4.1. GEFIs for Measuring Inorganic Ions
Ca
2+ is one of the most important secondary messengers in cell signaling pathways. It plays key roles in numerous cellular processes such as fertilization, development, learning, and memory [
107]. At present, many excellent small-molecule calcium indicators with suitable properties in terms of kinetics and fractional fluorescence change are available [
108]. However, the process of introducing these molecules into tissues is invasive and nonspecific. Furthermore, loading of molecules is destructive and incompatible with long-term imaging. The use of FP-based indicators can overcome these problems. Various genetically encoded calcium indicators (GECIs) with a wide range of colors; different K
d, appropriate for each cell compartment; and pKa, are available (Table 1).
The first cpFP-based GECI (G-CaMP: the chimeric protein consists of M13-peptide, CaM, and cp148GFP) was developed by Nakai and co-workers [
65]. Almost at the same time, Pericams were constructed from cp145EGFP, M13-peptide, and CaM [
66]. The subsequent version of the Ca
2+ sensor had a larger dynamic range (8.5-fold vs. 4-fold). Moreover, Nagai et al. showed that the introduction of subtle mutations in the amino acids close to the chromophore remarkably changed the Ca
2+-dependent behavior of biosensor. The substitution of phenylalanine for tyrosine at residue 203 (Y203F) led to the creation of a ratiometric sensor—ratiometric-Pericam [
66]. The mechanism underlying this type of sensors is the interaction between the M13 and CaM mediated by Ca
2+ binding. This conformational shift modulates the environment of the cpGFP chromophore by allowing the formation of new domain interfaces, rearrangement of protein side chains, and reduction of solvent access to the chromophore, leading to an increased fluorescence owing to water-mediated cooperation between the chromophore and R377 of CaM [
109,
110]. At present, the GCaMP family of intensiometric Ca
2+ sensors is one of the most widely used tools for monitoring Ca
2+ concentration fluctuations, especially in neurons [
111].
More than any other cell types, neurons are critically dependent on spatially and temporally controlled Ca
2+ dynamics. This is achieved via an exquisite level of compartmentalization of Ca
2+ storage and release from various organelles. Thus, further evolution of calcium sensors developed in the direction of increasing sensitivity to neuronal activity [
112,
113,
114]. GECIs have been used to measure the dynamics of large populations of neurons over weeks, even during learning [
112,
114,
115]. They can track activity in local and long-range axonal projections and provide information about the activity of neurons that are inaccessible to electrophysiological methods [
116].
Large-scale mutagenesis and screening approaches allowed the generation of excellent Ca
2+ sensors called G-CaMP6 and G-CaMP8 with greater dynamic range (11.4 and 38, respectively) and sensitivity (K
d = 158 and 200 nM, respectively) than those of the respective small-molecule-based probes [
113]. Moreover, GECIs tagged with target peptide sequences allowed the imaging of Ca
2+ dynamics in specific organelles [
117]. For instance, the spatiotemporal properties of calcium-measuring organelle-entrapped protein indicators (CEPIA) with greater K
d (368–558 µM) than that of G-CaMP6s (144 nM) allowed the resolution of Ca
2+ import into individual mitochondria while simultaneously measuring ER and cytosolic Ca
2+ levels [
118].
Expanding the color options for Ca
2+ sensors led to the creation of blue- and red-emitting variants [
119]. Since GFP and RFP chromophores are significantly different, the development of red calcium indicators has progressed slower than that of the green analogs. Nevertheless, R-GECO, based on cp-mApple [
119], R-CaMP2 [
120], and photoconvertible calcium sensor based on cp-mMaple, named GR-GECO [
121], have been successfully applied for in vivo imaging owing to their lower phototoxicity and autofluorescent background and deeper tissue penetration, allowing the labeling of specific types of cells such as neurons and astrocytes and targeting to subcellular compartments. Importantly, the mechanism of the red calcium sensor R-GECO1 is slightly different from that of GCaMP, which has been described above. In R-GECO1 Lys78 from strand eight of cpmApple, adjacent to the circular permutation site, forms an ionic interaction with the phenolate oxygen of the chromophore and is stabilized by a hydrogen bond from Ser62, immediately following the M13pep-cpmApple linker [
122]. Conformational rearrangements in the sensory unit affect these interactions providing a molecular switch.
Despite all these advantages of calcium sensors, they have one main drawback—all calcium indicators act as calcium buffers. Therefore, the expression of any type of GECI may unintentionally change the spatiotemporal dynamics of this essential secondary messenger [
123]. Recently, several attempts have been made to reduce the buffering activity of FRET-based GECIs. One of them involves the use of troponin C protein from swim bladder and white muscle of
Opsanus tau [
124]. The general idea was to develop a sensory unit with one or two calcium binding sites per indicator molecule. To that end, a minimal calcium binding motif was engineered on the basis of EF-hand from C-terminal domain of troponin C. The resulted probes (Twitch sensors) bind less calcium ions compared to their counterparts that can lead to reduced ion buffering during long-term expression. Moreover, because troponin C is muscle-specific, GECIs such as Twitch sensors would not be affected by interactions with endogenous proteins when expressed in non-muscle tissues.
Obtaining the crystal structures of Ca
2+-bound bright states for some variants such as GCaMP2 [
109,
110], GCaMP5 [
114], and GCaMP6 [
125] might provide a structural basis for Ca
2+-induced fluorescence changes and promote the rational design of improved GECI. Detailed information is listed in
Table 1.
At present, the repertoire of genetically encoded fluorescent sensors has been expanding for not only monitoring common metabolites and secondary messengers, but also a rather unusual class of molecules present in our body—transition metals (TMs). TMs play essential roles in numerous biological processes, including respiration, gene transcription, enzymatic catalysis, and cell signaling; however, their excessive or inappropriate accumulation may become toxic [
133].
Most genetically-encoded TM sensors for zinc and copper, as well as heavy metals, including lead and cadmium, were developed using the metal-binding motif in combination with one or two FPs (Table 2). The evolution of TM sensors shows that the first genetically encoded sensors were FRET sensors. The Cu(I) sensor was created by introducing cysteine-rich metallothionein-like Cu(I)-sensing domains of Atm1 between a FRET pair [
134]. Subsequently, Choi et al. developed a new genetically encoded fluorescent Cu(II) sensor GCS-2 by the rational insertion of the Cu-binding tripeptide ATCUN (the amino terminal and Cu- and Ni-binding motif) into cpGFP [
135]. This sensor has been used to detect dynamic Cu(II) fluctuations on the surfaces of live mammalian cells, representing, to our knowledge, the first report for Cu(II) imaging by using a genetically encoded fluorescent sensor.
The situation is the same for Zn(II) sensors. When several versions of Zn(II) FRET sensors had been constructed (ZapCY1 [
136] and ZapOC2 [
137]), Palmer et al. constructed a single FP-based, genetically encoded Zn(II) sensor GZnP1(Kd = 580 nM) that consisted of two zinc fingers from
Saccharomyces cerevisiae Zap1 fused to a cpGFP [
138]. ZnGreen2 (K
d of 20 mM for Zn(II)) was constructed by connecting a zinc hook peptide from Rad50 to each of the two termini of cp-monomeric teal FP [
139]. It has lower affinity for zinc, allowing the monitoring of intracellular Zn
2+ dynamics.
The rapid increase in biosensors of this type has revealed many interesting features of TMs. These biosensors help to monitor the actual concentrations of Cu and Zn(II) in different cell compartments, to detect heavy metals in organisms and to study their interaction with other proteins. However, genetically encoded TM sensors still have several limitations in brightness, dynamic range, and kinetics, unlike GECIs, and need to be improved.
Furthermore, a unique class of biosensors was engineered using the so-called “Matryoshka technology”—it is a generalized platform to create dual-FP biosensors with large dynamic ranges by using a single FP cassette, named GO (green–orange) Matryoshka [
73]. The cassette nests a stable reference FP (large Stokes shift mOrange) within a reporter FP (cp-GFP). GO-Matryoshka yields green and orange fluorescence under blue excitation. This technology allowed the conversion of existing single-emission biosensors into ratiometric versions, namely, calcium sensors (MatryoshCaMP6s) and ammonium transport activity sensors (AmTryoshka1;3). The Matryoshka approach provides an alternative design for ratiometric sensors with a large dynamic range and is particularly advantageous when terminal fusion of FPs is not tolerated. Detailed information is listed in
Table 2.
