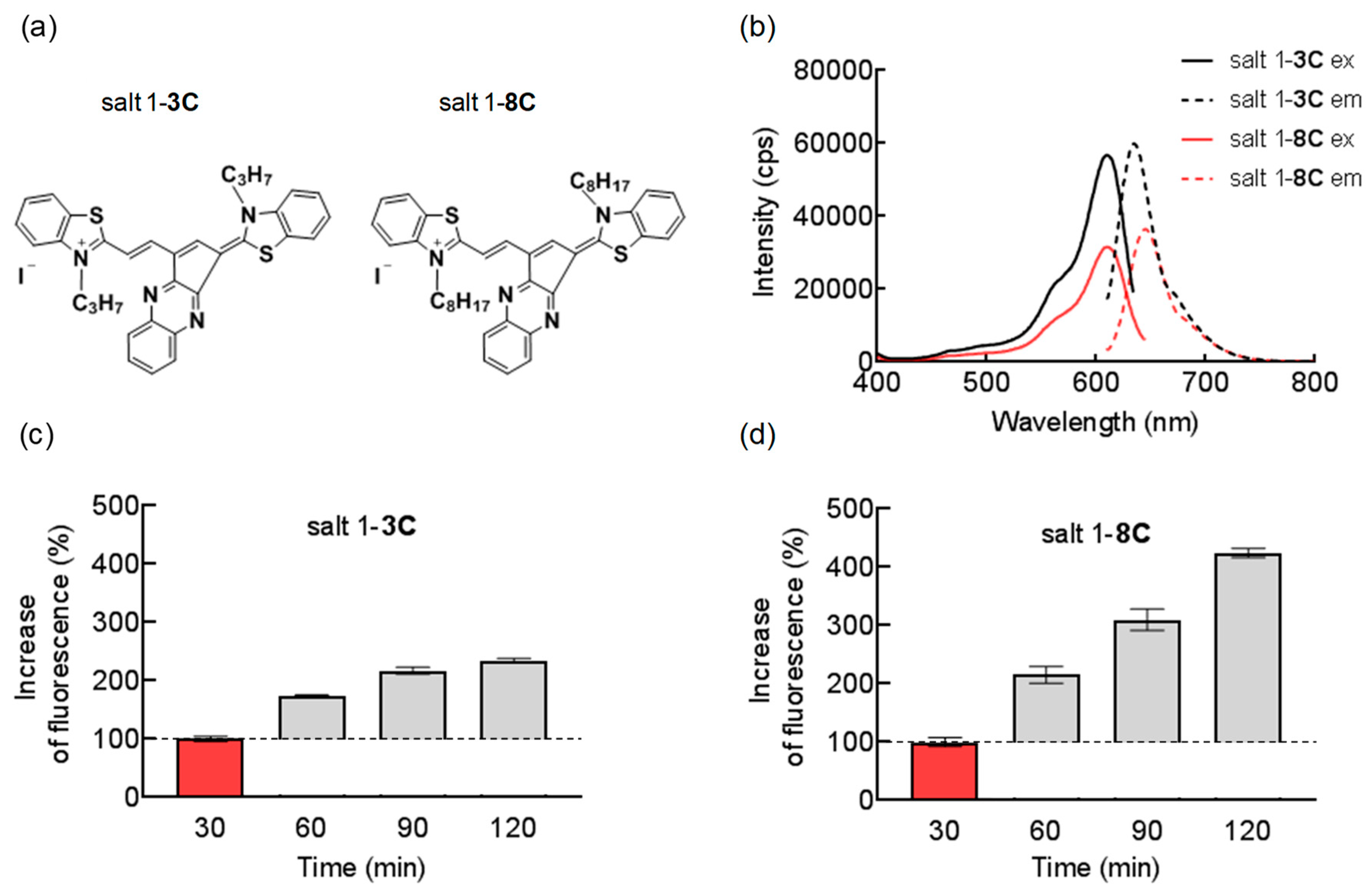
2.1. Chemical and Biological Properties of Salts
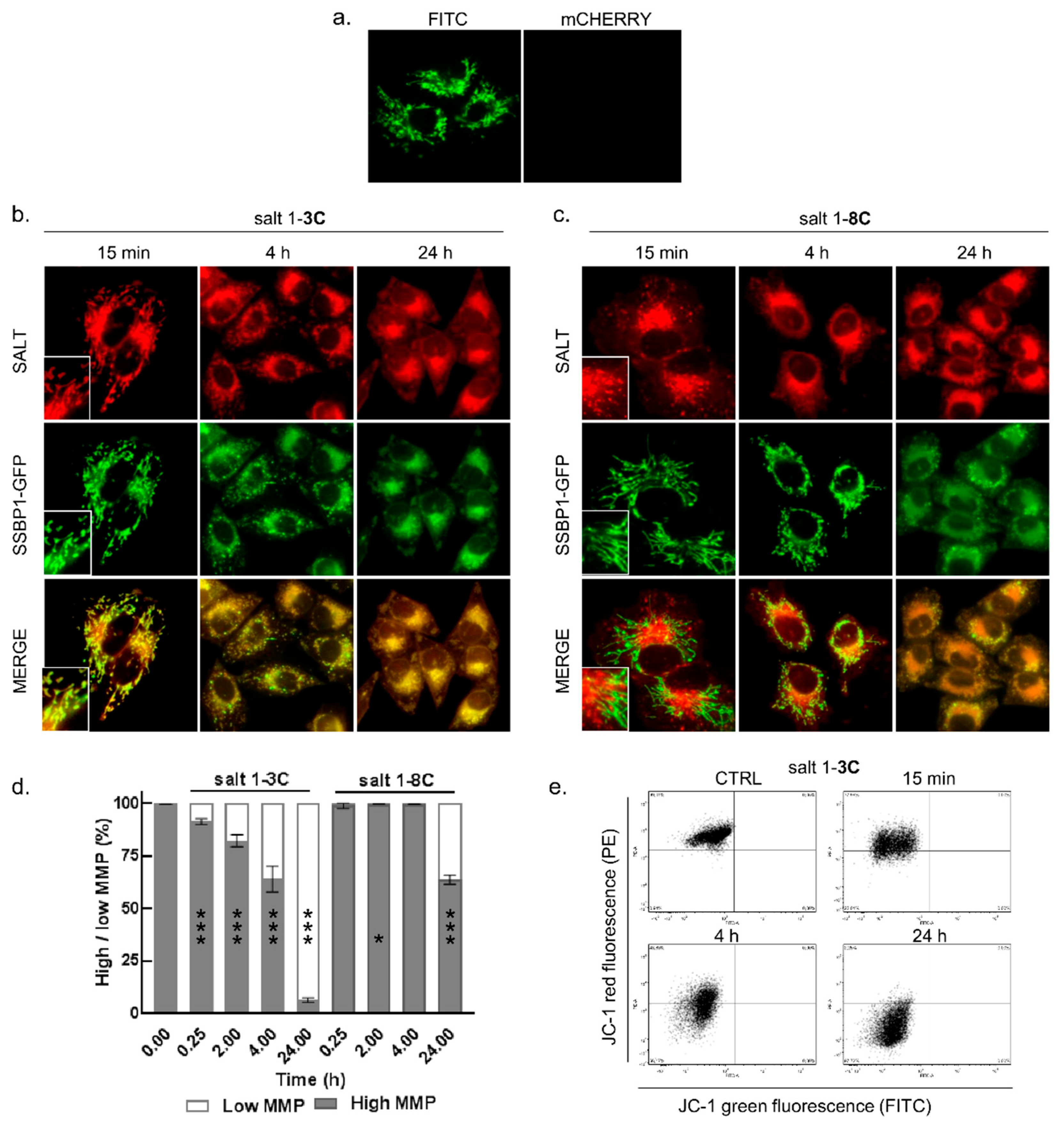
2.2. Mitochondrial Localization of Salts
2.3. Salt 1-3C Lowers Mitochondrial Membrane Potential
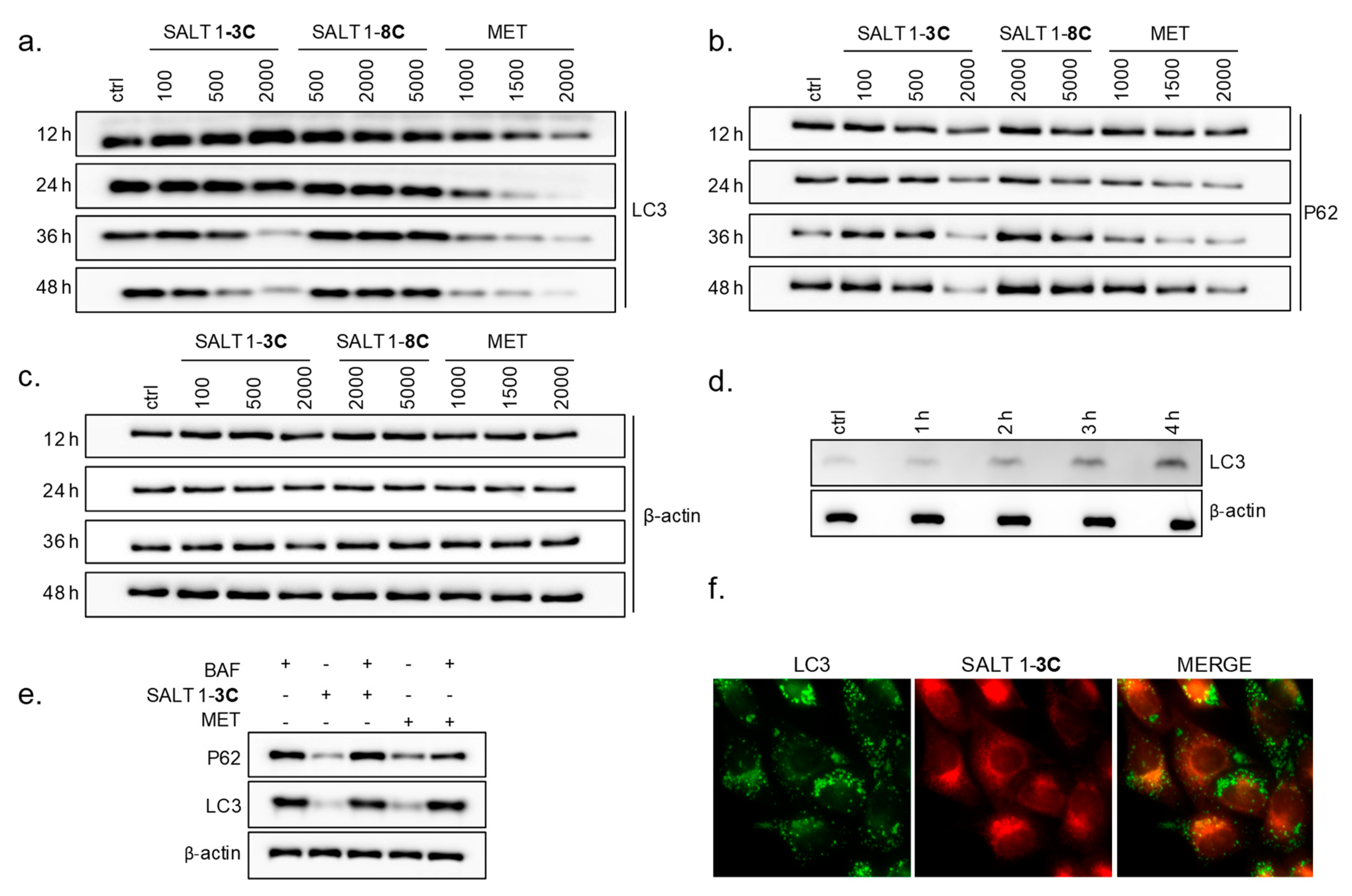
2.4. Salt 1-3C Leads to Altered Autophagy Processes
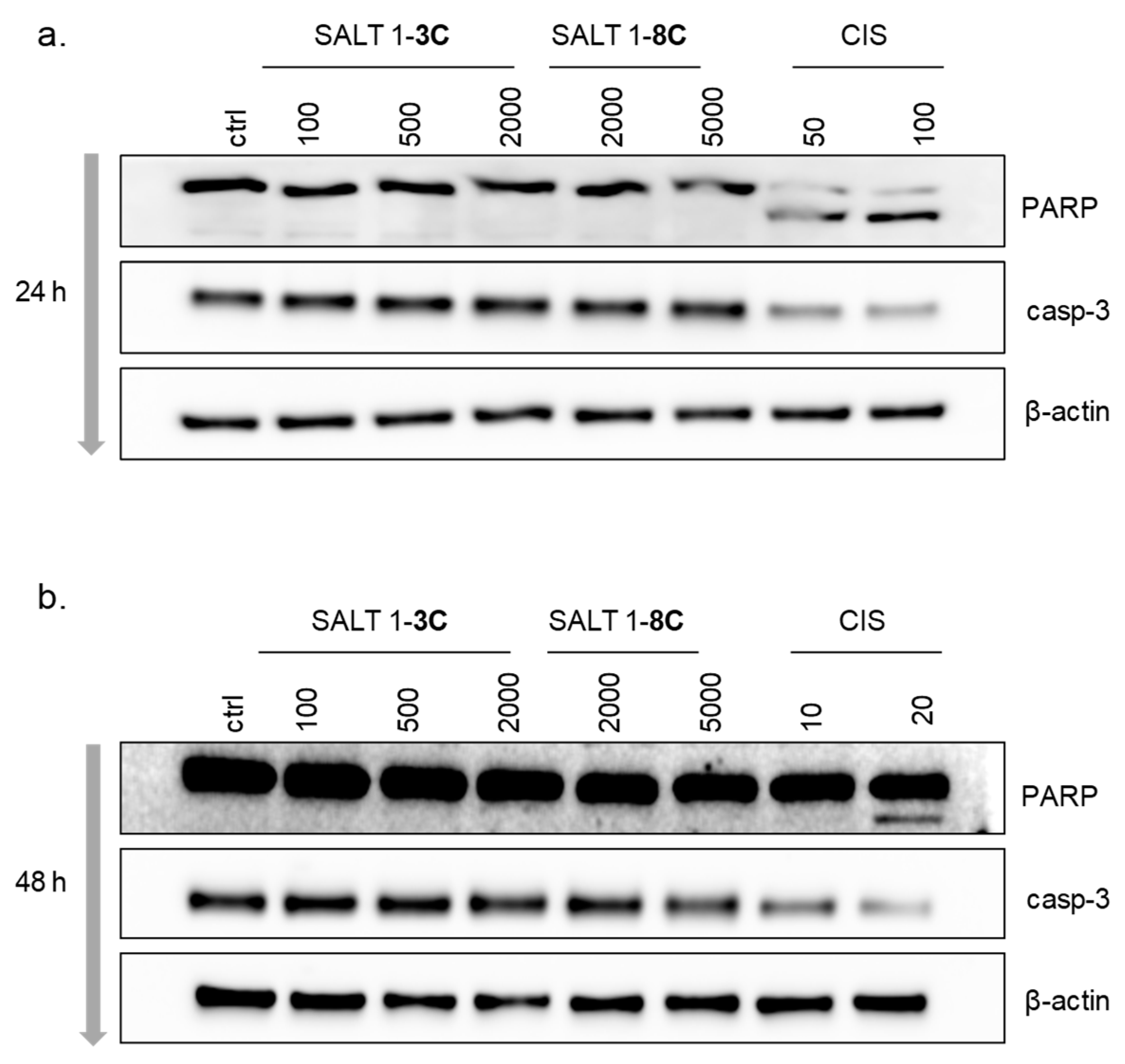
2.5. Salts 1-3C and 1-8C do not Induce Apoptosis
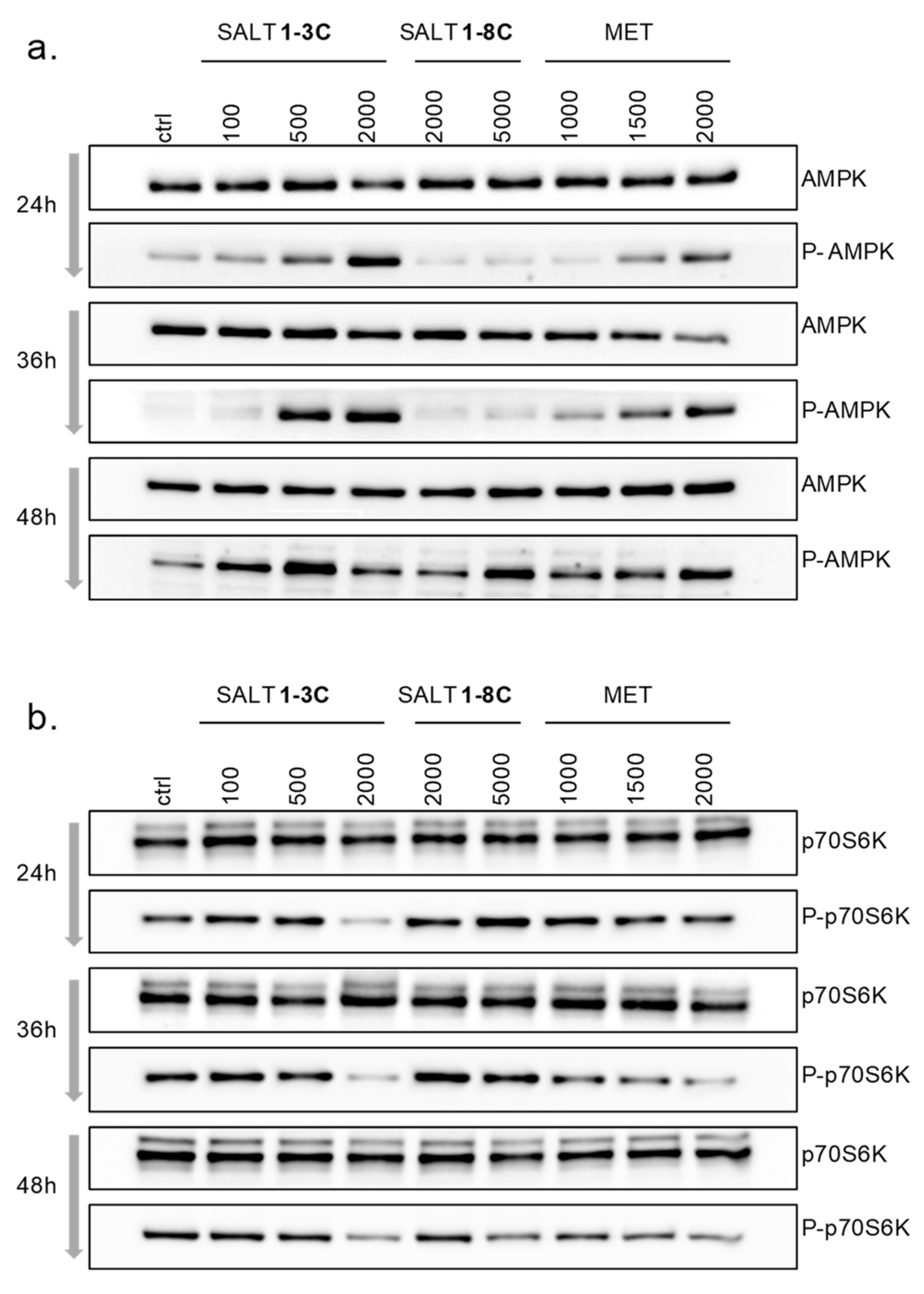
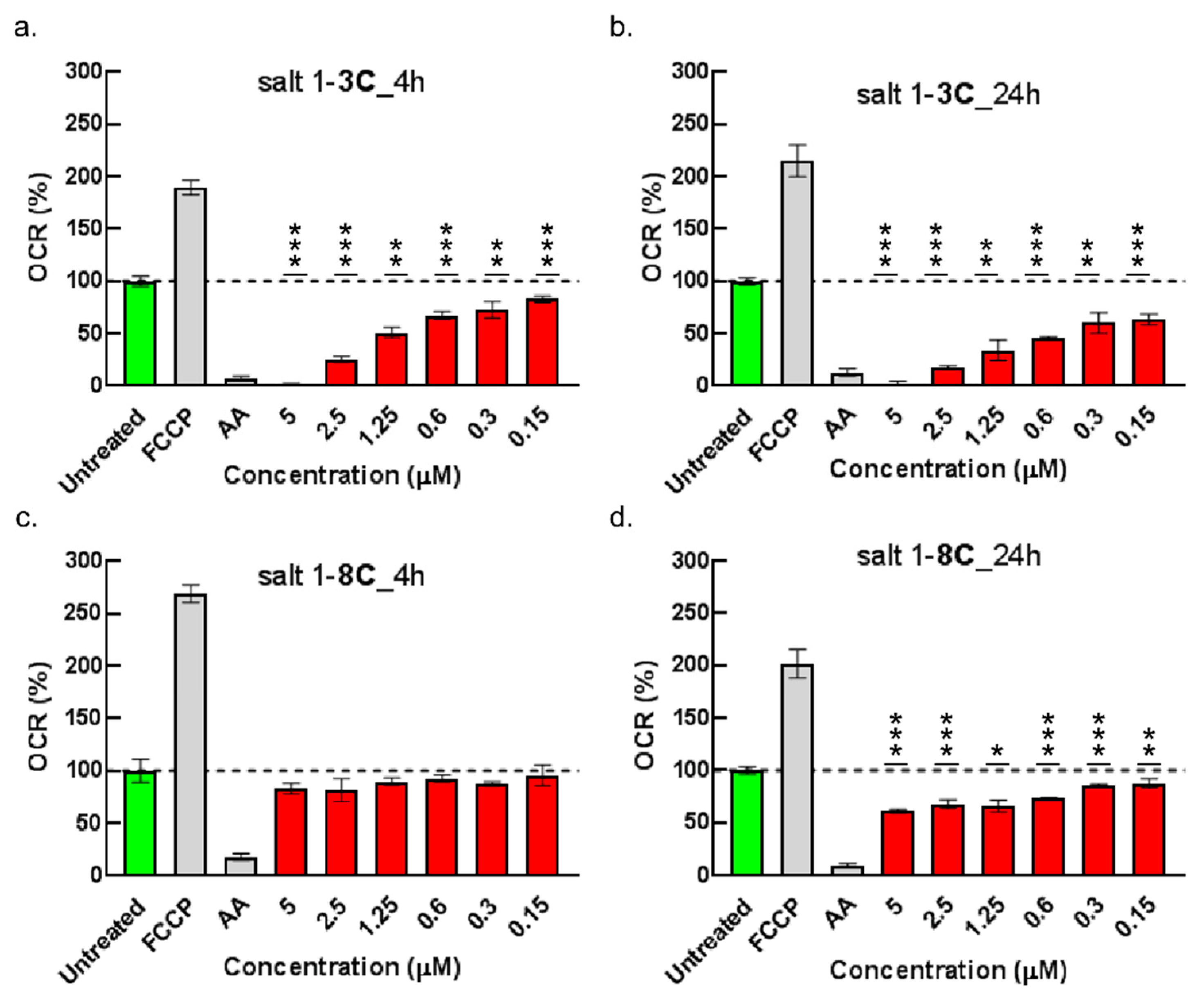
2.6. Impact on Mitochondrial Bioenergetics through AMP-Activated Protein Kinase–Mammalian Target of Rapamycin (AMPK–mTOR) Signaling
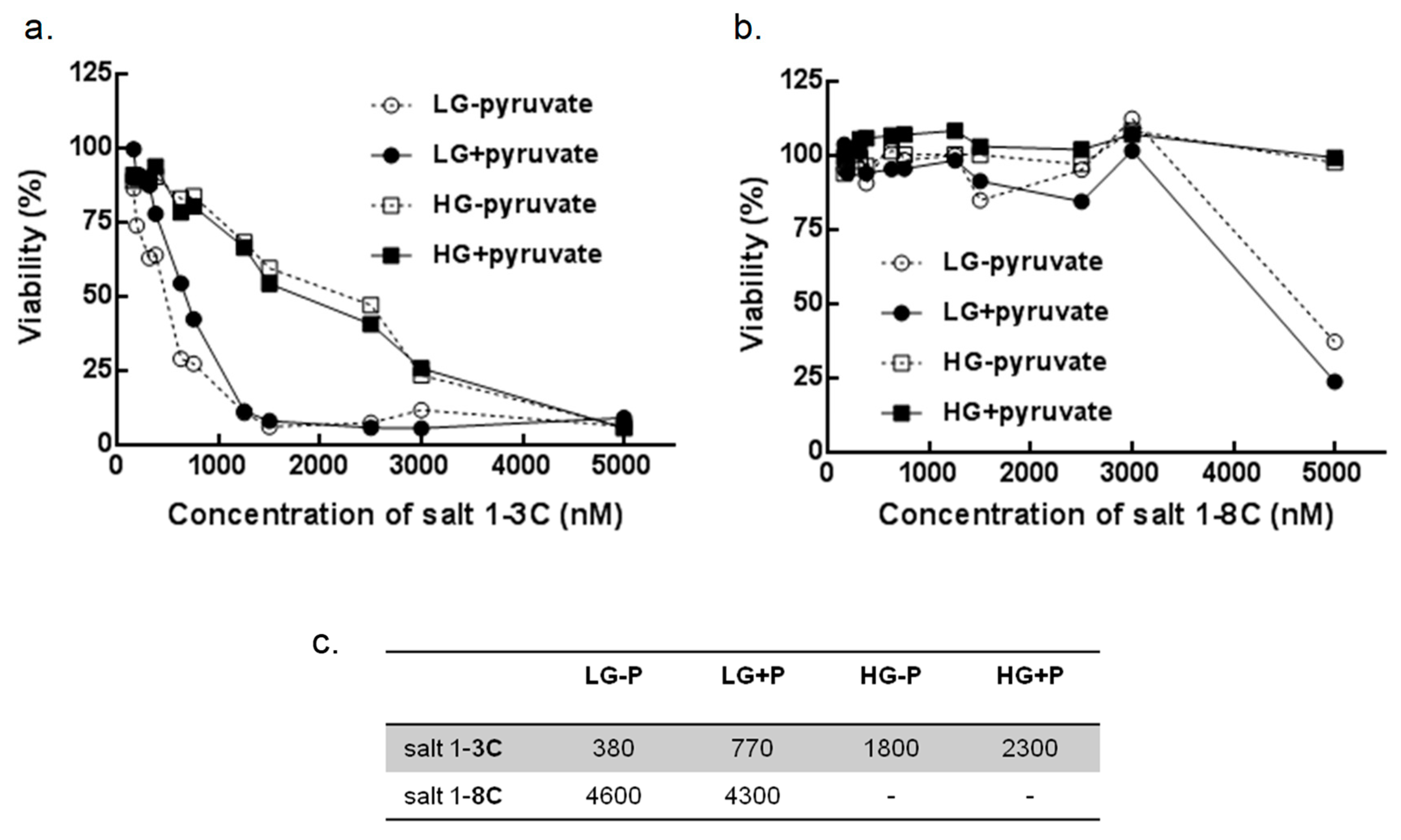
2.7. Glucose and Pyruvate Deprivation Influence Cytotoxicity
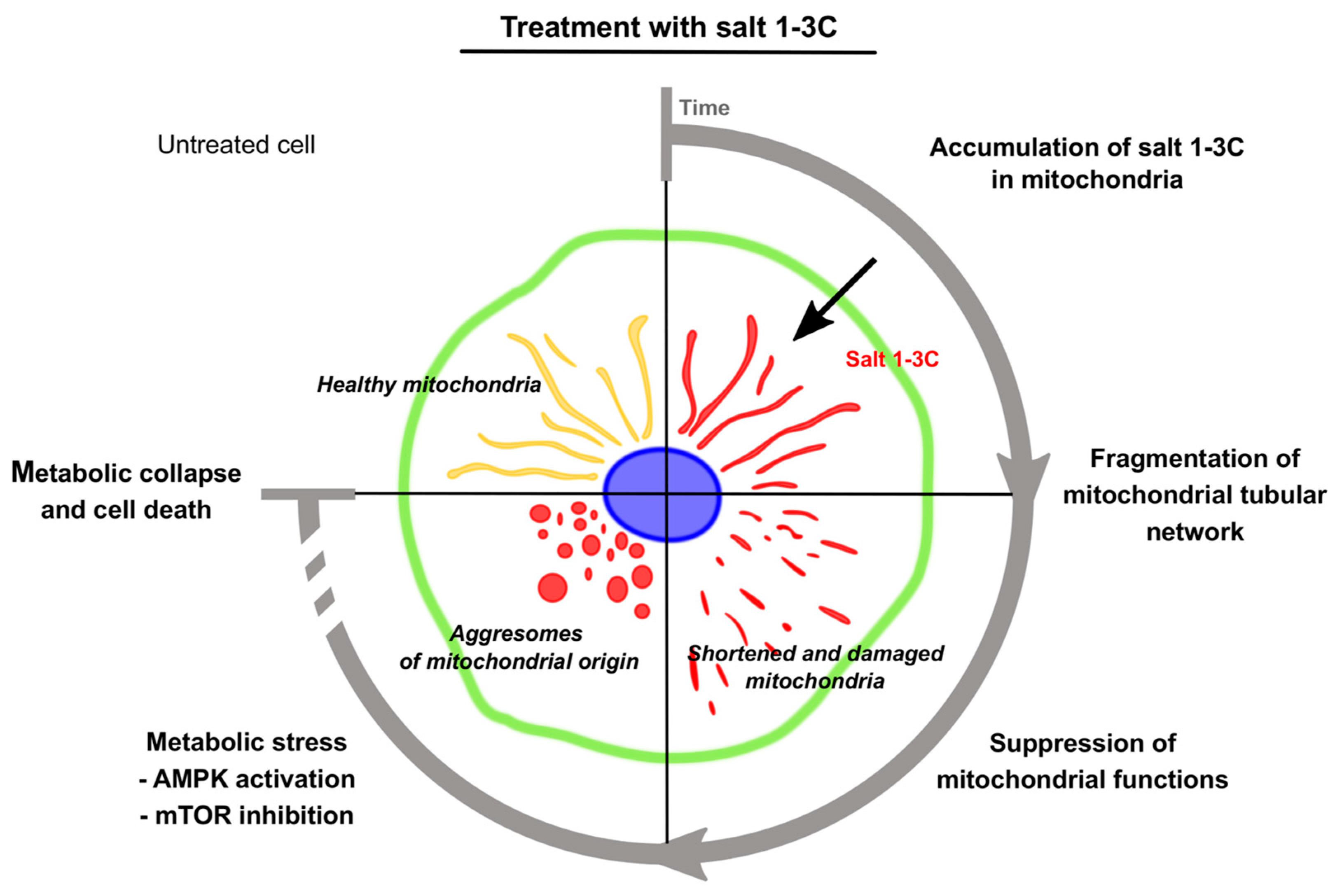
3. Discussion
4. Materials and Methods
4.1. Synthesis of Salt 1-3C and Salt 1-8C
4.2. Fluorescence Spectra
4.3. Cell Culture and Reagents
4.4. Flow Cytometry Analysis of Uptake Kinetics and Mitochondrial Membrane Potential
