Oral Bacteria and Intestinal Dysbiosis in Colorectal Cancer

 ,
,  ,
,
Abstract
:1. Introduction
2. Oral Microbiota: An Overview
2.1. The Composition of the Oral Microbiota
2.2. Oral Microbiota Effects in Health and Disease
3. The Concept of Intestinal Dysbiosis in Colorectal Cancer (CRC)
4. Oral Bacteria and Intestinal Dysbiosis
5. Oral Bacteria Detected in CRC
6. Possible Mechanisms of Oral Microbiota Involvement in CRC Dysbiosis
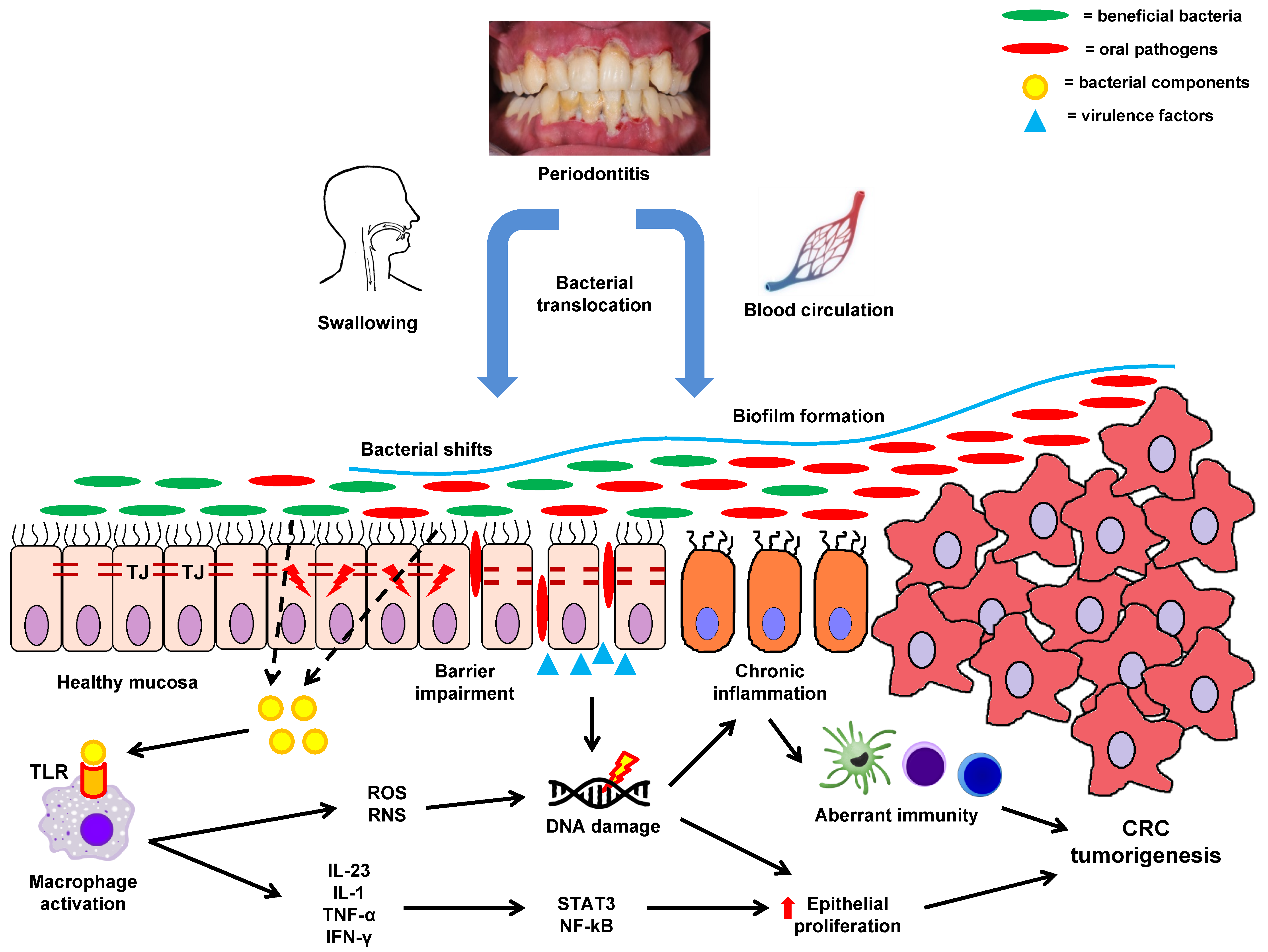
6.1. Dissemination of Oral Bacteria into the Intestinal Environment
6.2. The Role of Oral Polymicrobial Biofilms in CRC
6.3. The Metabolic Properties of Oral Bacteria in the Colon
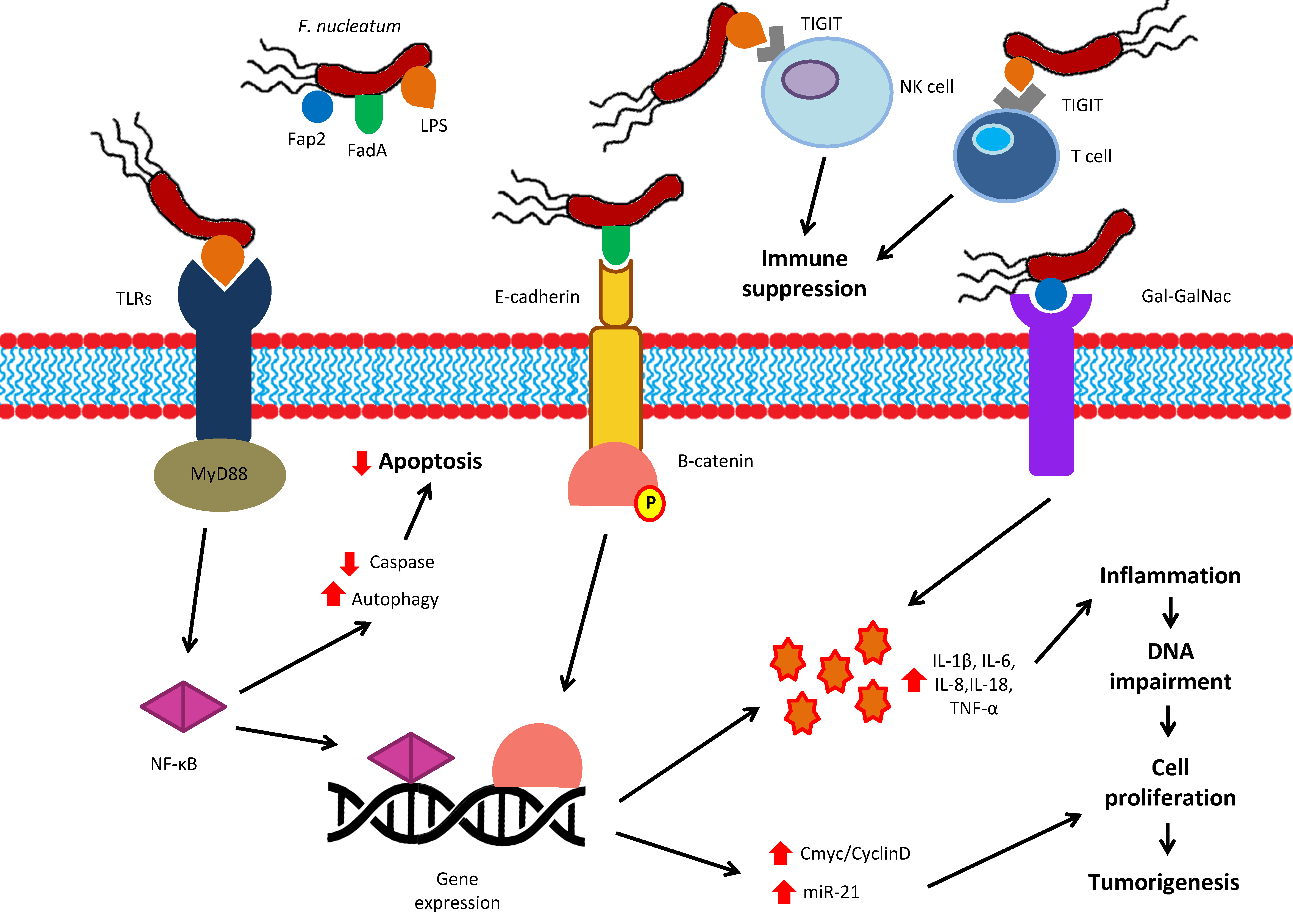
6.4. Virulence Factors of Oral Bacteria Inhibit Apoptosis and Modulate Inflammation and Immune Response in the Colon
7. Conclusions
Funding
Conflicts of Interest
References
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra265. [Google Scholar] [CrossRef] [PubMed]
- Maffei, V.J.; Kim, S.; Blanchard, E.t.; Luo, M.; Jazwinski, S.M.; Taylor, C.M.; Welsh, D.A. Biological Aging and the Human Gut Microbiota. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1474–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Jobin, C. The struggle within: Microbial influences on colorectal cancer. Inflamm. Bowel. Dis. 2011, 17, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed]