4.2. Genetically Encoded Voltage Indicators
Living cells precisely regulate their membrane potentials as they orchestrate biochemical events crucial for normal metabolism. Ion fluxes, enzyme activity, as well as protein/protein and protein/lipid interactions strongly depend on the difference between intercellular and extracellular potentials, which not only is important for maintaining the microenvironment that is tuned for appropriate cellular machines functioning, but also provides an opportunity to set regulation switches for adjusting intracellular biochemical processes in response to environmental changes [
140]. This principle is implemented to a greater extent in the nervous system where neurons transmit signals to each other by affecting membrane potentials of target cells, eventually generating complex animal behavior [
141]. Detailed investigation of this issue has been performed using traditional approaches such as patch-clamping and voltage chemical dyes; however, despite their contribution, to our knowledge, these methods are invasive and often do not provide the desired spatial resolution. Therefore, developing instruments that would enable the registration of voltage shifts in compartments, such as small-caliber dendrites, spines, and boutons for individual cells in the real-time mode, are required. Such studies can be conducted by implementing GECIs, but calcium fluxes might not necessarily reflect membrane potential changes because of different temporal parameters and kinetic constants of GECIs are considerably slow for detecting fast electrochemical events. These drawbacks were overcome by developing genetically encoded voltage indicators (GEVIs) (Table 3). The first versions—namely, Flash [
35], VSFP1 [
142], and SPARC [
36]—were engineered by introducing one or two FPs into the voltage-sensing domains (VSDs) of potassium or sodium channels; however, their plasma membrane targeting in neurons was found to be poor. The discovery of the non-ion channel protein
Ciona intestinalis voltage sensor-containing phosphatase (Ci-VSP) [
143] revolutionized this field as the replacement of VSDs from K2.1 potassium channel in FRET-based sensor VSFP1 with Ci-VSD resulted in the VSFP2.1 version that showed excellent subcellular localization [
144]. The latter was subsequently improved; in one version (VSFP3.1), which is especially important in the context of this review, the FRET-pair was substituted with a single fluorescent domain that narrowed the optical channel occupied by the probe, facilitating multiparameter imaging [
145].
The success of cpFP-based GECIs became an inspiration for attempts to engineer GEVIs that would function on the same principle with the hope that the flexible nature of a cpFP could improve the dynamic range and/or kinetic properties of the indicator. VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180) were the first functional voltage probes in which cpFPs were used in their architecture [
146]. In these sensors, cpmKate(180), a permuted version of mKate with a break point in one of the disorganized loops, was fused to the Ci-VSD C-terminus with a linker of varying length. The authors attempted to use other linkers and another permutation position (144) located on the beta-barrel surface; however, the fluorescence intensity of such versions was low. Both the probes were characterized by considerably poor dynamic range (approximately 1.9% and 1.2%, respectively) and kinetic constants lower than for their contemporaries (dozens of ms). Nevertheless, the half-response voltages of VSFP(C)cpmKate(180) and VSFP(D)cpmKate(180) were about −80 to −90 mV, which is closer to the physiological range, unlike that of many cpFP-based probes that were developed later. Although cpmKate(180) has a permutation point not in the close proximity to the chromophore, the authors confirmed that the cpFP-based design could be implemented in the development of GEVIs.
The authors also attempted to engineer a cpEGFP-based indicator; however, they reported that all obtained chimeras were characterized by low brightness as emission parameters of cpFPs strongly depend on the fusing partner [
146]. Several years later, Barnett et al. developed such a probe and named it ElectricPk [
147]. In general, it replicates the design described above with an important difference that the permutation position is located on the beta-sheet. This was achieved by screening through a library of total 90 chimeras that varied by the size of the hole on the cpFP surface and the composition of linkers. Notably, the probe performance increased by decreasing the hole size probably because of the better sensory and reporter unit coupling. Despite being the best version, ElectricPk was characterized by a very small dynamic range of −1.2%, although its kinetic properties exceeded those of other GEVIs available at that time (τ
on approximately 2.24 ± 0.58 ms; τ
off approximately 2.09 ± 0.74 ms). The authors hypothesized that this could be attributed to the fact that Ci-VSP undergoes conformational rearrangements, revealing different time scales, and ElectricPk is able to catch the fast components, unlike the probes with other types of architecture [
147].
Another sensor where cpFP is fused to the C-terminus of Ci-VSD is FlicR1, which emits in the red region and is favorable for in vivo imaging [
94]. That study was inspired by experimental evidence suggesting that the R-GECO1 sensing mechanism relies on the interaction of the Lys80 chain located on the beta-barrel surface with the chromophore group [
122], unlike the GCaMP sensing mechanism in which the key residue is Arg from CaM [
109]. Therefore, the working hypothesis was that cp-mApple from R-GECO1 represents a self-contained reporter module that could be combined with varying sensory units. The probe was engineered by inserting cpFP close to the S4 helix to enable efficient signal transition between the latter and the key lysine. Linker optimization and further random mutagenesis of the entire gene resulted in the improvement of brightness and increase in sensitivity, which was attributed to Val207 substitution. The importance of this residue was discovered accidentally, as the Val207Ala mutation led to better performance; after all possible substitutions were tested, the Val207Phe variant was found to be the best one. FlicR1 emission increases by approximately 6.6% per 100 mV; its fast kinetic components (τ
on approximately 3.0 ± 0.2; τ
off approximately 2.8 ± 0.3) are slightly greater than those of ASAP1 (mentioned below); however, their contribution to the total response was higher; therefore, both probes showed similar performance in the original study [
94].
The first GEVI that combined fast kinetics and a large dynamic range was ASAP1 [
86]. Crystal structures of Ci-VSP indicate that membrane potential shifts induce conformational changes in the region between the third (S3) and fourth (S4) transmembrane segments of the protein [
148]. St-Pierre et al. used this information and introduced an FP into the homologous region of
Gallus gallus VSD (Gg-VSD), containing R153Q substitution that drives Ci-VSP response to a less negative range of potentials [
86]. The S3-S4 loop of Gg-VSD is shorter than that of Ci-VSD; therefore, it was suggested to ensure better conformational coupling between sensory and reporter units [
86]. With the same objective, cpGFP was initially chosen because of its enhanced flexibility compared to that of intact GFP and was later replaced with cpSFGFP-OPT to increase the brightness and dynamic range. The dynamic range decreased when VSDs from
Danio rerio or
Xenopus laevis were tested as sensory units, and the variant with Ci-VSD showed poor plasma membrane localization. The resultant probe was named ASAP1. Its emission decreased by 17% per 100 mV, and its fast kinetic constants (τ
on approximately 2.1 ± 0.2 ms; τ
off approximately 2.0 ± 0.1 ms, contributing to 60% and 43% of response, respectively) exceeded the analogous parameters of ArcLight by 7 and 22.5 times, respectively [
86]. However, the half-response voltage of the probe is about −135 mV [
149], which lies relatively far from the physiological range. Further studies led to the development of improved ASAP versions. Accelerated ASAP1 kinetics apparently arises from the insertion of cpSFGFP-OPT, and the probe shows a split in both electrophysiological and optical signals, indicating the presence of an intermediate state, which is not observed in GgVSD R153Q [
150]. The rational design in ASAP-Y allowed the discovery of a key substitution L158Y that eliminates the intermediate state, preserving accelerated kinetics and a large dynamic range [
150]. In another study, ASAP2f was developed by linker optimization (A147S and DA148 mutations), resulting in increased response amplitude with similar temporal properties [
97]. The R415Q substitution in the S4 transmembrane segment is responsible for voltage sensing and led to a probe named ASAP2s with altered kinetic constants (τ
on approximately 5.2; τ
off approximately 24.1) and significantly increased dynamic range (−39% per 100 mV) [
98]. Combination of slower turn-off rate and higher response amplitude provided 90% improvement in the integral signal intensity compared to that with the initial sensor [
98]. The half-voltage response for this version lies closer to the physiological range (−105 mV) [
149]. Thus, ASAP1, ASAP-Y, and ASAP2f are better at resolving fast electrochemical events, whereas ASAP2s provides increased sensitivity. Finally, in a recent study, ASAP3 was developed [
149] by combining mutations from ASAP2f and ASAP2s with the following random mutagenesis of the 146–151 residue region of cpFP as alterations in this fragment were suggested to affect His151 that normally stabilizes the protonated form of the chromophore. A novel protocol for multi-well library screening based on direct PCR products transfection and electroporation as a dynamic range test allowed the identification of an indicator version that showed a signal change of −50% per 100 mV. To our knowledge, this is the greatest response amplitude among all GEVIs published to date. Moreover, ASAP3 has a half-response voltage of −88 mV, which is closer to the physiological range compared to that of other family members and shows considerably rapid kinetics (τ
on approximately 3.7 ± 0.1 ms; τ
off approximately 16.0 ± 0.3 ms, constants of the fast component) [
149].