4.5. Generation of A-375-SSBP1-GFP and Live Cell Imaging
4.6. Western Blotting
4.7. Oxygen Consumption Assay
4.8. Immunofluorescence Microscopy
4.9. Viability/Metabolic Activity Assay
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| bafA1 | bafilomycin A1 |
| BSA | bovine serum albumin |
| casp-3 | caspase-3 |
| DMEM | Dulbecco´s modified Eagle´s medium |
| FCCP | carbonyl cyanide-p-triflouromethoxyphenyl-hydrazone |
| GFP | green fluorescent protein |
| LC3 | microtubule-associated protein light chain 3 |
| MET | metformin |
| mTOR | mammalian target of rapamycin |
| OCR | oxygen consumption rate |
| OXPHOS | oxidative phosphorylation |
| P-AMPK | (Thr172) phosphorylated AMPK |
| PARP | poly(ADP-ribose) polymerase |
| PBS | phosphate buffered saline |
| p62/SQSTM1 | sequestosome 1 |
| p70S6K | p70 S6 kinase |
| P-p70S6K | (Ser434) phosphorylated p70S6K |
| SSBP1 | single-stranded DNA-binding protein, mitochondrial |
References
- Saraste, M. Oxidative phosphorylation at the fin de siecle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y. Regulation of cell death by the Abl tyrosine kinase. Oncogene 2000, 19, 5643–5650. [Google Scholar] [CrossRef] [PubMed]
- Mazel, S.; Burtrum, D.; Petrie, H.T. Regulation of cell division cycle progression by bcl-2 expression: A potential mechanism for inhibition of programmed cell death. J. Exp. Med. 1996, 183, 2219–2226. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lin, Q.; Sun, L.; Feng, C.; Zhang, P.; Yu, B.; Chen, Y.; Wen, Y.; Wang, H.; Ji, L.; et al. A mitochondrial targeted two-photon iridium(III) phosphorescent probe for selective detection of hypochlorite in live cells and in vivo. Biomaterials 2015, 53, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Johnson, L.V.; Walsh, M.L.; Chen, L.B. Localization of mitochondria in living cells with rhodamine 123. Proc. Natl. Acad. Sci. USA 1980, 77, 990–994. [Google Scholar] [CrossRef]