- Wade, W.G. The oral microbiome in health and disease. Pharmacol. Res. 2013, 69, 137–143. [Google Scholar] [CrossRef]
- Aas, J.A.; Paster, B.J.; Stokes, L.N.; Olsen, I.; Dewhirst, F.E. Defining the normal bacterial flora of the oral cavity. J. Clin. Microbiol. 2005, 43, 5721–5732. [Google Scholar] [CrossRef]
- Marcotte, H.; Lavoie, M.C. Oral microbial ecology and the role of salivary immunoglobulin A. Microbiol. Mol. Biol. Rev. 1998, 62, 71–109. [Google Scholar]
- Dowd, S.E.; Wolcott, R.D.; Sun, Y.; McKeehan, T.; Smith, E.; Rhoads, D. Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS ONE 2008, 3, e3326. [Google Scholar] [CrossRef]
- Kolenbrander, P.E.; Andersen, R.N.; Blehert, D.S.; Egland, P.G.; Foster, J.S.; Palmer, R.J., Jr. Communication among oral bacteria. Microbiol. Mol. Biol. Rev. 2002, 66, 486–505. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Ye, Y.; Zhou, Y.; Fodor, A.A. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE 2012, 7, e34242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Gao, H.; Mihindukulasuriya, K.A.; La Rosa, P.S.; Wylie, K.M.; Vishnivetskaya, T.; Podar, M.; Warner, B.; Tarr, P.I.; Nelson, D.E.; et al. Biogeography of the ecosystems of the healthy human body. Genome Biol. 2013, 14, R1. [Google Scholar] [CrossRef] [PubMed]
- Hull, M.W.; Chow, A.W. Indigenous microflora and innate immunity of the head and neck. Infect. Dis. Clin. North Am. 2007, 21, 265–282. [Google Scholar] [CrossRef] [PubMed]