Previously, another strategy for cpFP-based GEVI development was published [
151]. The VSD-FR189–188 probe consists of cpFusionRed(189–188) and VSD from VSFP-Butterfly1.2, which serves as a linker joining native ends of the FP. VSFP-Butterfly1.2 is a FRET-based sensor with mCitrine and mKate fused to its N- and C-ends [
152]. It works on the principle that shifts in the membrane potential induce conformational rearrangements in the VSD, leading to a change in the energy transfer efficiency between the FRET pair. In VSD-FR189–188, the same chain of biophysical events affects the relative positions of cpFP halves modulating the emission intensity [
151]. This molecular switch, in a sense, resembles the bimolecular fluorescence complementation technique; however, in this case, rejoining of the split parts resulted not from the association of two macromolecules but from intramolecular rearrangement. VSD-FR189–188 is characterized by a small dynamic range (not more than 3%) and slow kinetics (approximately 26 ms). This probe was recently improved by linker shortening that gave 25–30-fold increase in the rate of response [
153]. New versions showed responsiveness comparable to that of the best FP-based GEVIs; however, further optimization of the dynamic range is required [
153]. Detailed information is listed in
Table 3.
4.3. GEFIs for the Visualization of Oxidation and Reduction Events
Maintaining a normal redox status is essential for the functioning of cellular systems. Changes in the redox state because of stress or intrinsic cellular activity may be involved in the regulation of various cellular processes. Disruption of the redox status is associated with increased formation of reactive oxygen species (ROS)—highly reactive particles that are generally considered as products of incomplete reduction of molecular oxygen in cells. ROS can cause oxidative stress and damage cellular DNA, lipids, and proteins [
154], thereby participating in the development of many pathologies such as cancer, ischemia, reperfusion injury, and some neurodegenerative diseases [
155]. However, ROS may also function as signaling molecules and participate in the regulation of some physiological processes [
156].
ROS are extremely reactive particles; therefore, their detection in cells is difficult. However, several ROS detection methods have been developed. For example, dichlorofluorescein derivatives that change optical parameters upon interaction with ROS are used for this purpose [
157]. The disadvantages of this approach include the low selectivity of dyes and the difficulties in ROS localization in certain cellular compartments. Genetically encoded biosensors have become a kind of breakthrough in measuring ROS concentration dynamics.
The most popular GEFIs for H
2O
2 registration are HyPer family probes. The first such indicator, HyPer [
90], was designed as a chimera consisting of cpYFP integrated into the regulatory domain of the transcription factor OxyR from
E. coli. In the presence of H
2O
2, the reduced form of OxyR is converted to its oxidized form. The key residue Cys199, which is located in the hydrophobic pocket of OxyR, reacts with H
2O
2 with a consequent conversion to a sulfenic acid derivative. After sulfenic acid is released from the hydrophobic pocket, it can form a disulfide bond with Cys208, causing remarkable conformational changes in OxyR. The probe shows ratiometric response with high amplitude of about 3-fold F500/f420 decrease and selectively reacts with H
2O
2. Subsequently, improved versions of indicators named HyPer-2 [
158] and HyPer-3 [
105] were developed. HyPer-2 was developed by introducing a point mutation into the OxyR domain; its dynamic range was twice that of HyPer. In addition, HyPer-2 showed slower oxidation and reduction than HyPer: both half-oxidation and half-reduction times of HyPer-2 were twice as those of HyPer. HyPer-3 was also developed by introducing a point mutation into the sensory unit of HyPer, leading to a greater dynamic range (about 6-fold) and faster kinetics of response, thereby combining the strengths of the previous versions of the biosensor. Because the fluorescent signal of HyPer is pH-dependent, its variant with Cys199 replaced for Ser (C199S), named SypHer, was developed as an appropriate pH control for using in parallel experiments. Several improved versions of SypHer such as SypHer2 [
159] and SypHer3s [
160] are available. SypHers are now used as pH indicators in living systems [
161].
A red variant of HyPer, named HyPerRed [
103], was designed by replacing cpYFP with the RFP cp-mApple. For this probe, peptide linkers between sensory and reporter units were optimized using random mutagenesis. HyPerRed is an intensiometric probe with about 2-fold amplitude response and is pH-sensitive; hence, HyPerRed-C199S should be used in parallel. Since the tissue penetration of light increases with an increase in wavelength, HyPerRed fits better for in vivo imaging. The red variant of the indicator also allows the visualization of H
2O
2 in multiparameter mode. Since HyPer reacts with H
2O
2, its possible antioxidant role is an important issue. However, in most experiments, HyPer showed no influence on the H
2O
2-dependent physiological processes [
162]. The only exception observed is the
Arabidopsis thaliana model [
163].
For the detection of another type of ROS, organic hydroperoxides (OHPs), a genetically encoded sensor, has also been developed [
164]. This probe is constructed by inserting cpVenus into conformationally mobile α-helix of the transcriptional factor OhrR that participates in controlling the OHP detoxification apparatus of bacteria. The probe shows higher selectivity to OHPs than to other types of ROS.
Methionine can be converted by biological oxidants such as ROS to the R and S diastereomers of methionine sulfoxide (MetO) and can be used as a marker of oxidative damage to proteins and metabolites. Genetically encoded fluorescent probes for R- and S-MetO have been developed [
165]. These probes have a rather interesting design: the cpYFP domain is located between
S. cerevisiae methionine sulfoxide reductase A or B (MSRs) and their specific thioredoxins (Trx1 or Trx3, respectively) with short amino acid linkers. MSRA or MSRB selectively interacts with R- or S-MetO, respectively, and a disulfide bond is formed between two redox active Cys of an MSR moiety. Next, specific Trx reduces this bond by its catalytic Cys, and a disulfide bond is formed between MSR and Trx, inducing conformational changes that cause a change in the fluorescent signal of the sensor. Both the probes show ratiometric pH-dependent response to their substrate with high dynamic range: at the optimal value of pH 7.5, MetSOx and MetROx showed about 6-fold ratio increase and decrease after oxidation, respectively. In vitro and in vivo experiments have shown that both sensors can detect changes in MetO levels from 1 to 1000 μM, which is in the physiological range of MetO. The oxidation of the sensors is reversible in cells, indicating that endogenous systems can reduce them.
Carbon monoxide (CO) plays an important role of a gasotransmitter in living systems and is involved in some pathological and physiological processes. A genetically encoded probe based on cpFP for CO detection, named COSer, has been reported [
166]. This probe is based on CO-sensing heme protein CooA from
Rhodospirillum rubrum. CooA is a transcriptional factor that consists of DNA- and CO-binding domains. The latter contains heme. The heme iron is a six-coordinate system with Pro2 as one of the axial ligands. CO displaces Pro2 and induces the twist of the long α-helix between Phe132 and Arg134, allowing the binding of CooA to DNA and activating the transcription of the
coo operon. The sensor was designed by introducing cpVenus sequence into the F132-R134 region of the CooA α-helix. COSer shows a ratiometric response with maximal amplitude of about 2.2-fold ratio increase after CO binding and is selective for CO over other small heme ligands.
NADH, along with its oxidized form NAD
+, is a key cofactor involved in numerous biochemical processes. The metabolic and redox states of cells depend on the [NAD
+]:[NADH] ratio. Many genetically encoded fluorescent probes for NADH, NAD
+, and [NAD
+]:[NADH] ratio have been reported. All indicators are based on bacterial redox-sensing repressor Rex [
167]. When the [NAD
+]:[NADH] ratio is high, Rex forms a stable complex with DNA and one NAD
+ molecule, thus repressing the transcription of target genes. If the NADH amount increases, it displaces NAD
+ in Rex since the latter has considerably higher affinity to NADH than to NAD
+. With this, Rex undergoes remarkable conformational changes and dissociates from the complex with DNA [
167].
Peredox sensor, which enables measuring the [NADH]:[NAD
+] ratio, was constructed by combining the pH-resistant FP cpT-Sapphire with
Thermus aquaticus Rex (T-Rex) [
70]. The fluorescent domain is located between two Rex subunits because the orientation of the T-Rex subunits in the dimer changes with the binding of NADH. For ratiometric signal, mCherry is fused to the C-terminus of the chimera. A version with YFP mCitrine instead of mCherry has also been reported [
71]. The major limitation of Peredox is the very high affinity to the substrate, which complicates its use under physiological conditions.