- Heller, A.; Brockhoff, G.; Goepferich, A. Targeting drugs to mitochondria. Eur. J. Pharm. Biopharm. 2012, 82, 1–18. [Google Scholar] [CrossRef]
- Onoe, S.; Temma, T.; Shimizu, Y.; Ono, M.; Saji, H. Investigation of cyanine dyes for in vivo optical imaging of altered mitochondrial membrane potential in tumors. Cancer Med. 2014, 3, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Briza, T.; Kejik, Z.; Cisarova, I.; Kralova, J.; Martasek, P.; Kral, V. Optical sensing of sulfate by polymethinium salt receptors: Colorimetric sensor for heparin. Chem. Commun. 2008, 1901–1903. [Google Scholar] [CrossRef] [PubMed]
- Rimpelova, S.; Briza, T.; Kralova, J.; Zaruba, K.; Kejik, Z.; Cisarova, I.; Martasek, P.; Ruml, T.; Kral, V. Rational design of chemical ligands for selective mitochondrial targeting. Bioconjugate Chem. 2013, 24, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Briza, T.; Kralova, J.; Dolensky, B.; Rimpelova, S.; Kejik, Z.; Ruml, T.; Hajduch, M.; Dzubak, P.; Mikula, I.; Martasek, P.; et al. Striking antitumor activity of a methinium system with incorporated quinoxaline unit obtained by spontaneous cyclization. Chembiochem 2015, 16, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. Methods Mol Biol 2015, 1266, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.S.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010, 15, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.T.; Hsu, W.M.; Lee, H.; Huang, W.P.; Liao, Y.F. The evolutionarily conserved interaction between LC3 and p62 selectively mediates autophagy-dependent degradation of mutant huntingtin. Cell. Mol. Neurobiol. 2010, 30, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J Exp Clin Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef]
- Perfettini, J.L.; Roumier, T.; Kroemer, G. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. 2005, 15, 179–183. [Google Scholar] [CrossRef]