Frex [
99] indicators for NADH were developed on the basis of the Rex repressor from
Bacillus subtilis (B-Rex). The design of the probes is similar to that of Peredox: the cpYFP domain is inserted between the full-length subunit and NADH-binding domain of Rex. The two best variants of the indicator obtained using single site-directed mutagenesis of the NADH-binding pocket are Frex, which responds to the analyte with a 9-fold increase in the fluorescent signal, and FrexH, which responds with a 3-fold decrease in fluorescence. These probes have differences in affinity to NADH; hence, one of them is preferred depending on the particular conditions. The main limitation of Frex family probes is their pH sensitivity; hence, a proper pH control is needed for their effective use in living systems.
RexYFP [
91] is a biosensor designed based on T-Rex; however, in this construction, the cpYFP domain is integrated into the flexible loop between the DNA- and NADH-binding domains of the sensory unit. This design has an important advantage over the one described earlier: the sensor molecule is smaller and targeting it to cellular organelles is easier. The brightness and maturation at physiological temperature of the sensor were optimized using random mutagenesis. The indicator is intensiometric, and its fluorescence intensity decreases about 2-fold after NADH binding. Like many cpYFP-based indicators, RexYFP is pH-dependent, and NADPH may also affect its signal in living cells, although the indicator has considerably lower affinity for NADPH.
Another probe for [NADH]:[NAD
+] ratio, SoNar, was constructed by inserting cpYFP into the surface loop of the NADH-binding domain of T-Rex, while the DNA-binding domain was truncated [
81]. Peptide linkers of the sensor were optimized, and the probe showed extremely high amplitude of ratiometric response (about 15-fold dynamic range in vitro). The signal of SoNar did not change in response to NADPH and other nucleotides.
Based on the principle of SoNar development, researchers have developed a group of iNap indicators for [NADPH]:[NADP
+] ratio registration [
100]. Comparing the ligand-binding pockets of NADH- and NADPH-binding proteins, the authors revealed the main differences in their structural organization. The data obtained were used to design mutations that could switch the substrate specificity of SoNar to NADPH. Thus, iNap1–iNap4 sensors with similar spectral properties and affinities to NADPH of about 2.0, 6.0, 25, and 120 μM, respectively, were created. The sensors showed high dynamic range of about 900%. The variant of the sensor with completely abolished ligand-binding ability, named iNapc, is proposed to be used for pH control.
A genetically encoded probe that reports only NAD
+ concentration, has also been developed [
168]. This sensor is designed by integrating cpVenus into DNA ligase from
Enterococcus faecalis. Point mutations were introduced in order to weaken NAD
+ consumption and allow the monitoring of NAD
+ within the predicted physiological range. The probe is reversible and allows ratiometric measurements with response amplitude of about 2-fold fluorescence signal decrease after NAD
+ binding.
A reporter for ratiometric monitoring of quinones in living systems has been developed. Quinones and their derivatives participate in many important biological processes such as electron transport in cell membranes and posttranslational modification of proteins. The QSer probe was the first genetically encoded fluorescent biosensor for detecting quinones in living cells [
169]. QSer was designed by inserting cpYFP between two monomeric subunits of QsrR, a transcriptional repressor from
S. aureus. A 9° rotation occurs between the α-helices of each monomer after quinone binding, promoting cpYFP conformational change. The probe is characterized by high response amplitude (approximately 3.5-fold) and is specific to quinone molecules containing fewer than three substituted groups.
In addition to the creation of classical fluorescent biosensors, cpFP can be used to obtain biosensors with unnatural amino acids (UAAs) by using a genetic code expansion technology. The idea of obtaining such indicators is based on the introduction of UAAs into the chromophore region of a FP by coexpressing the orthogonal tRNA/synthetase pair. For example, an orthogonal tRNA/synthetase pair exists that enables the genetic encoding of
p-azidophenylalanine in response to the amber (TAG) codon in
E. coli and mammalian cells. UAAs chemically react with the metabolite and change the fluorescent signal. The reason for using exactly circular permutants of FP is that the chromophore of such proteins is more spatially available compared to those of native FPs. This method was used to develop sensors for the detection of hydrogen sulfide [
170,
171] and peroxynitrite [
172]. Various redox biosensors are listed in
Table 4.
4.4. GEFIs for Measuring Organic Metabolites
For better understanding the mechanisms of various physiological and pathological processes, determining how the metabolic fluxes of a system change is important. Different ways are available to register the changes in the concentration of a particular metabolite, such as chromatography–mass spectrometry and colorimetric and fluorometric enzyme assays [
176,
177]. In addition to the methods for assessing the changes in the concentration of a single metabolite, methods are available for analyzing the total metabolome. The most widely used high-throughput experiments are mass spectrometry and NMR spectroscopy-based metabolomics [
178]. Although genetically encoded biosensors do not allow simultaneous analysis of numerous samples, they can be used to register the dynamics of metabolite concentrations in real time in living systems (Table 5).
Glucose is one of the main sources of energy for metabolic processes in living organisms. To date, relatively few biosensors are available that can allow the monitoring of the dynamics of glucose concentration in real time. Several FRET-based sensors, for example, series of FLIPglu probes [
179,
180,
181,
182] and glucose indicator proteins [
183,
184] are available. Along with these FRET-based sensors, cpFP-based sensors have also been designed.
The series of FGBP probes with differences in affinity to glucose was created by inserting cpYFP into the glucose/galactose-binding protein of
E. coli [
93]. Subsequently, the probe with physiologically relevant value of Kd, FGBP
1mM, was selected for further characterization. FGBP
1mM is a ratiometric probe, and its main advantage is the high amplitude of the sensor response (up to 7-fold), which is considerably more than that for FRET-based sensors. Another glucose sensor, iGlucoSnFR [
185], was designed by combining the cpGFP with a glucose/galactose-binding protein of
Thermus thermophilus. Peptide linkers were optimized by random mutagenesis, and several point mutations were introduced in order to decrease the affinity to glucose. The probe showed intensiometric response with about 3-fold fluorescence increase after glucose binding and had Kd of 7.7 mM, which is physiologically relevant. iGlucoSnFR-TS [
186], also called Sweetie-TS, was created by replacing cpGFP with a pH-stable FP cpT-Sapphire. This probe is considered as a fluorescence lifetime sensor that allows the conversion of measurements into actual concentrations in vivo. Peptide linkers between reporter and sensory units were optimized, and the probe with high fluorescence lifetime change and low pH sensitivity was selected from the library of mutants. The fluorescence lifetime change of the sensor is about 0.38 ns in HEK273T cells for a range of glucose concentrations (0.01 to 30 mM), and the signal is minimally affected by pH in the range of 7.1–7.4, making it a suitable sensor for measuring cytosolic glucose concentration.
Among the sensors for critically important cell metabolites, there are indicators that report the [ATP]:[ADP] concentration ratio [
80,
104]. In this case, competition between ATP and ADP for the ligand-binding center allows the sensor to report precisely the ratio of the concentrations of these two metabolites. The first such reporter, named Perceval, was designed by inserting the cpmVenus sequence into the flexible T loop of bacterial regulatory protein GlnK1 from
Methanococcus jannaschii [
80]. The original version of the sensor was optimized using semi-random mutagenesis targeting residues involved in ATP binding and T loop conformational rearrangement in order to increase the specificity, accelerate the kinetics of the response to change in [ATP]:[ATD] ratio, and improve the affinity of the instrument to be applicable in living systems. Subsequently, an improved version of the indicator, called Perceval HR, was released [
104]. This sensor has significantly greater response amplitude (more than 8-fold change compared to the initial 2-fold) and can be effectively used under physiological conditions.
The cpFP-based indicators that register a change in the concentration of ATP only have also been developed [
187,
188]. However, several FRET-based sensors for ATP have been already reported, for example, ATeam probes, optimized for use under various conditions [
60,
189,
190]. The major limitation of such probes is that the presence of two FPs in a sensor molecule can cause problems associated with the simultaneous maturation of two chromophores and degradation of the sensor inside the cell. Thus, malfunction of sensor molecules can occur, and their concentration may depend on the cell growth rate. These disadvantages may be overcome by generating single FP probes. The first single cpFP-based ATP indicator was the ratiometric probe QUEEN [
187]. It was designed by integrating the cpEGFP sequence into the F
0F
1-ATP synthase epsilon subunit. Three variants of QUEEN with different affinity, one of which (QUEEN-2m) has physiologically relevant Kd value, and one is insensitive to ATP (QUEEN-NA), have been reported. Recently, a series of biosensors for ATP with different properties, named iATPSnFRs, was published [
188]. The design of these probes was based on that of QUEEN with the following differences: cpEGFP was replaced with cpSFGFP, peptide linkers were optimized, and some point mutations were introduced into the sensory unit. The resulting biosensors were characterized by large ATP-dependent fluorescence intensity increase: dF/F for iATPSnFR
1.0 is 2.4 and for iATPSnFR
1.1 is 1.9, which is considerably more than that for QUEEN and ATeam. The probes function as single-wavelength sensors with little sensitivity to ATP metabolites.
GTP plays the role of an energy source in organisms as well as participates in cellular signaling, including the activation of G-proteins, which are signal transducers that transmit signals from various hormones, neurotransmitters, and chemokines [
191]. A group of GTP sensors, named GEVALs (GTP evaluators), has recently been reported [
101]. The indicators were created by inserting cpYFP into the flexible region of the FeoB G-protein domain. The sensor was further optimized by introducing mutations to weaken or eliminate GTP binding, and following versions with different substrate-binding constants were obtained: GEVAL30, GEVAL260, GEVAL530, GEVAL1150, and GEVAL2300, where the number in the sensors’ names shows K
eff in μM. The response amplitude of all versions was about 2-fold F400/F485 increase after GTP binding. Furthermore, a pH-control version, GEVALNull, has been reported. These probes are equally selective for GTP and dGTP.
Citrate is one of the most important cell metabolites since it is the starting point of the tricarboxylic acid cycle. It participates in fatty acid biosynthesis by supplying acetyl-CoA equivalents in the cytosol as well as regulates fatty acid synthesis and glycolytic/gluconeogenetic switch by the allosteric control of the enzyme phosphofructokinase. The FRET-based biosensor for citrate detection has been previously reported [
192]. Subsequently, the first citrate sensor based on cpFP was developed [
92]. This sensor was designed by inserting the cpGFP domain in the
Klebsiella pneumoniae CitA protein, a highly specific citrate receptor. Some point mutations previously described for accelerating chromophore maturation and mutations optimized for ratiometric Pericam were introduced into the cpGFP sequence. The sensor response is about 2-fold, which is almost twice than that for the previous FRET indicators.
Maltose, a disaccharide, is split into two glucose molecules in living organisms under the action of maltase enzyme. It is used in the manufacture of confectionery products, as well as in food products for children because of its low allergenicity. Genetically determined absence of maltase in humans causes maltose intolerance, requiring its elimination from the diet and supplementation with maltase. A simple cpFP-based maltose indicator [
193] was designed by inserting an improved cpGFP with better brightness, spectral properties, and fluorophore maturation rate, as well as bearing the mutation S65T, into linker loop between the N- and C-terminal domains of
Thermotoga maritima MBP. The sensor is intensiometric and shows an approximate 20% amplitude response. A family of maltose indicators has been described in another study [
194]. The maltose-binding protein from
E. coli has been chosen as a sensory domain. These sensors differ in not only affinity to the substrate but also reporter unit: sensors based on cpCFP, cpBFP, cpGFP, and cpYFP have been developed. The authors also showed that the ligand-binding specificity of the sensor can be changed from maltose to sucrose by introducing four point mutations previously shown to confer MBP with an affinity for sucrose.
A sensor for recording the dynamics of phosphonate concentrations in living systems has also been described [
96]. The most abundant natural phosphonate is 2-aminoethylphophonate (2-AEP), which is a precursor in the biosynthesis of phosphonolipids, phosphonoproteins, and phosphonoglycans. Previously, a fluorescent biosensor was constructed by the covalent attachment of environmentally sensitive fluorescent dyes to introduced cysteines of EcPhnD (periplasmic-binding protein of phosphonate uptake and utilization system from
E. coli) [
195]. However, in that study, the authors relied on the structure of the EcPhnD homologue, sulfate-binding protein, although it shares only 37% sequence identity with PhnD. Subsequently, the crystal structure of EcPhnD was determined, and a biosensor designed by integrating cpGFP in the EcPhnD sequence was developed [
96]. This sensor has been optimized by screening of linker mutants. Interestingly, the variant with the deletion of two amino acid residues of the sensory domain right before the cpGFP sequence, obtained by linker mutagenesis, was selected. The sensor has 2.6-fold amplitude response to 2-AEP and was suitable for the screening of phosphonates in living systems.
Histidine is an essential amino acid and a precursor for histamine; it is of vital importance for neurotransmission and neuromodulation [
196]. The FRET-based biosensor for histidine detection based on bacterial periplasmic-binding protein (PBP) HisJ, which specifically binds to histidine, and its analogue with circularly permuted HisJ subunit, have been reported [
197]. Subsequently, a single FP-based sensor, named FHisJ, was designed by integrating the cpYFP domain into the flexible regions of HisJ [
95]. This indicator has a high amplitude response (520%), which is about 8-fold higher than that for the FRET permuted biosensor, and the value of its dissociation constant is 22 μM, which is physiologically relevant. Detailed information is listed in
Table 5.
4.5. GEFIs for Cellular Signaling Visualization and Neurotransmitter Measurement
One of the most important properties of living cells is their ability to adapt to environmental perturbations. In order to maintain their metabolism adjusted for current needs, cells have developed complicated signaling systems in the course of evolution [
198]. These systems provide metabolic switches that allow either metabolism buffering or shifting depending on the precise situation with different degrees of biochemical pathways rearrangements and varying temporal dynamics. The fastest events include manipulating with ion concentrations and proceed at millisecond rates, whereas the slowest rely on affecting gene activity and require hours to be completed. This area of investigation is essential in modern research as deciphering signaling intercalations is of remarkable importance for understanding development, metabolic diseases, tumorigenesis, nervous system functioning, and consequent drug development. Hence, the real-time kinase activity dynamics, G-protein-coupled receptor activation, secondary messengers, and neurotransmitter concentration shifts at single cell resolution have been visualized by using genetically encoded indicators. To date, numerous indicators of varying colors have been developed, and some of them involve the use of cpFPs (Tables 6–8).
Secondary messengers from inositol phosphate family orchestrate signal transduction in many pathways that are involved in cell differentiation, proliferation, neural plasticity, and muscle contraction [
199]. They are often recognized by pleckstrin homology (PH) domains; however, the latter often lack conformational mobility sufficient for FRET-based probe development. Inspired by successful engineering of a split phospholipase Cδ1 PH domain that retained affinity and selectivity for the natural ligand [
200], Sakaguchi et al. developed a genetically encoded probe for Ins(1,3,4,5)P
4 by inserting cpGFP into a mobile loop of the PH domain from Bruton’s tyrosine kinase [
67]. In this probe, the flexible nature of cpFP provides efficient conformational coupling of sensory and reporter units, converting small protein rearrangements into an optically detectable signal. The authors speculated that ligand binding disrupts His148 interaction with the chromophore that shifts the acid-base equilibrium toward the deprotonated form. This results in a 1.5-fold ratiometric response.
Another important secondary messenger related to phospholipids is diacylglycerol (DAG). It is mostly generated in the course of phospholipase C reaction and serves as a key inducer of protein kinase C (PKC) [
201]. Given that the activation of the latter usually requires Ca
2+ influx, GECIs have been successfully implemented for real-time imaging of this signaling pathway. However, this approach has a significant drawback coming from the fact that Ca
2+ ions play a role in other biochemical processes. The need for clearly separating these factors led to the development of cpGFP-based DAG probes [
202]. They utilize conformational rearrangements occurring in PKCδ under ligand binding, namely, the loss of the contact between enzyme and pseudosubstrate domains of the protein. This isoform was selected because experimental evidence suggests that its C2-domain is insensitive to Ca
2+ [
203], and the C1-domain shows high affinity for analyte [
204]. Among 30 engineered variants, Tewson et al. selected Upward and Downward DAG sensors that differed by the direction of the response, which was about 40% in HEK293 cells for both the probes. This response magnitude was higher than that for the contemporaries [
202]. Subsequent optimization led to the development of Upward and Downward DAG2 versions with improved dynamic range [
26]. Moreover, the first prototypes based on cpmNeon, a brighter protein were reported [
205]. Interestingly, Upward and Downward DAG2 probes showed different response kinetics: return to the baseline was faster for the Upward DAG2 probe [
26]. The authors suggested that this might be because of photobleaching; therefore, intensiometric probes with different directions of response can be good controls for each other. Notably, other designs for DAG sensors have been reported. One type is based on the translocation of activated PKC-FP chimeras to the membranes [
206] and another is FRET-based [
207]. Translocation readout is often not sufficiently quantitative and requires high-resolution imaging for good results, whereas FRET-based probes show low maximal amplitude.