- Arnoult, D. Mitochondrial fragmentation in apoptosis. Trends Cell Biol. 2007, 17, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef]
- Kisfalvi, K.; Eibl, G.; Sinnett-Smith, J.; Rozengurt, E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009, 69, 6539–6545. [Google Scholar] [CrossRef] [PubMed]
- Bartolome, A.; Garcia-Aguilar, A.; Asahara, S.I.; Kido, Y.; Guillen, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 2017, e00441-17. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Dranka, B.P.; Benavides, G.A.; Diers, A.R.; Giordano, S.; Zelickson, B.R.; Reily, C.; Zou, L.; Chatham, J.C.; Hill, B.G.; Zhang, J.; et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 2011, 51, 1621–1635. [Google Scholar] [CrossRef]
- Ross, M.F.; Kelso, G.F.; Blaikie, F.H.; James, A.M.; Cocheme, H.M.; Filipovska, A.; Da Ros, T.; Hurd, T.R.; Smith, R.A.; Murphy, M.P. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochem. Biokhimiia 2005, 70, 222–230. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Ceskova, P.; Vojtesek, B. Hsp90 is essential for restoring cellular functions of temperature-sensitive p53 mutant protein but not for stabilization and activation of wild-type p53: Implications for cancer therapy. J. Biol. Chem. 2005, 280, 6682–6691. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krejcir, R.; Krcova, L.; Zatloukalova, P.; Briza, T.; Coates, P.J.; Sterba, M.; Muller, P.; Kralova, J.; Martasek, P.; Kral, V.; et al. A Cyclic Pentamethinium Salt Induces Cancer Cell Cytotoxicity through Mitochondrial Disintegration and Metabolic Collapse. Int. J. Mol. Sci. 2019, 20, 4208. https://doi.org/10.3390/ijms20174208
Krejcir R, Krcova L, Zatloukalova P, Briza T, Coates PJ, Sterba M, Muller P, Kralova J, Martasek P, Kral V, et al. A Cyclic Pentamethinium Salt Induces Cancer Cell Cytotoxicity through Mitochondrial Disintegration and Metabolic Collapse. International Journal of Molecular Sciences. 2019; 20(17):4208. https://doi.org/10.3390/ijms20174208
Chicago/Turabian StyleKrejcir, Radovan, Lucie Krcova, Pavlina Zatloukalova, Tomas Briza, Philip J. Coates, Martin Sterba, Petr Muller, Jarmila Kralova, Pavel Martasek, Vladimir Kral, and et al. 2019. "A Cyclic Pentamethinium Salt Induces Cancer Cell Cytotoxicity through Mitochondrial Disintegration and Metabolic Collapse" International Journal of Molecular Sciences 20, no. 17: 4208. https://doi.org/10.3390/ijms20174208
APA StyleKrejcir, R., Krcova, L., Zatloukalova, P., Briza, T., Coates, P. J., Sterba, M., Muller, P., Kralova, J., Martasek, P., Kral, V., & Vojtesek, B. (2019). A Cyclic Pentamethinium Salt Induces Cancer Cell Cytotoxicity through Mitochondrial Disintegration and Metabolic Collapse. International Journal of Molecular Sciences, 20(17), 4208. https://doi.org/10.3390/ijms20174208