Many biochemical events are induced by cyclic nucleotides cAMP and cGMP generated by adenylate cyclase and guanylate cyclase, respectively. The drawbacks of the already available FRET-based biosensors for cGMP visualization were overcome by Nausch et al., who engineered cpEGFP-based probes by inserting the latter into the regulatory domains of protein kinases G (PKG) I α or β [
87]. This resulted in the development of three indicators, namely, α-, β-, and δ-FlincGs, that differed by affinity, cGMP/cAMP selectivity, and dynamic ranges. δ-FlincG was obtained from the α-version by removing the entire PKGIα N-terminal domain, and it showed a 3.5-fold ratiometric excitation response, unlike the other variants that responded intensiometrically despite having both 410 nm and 480 nm excitation maxima in their spectra. Moreover, the responses of α- and β-FlincGs were reduced during cell imaging compared to those obtained in vitro; the authors hypothesized that it can be explained in the terms that unlike the δ-version, the α- and β-FlincGs contain dimerization interfaces driving them to interact with cellular PKGs. The affinity of δ-FlincG to cGMP was shown to be similar to the wild-type protein; therefore, it cannot likely act as a metabolic sink, and its pKa was estimated to be 6.1, making the probe relatively resistant to pH shifts in the physiological range. However, subsequent studies on the issue [
208] questioned these data and provided a different pKa value of 7.5; moreover, the authors showed that the alteration of the C-terminus remarkably affects the performance of the sensor, in contrast to that mentioned in the previous study [
87]. Further optimization led to the development of FlincG2 and FlincG3 [
208]. The former retains the δ-FlincG sequence except for the C-terminal part, whereas the latter has M335K substitution in the cpFP and N-terminal His-tag that was shown to increase brightness. The affinity of FlincG3 to cGMP was lower than that of the δ-version, which might be preferable in some situations, but the ratiometric response characteristic was lost. Detailed information is listed in
Table 6.
The first published cpFP-based probe for cAMP measurement was cADDis [
205]. It was engineered in order to develop a sensor with a good signal-to-noise ratio for multi-well plate automatic assays. The crystal structure of guanosine exchange factor EPAC2 revealed that ligand binding induces twisting in a hinge that connects the catalytic and regulatory regions [
210]. While testing prototypes varying by cpGFP insertion position inside this sequence and linker composition a chimera with a dynamic range of about 35% [
205] was found that became the final version. Further characterization revealed that cADDis shows optimal sensitivity at analyte concentrations between 10 and 100 μM.
Another green cAMP probe based on cpFP was recently published [
79]. cAMPr was developed by connecting the catalytic and regulatory Iα subunits of protein kinase A with cpGFP as a linker. The authors suggested that, in the ligand-free state, the subunits would be connected to each other, and relaxation induced by cAMP would be transmitted to the reporter domain affecting its fluorescence. Membrane localization and interaction with wild-type enzyme was avoided by removing the dimerization/docking domain from the regulatory subunit. Next, the second cAMP-binding site was deleted to decrease the affinity of the probe, expanding its field of application as cAMP concentration can vary in a wide range. Further linker optimization led to the development of the final version with a dynamic range of about 50%.
Ohta et al. focused on the high affinity and selectivity of PKA RIα and engineered a red cAMP probe named R-FlincA [
209]. In this case, the architecture of the sensor was different as cp146mApple was directly inserted into the high-affinity cAMP-binding motif of this protein. To our knowledge, R-FlincA is characterized by a very large dynamic range of 860%, which is the highest among cAMP probes, and the best cAMP/cGMP selectivity. Its dissociation constant for analyte is about 0.3 µM, which surpasses the corresponding values for Flamindo2 and Pink-Flamindo (3.2 µM and 7.2 µM, respectively) [
209]. The main drawbacks of this probe are pH sensitivity and low brightness.
Many signaling pathways rely on the activation of protein kinases (PKs) that are capable of phosphorylating target molecules affecting their functions. Different classes of enzymes and transcriptional factors are subjected to such regulation. To date, numerous genetically encoded probes have been used for visualizing PK activity; however, most of them utilize FRET technology, and only few are based on cpFPs [
211]. This design based on cpFPs was first implemented in sinphoses, which represent a family of probes for insulin-induced protein phosphorylation [
212]. These probes were constructed by joining the SH2n domain from the p85 regulatory subunit of phosphatidylinositol 3-kinase and Y941 domain from IRS-1 with cpECFP, cpEGFP, or cpCitrine as linkers. Efficient interaction with insulin receptor was confirmed by fusing a PH domain and a phosphotyrosine-binding (PTB) domain derived from IRS-1 to the N-terminus of the probes. When insulin receptor is activated, it phosphorylates Y941, converting it to a binding partner for SH2n with subsequent spatial reorganization of the sensor that can be detected by cpFPs. Sinphoses are characterized by maximal amplitudes of about 10–15%, with the cyan version being the only one relatively resistant to pH shifts. Recently, the color options for cpFP-based PK sensors have been complemented by ExRai family members that utilize cpGFP as a reporter unit [
28]. Basically, these probes have the same design, in which the phosphoaminoacid-binding domain is represented by FHA1 from AKAR, and PKA, PKB, or PKC substrate serves as the phosphate acceptor moiety, generating ExRai-AKAR, ExRai-AtkAR, and ExRai-CKAR, respectively. Unlike GCaMP3 that contains the same reporter unit, all the above probes showed ratiometric excitation response, indicating that in ExRais the wtGFP chromophore behavior is ‘rescued’ by fusion partners. ExRai-AtkAR and ExRai-CKAR were characterized by the greatest dynamic range among other genetically encoded probes for the same biochemical events. The maximal response of ExRai-AKAR was more than 2-fold, that is, 3- and 1.7-times higher than that of AKAR4 and AKAR3ev, respectively [
28]. The authors also showed that the ExRai family is color-tunable by converting ExRai-AKAR and ExRai-CKAR to blue-shifted versions by substituting cpGFP for cpCFP or cp-T-sapphire. Detailed information is listed in
Table 7.
One more field of interest is to register the real-time dynamics of neurotransmitter release in neuronal tissue. Such data remarkably contribute to our understanding of neurogenerative diseases, physiological regulation, and behavior and facilitate rational drug development. Traditional approaches to investigate this issue are microdialysis, current measurement by using a microelectrode, and enzyme coupling. Despite their contribution, to our knowledge, these approaches lack the desired spatial and temporal resolution; genetically encoded biosensors can overcome these obstacles. Thus, iGluSnFR, a cpFP-based probe for glutamate, which is one of the most widespread neurotransmitters both in vertebrates and invertebrates, was developed [
84]. The authors had previously engineered sensors by using periplasmic bacterial proteins (PBPs) as sensory units—namely,
E. coli maltose-binding protein MalE [
194] and
E. coli phosphonate-binding protein PhnD [
96] (discussed before)—and implemented this technique to
E. coli GltI protein. The cpGFP insertion points were selected on the basis of global structural similarity of a homologous protein from
Shigella flexneri to the resolved structure of MalE. Primer optimization resulted in the final version that showed approximately 450% response under glutamate binding. Finally, IgG kappa secretion tag, Myc tag, and platelet-derived growth factor receptor domain were added for desired subcellular targeting. iGluSnFR has high affinity and is selective with the exception for aspartate. cpGFP was substituted with cpSFGFP, and a version with improved brightness, named SF-iGluSnFR, was developed [
102]. Additional modifications that affect the kinetic properties of the probe were discovered. Considering that mutations of residues in the ‘hinge’ of PBPs can allosterically alter affinity, a version with A184S substitution was engineered. It showed a lower dissociation constant and slower off-rate, thereby allowing better integral signal collection. S72A afforded the opposite effect that might be beneficial for large synapse imaging where glutamate clearance is limited. By introducing mutations to the reporter unit, the authors developed SF-Azurite-iGluSnFR and SF-Venus-iGluSnFR with different spectral properties (linker optimization was required for the former). Finally, in another study, a red version—R-iGluSnFR1—was presented [
58]. cpmApple was chosen as a reporter unit for the same reason as in the case of FlicR1 [
94] (see before). After linker optimization and seven rounds of random mutagenesis, the authors obtained the final version with a dynamic range of 4.9-fold. Its pKa values were 5.1 and 8.3 for free and bounded configurations, respectively, indicating that a molecular switch may consist in the glutamate-induced conformational rearrangement, preventing the stabilization of the deprotonated state by the key lysine residue. Unlike the original version, R-iGluSnFR1 is sensitive to not only glutamate and aspartate, but also asparagine. In the same study, both sensors were converted to non-permutated forms bearing Camgaroo-like topology [
58].
Another probe based on PBP is iGABASnFR; it can allow GABA monitoring [
85]. Initially, the authors attempted to utilize the well-described Atu2422 and Atu4243 proteins from
Agrobacterium tumefaciens as sensory units; however, the former lacked selectivity and affinity for the target analyte, and the latter failed to provide good membrane localization. Next, a protein homologous to Atu4243 from
Pseudomonas fluorescens was detected using genome screening; insertion points for cpSFGFP were selected on the basis of structural similarity. F101L substitution was introduced into the hinge region to allosterically modulate affinity, and another mutation N260A significantly increased the brightness in HEK cells possibly because of the potential interaction of this residue with cpFP or its capacity to alter the glycosylation site. Linker re-optimization and F145W substitution in cpSFGFP led to the final version with high selectivity, sensitivity, and a dynamic range of 250%. Other mutations, namely, F102G and 102Y.Y137L combination, provided lower dissociation constant and increased maximal response amplitude of up to 450%.
Lastly, cpFP-based probes utilizing G-protein coupled receptors (GPCRs) as sensory modules are described. All these probes share topological similarity in cpFP insertion position located in the internal loop 3 region that in β-2 adrenergic receptor undergoes significant conformational rearrangement under ligand binding that might provide a molecular switch for sensor development [
213,
214,
215,
216]. A family of dLight probes for dopamine visualization was engineered by introducing cpGFP into dopamine receptors D1, D2, or D4 at different positions with six resulting variants, covering a broad spectrum of affinity (dissociation constants from 4.1 nM to 2.3 µM) and dynamic ranges (from 170% to 930%) [
82]. Although dLight1.1 and dLight1.2 are optimized variants combining a balance between response amplitude and affinity, the authors claimed that other versions are suitable for specific purposes, for example, dLight1.3 and dLight1.4 are useful for measuring synaptic release and tonic dopamine transients, respectively. Such sensor design is applicable to other GPCRs that was shown by developing a range of probes based on β-1 and β-2 adrenergic receptors, κ- and μ-type opioid receptors, α-2 adrenergic receptors, 5-hydroxytryptamine (serotonin) receptor-2A, and melatonin type-2 receptor, but without further optimization [
82].
Another family of cpFP-based probes for dopamine visualization is GRAB
DA [
88]. These probes were developed independently from dLight sensors on the basis of the same principles mentioned above. Each human DA receptor subtype was tested, and DRD2-cpEGFP chimera was selected as it provided good membrane trafficking and showed high affinity for analyte. After additional mutations were introduced, two versions were selected that differed in their dissociation constants (130 nM and 10 nM for GRAB
DAm and GRAB
DAh, respectively) and shared a dynamic range of about 90%, which is smaller than the corresponding value for dLights. However, compared to the latter, GRAB
DA sensors were optimized for brightness [
88]. Both GRAB
DA and dLights are selective with the exception for norepinephrine; however, considering the difference in affinities and physiological concentrations of dopamine and norepinephrine, this feature is not a limitation for their in vivo implementation. As FRET-based dopamine probes are characterized by a dynamic range of not more than 10%, both sensor families represent a remarkable breakthrough in this field. The spatial structure of DRD2 receptor has been resolved recently, and this might provide useful information for the rational optimization of these indicators [
217].
GACh probes for acetylcholine measurements were engineered on the same principle [
83]. Human muscarinic acetylcholine receptors 1–5 were tested as candidates for sensory unit with their internal loop 3 substituted with modified homologous region from β-2 adrenergic receptor. This was required because the initial length of this sequence in muscarinic acetylcholine receptors is very large; the authors intended to avoid the possible problems associated with expression and trafficking resulting from a lengthy cpGFP-containing loop. M
3R chimera showed excellent membrane targeting and could respond to the analyte when expressed in HEK293T cells with amplitude of 30%. Linker optimization allowed to increase the dynamic range of the probe to 90%.
Finally, a family of norepinephrine-sensing cpFP-based probes, named GRAB
NE, was developed [
89]. Among all tested candidates for the sensory unit, α-2 adrenergic receptor showed the best performance. Two versions—namely, GRAB
NEm and GRAB
NEh—differing by the presence of a mutation T6.34K in a region close to a conservative site E6.30 were developed; this mutation lowers the dissociation constant of the latter. Thus, the maximal response amplitudes and dissociation constants are 230% and 930 nM for GRAB
NEm and 130% and 100 nM for GRAB
NEh, respectively. Both the probes could not distinguish between epinephrine and norepinephrine; however, the authors claimed that it was not a concern in mammalian central nervous tissue as most human adrenergic receptors also respond non-discriminately to both ligands [
89]. Therefore, GRAB
NE probes reveal the regions where noradrenergic or adrenergic signaling occurs.
Notably, none of the cpFP- and GPCR-based probes disrupted the normal signaling pathways, which is experimentally proven. The theoretical basis underlying this observation is that internal loop 3 region is crucial for G-protein/arrestin and GPCR interactions, and a bulky moiety of cpFP hinders such coupling [
191,
218]. In addition to the approaches for neurotransmitter visualization already mentioned above, several other methods exist that utilize fluorescent reporters for either direct analyte detection or GCPRs activation. In some cases, cpFP-based probes provide several advantages over such reporters: FRET-based sensors usually do not have a large dynamic range, CNiFERs [
219] technology is invasive and lacks spatial resolution, and TANGO assay [
220] requires long time for signal amplification. Detailed information is listed in
Table 8.

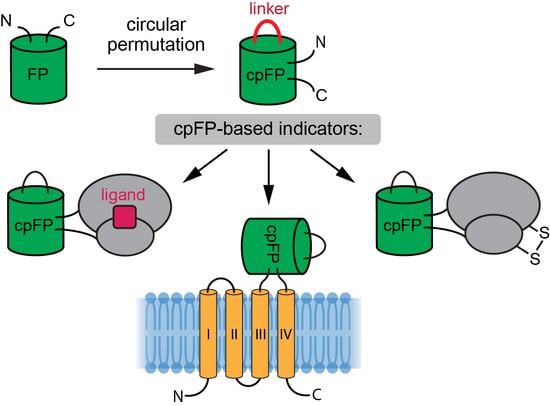
 ,
, 



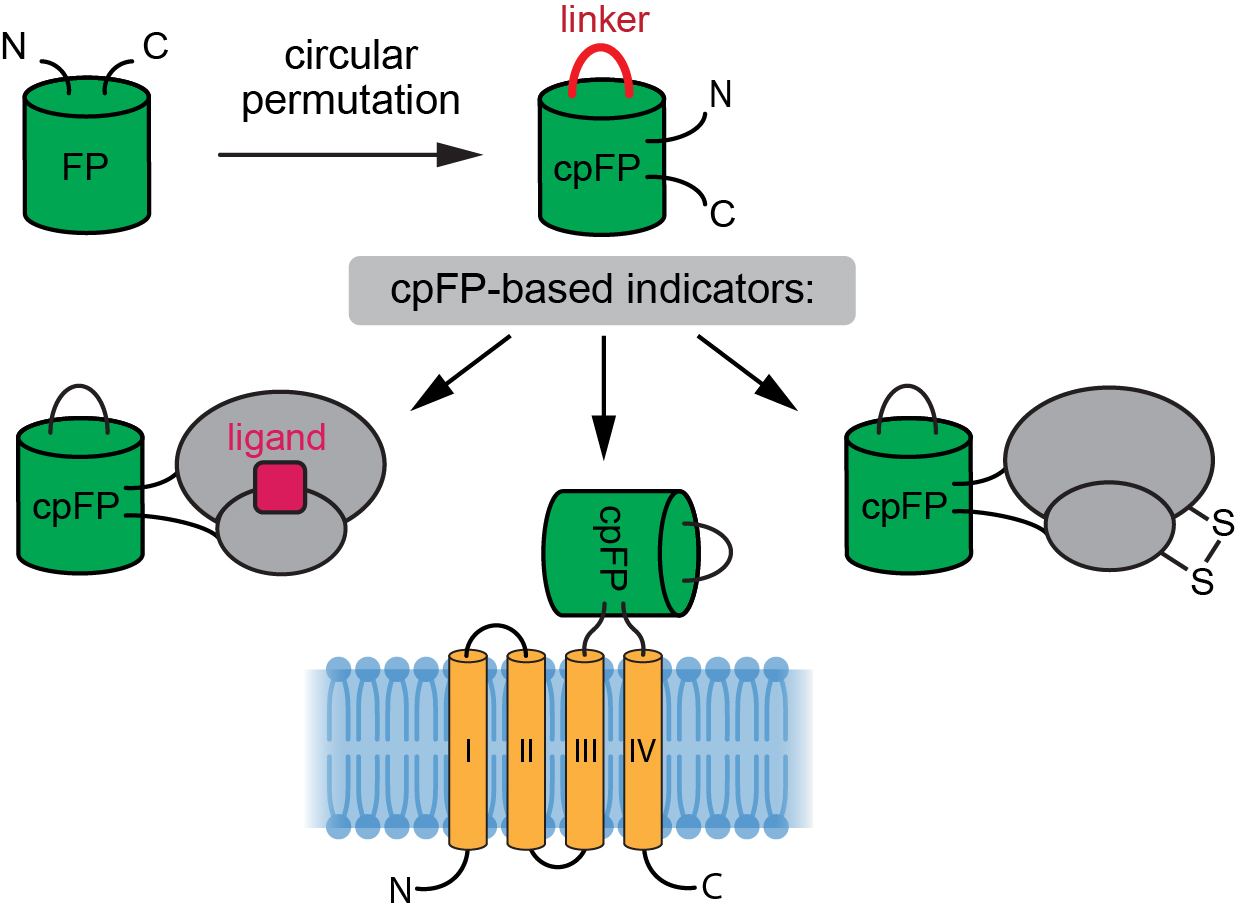{kind=link}
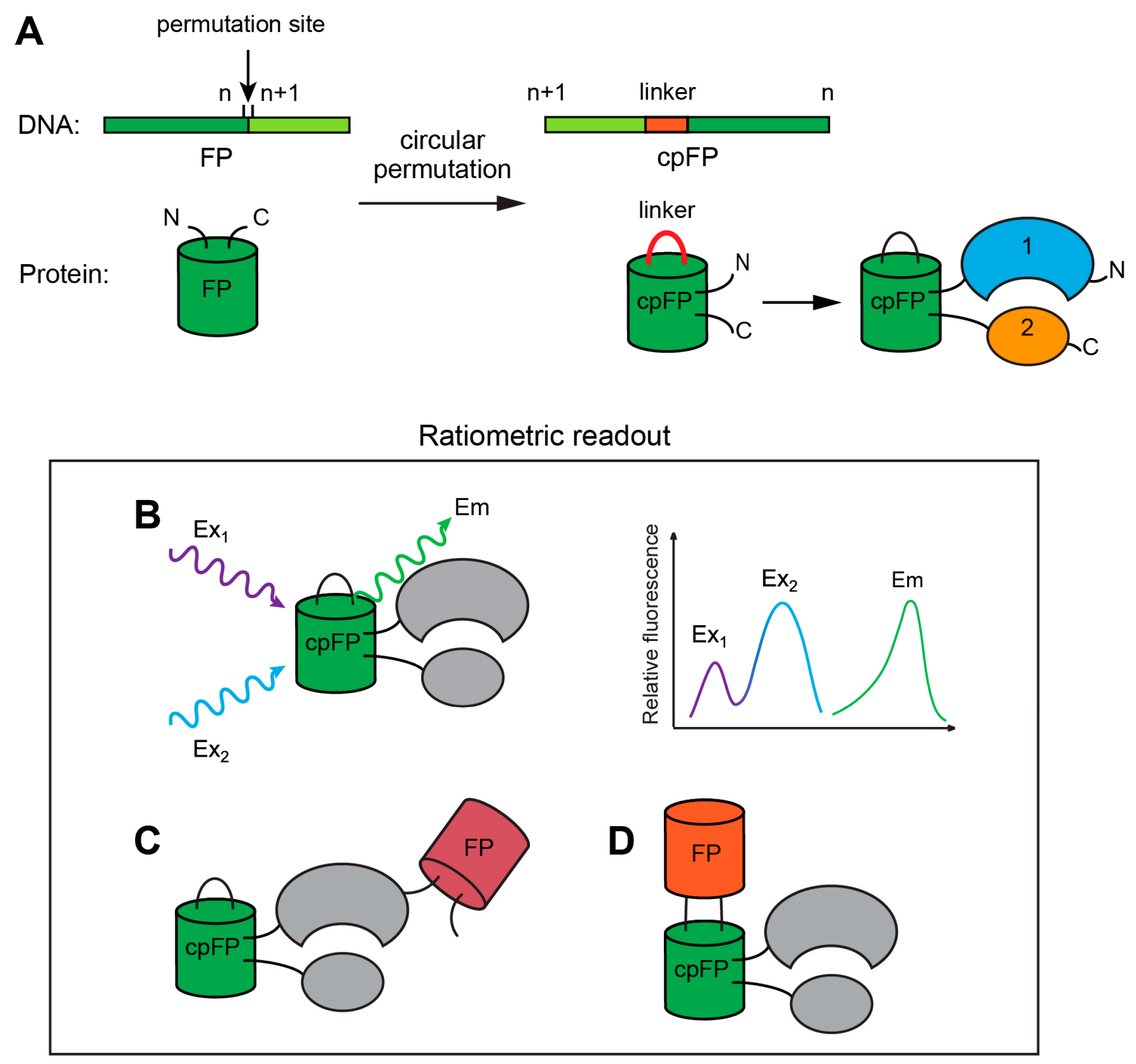{kind=link}
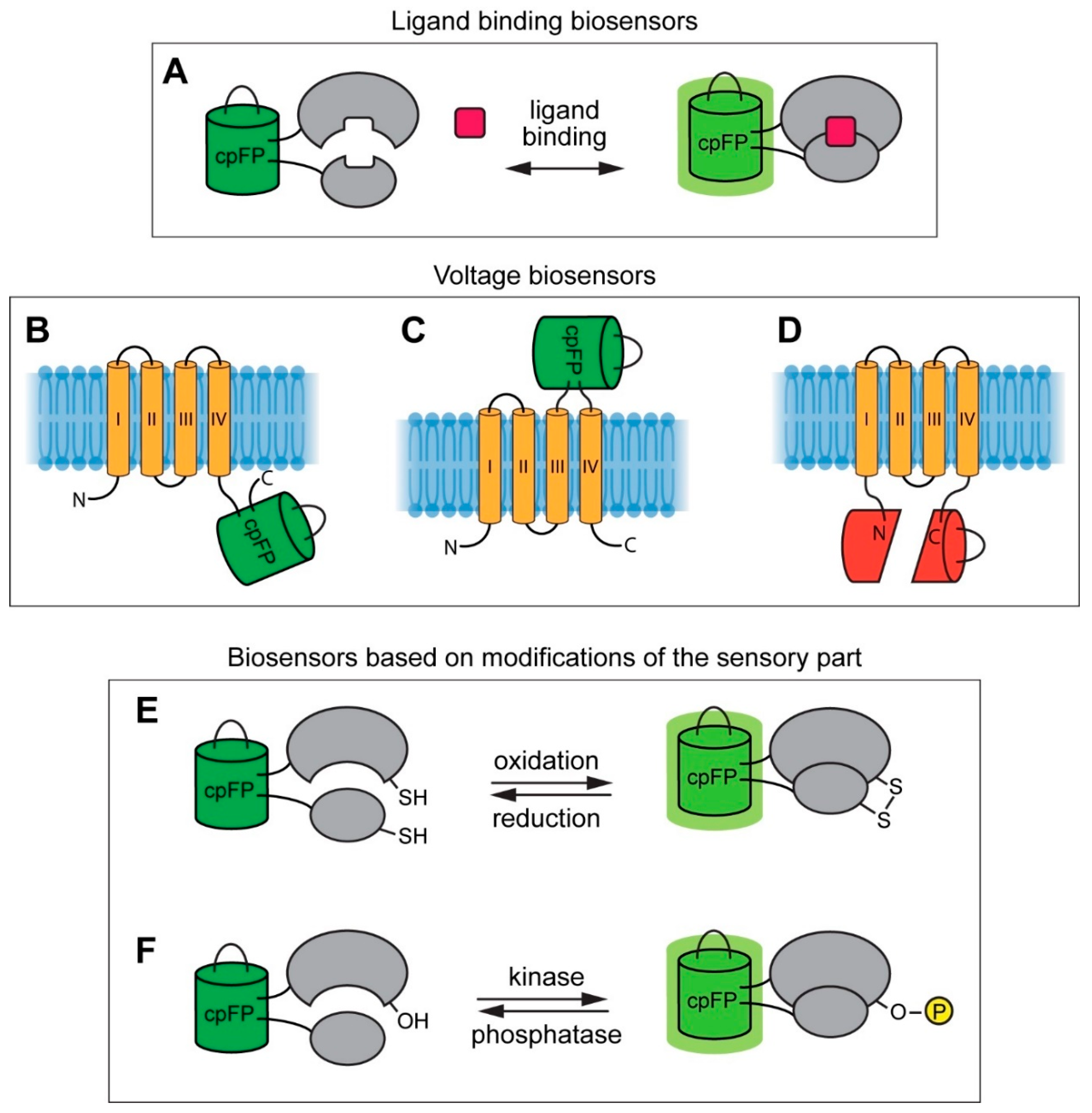{kind=link}