- Teles, R.; Teles, F.; Frias-Lopez, J.; Paster, B.; Haffajee, A. Lessons learned and unlearned in periodontal microbiology. Periodontol 2000 2013, 62, 95–162. [Google Scholar] [CrossRef]
- Mager, D.L.; Haffajee, A.D.; Devlin, P.M.; Norris, C.M.; Posner, M.R.; Goodson, J.M. The salivary microbiota as a diagnostic indicator of oral cancer: A descriptive, non-randomized study of cancer-free and oral squamous cell carcinoma subjects. J. Transl. Med. 2005, 3, 27. [Google Scholar] [CrossRef]
- Danser, M.M.; Gomez, S.M.; Van der Weijden, G.A. Tongue coating and tongue brushing: A literature review. Int. J. Dent. Hyg. 2003, 1, 151–158. [Google Scholar] [CrossRef]
- Zaura, E.; Nicu, E.A.; Krom, B.P.; Keijser, B.J. Acquiring and maintaining a normal oral microbiome: Current perspective. Front. Cell. Infect. Microbiol. 2014, 4, 85. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Vollaard, E.J.; Clasener, H.A. Colonization resistance. Antimicrob. Agents Chemother. 1994, 38, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, A.; Edlund, C.; Nord, C.E. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect. Dis. 2001, 1, 101–114. [Google Scholar] [CrossRef]
- Kapil, V.; Milsom, A.B.; Okorie, M.; Maleki-Toyserkani, S.; Akram, F.; Rehman, F.; Arghandawi, S.; Pearl, V.; Benjamin, N.; Loukogeorgakis, S.; et al. Inorganic nitrate supplementation lowers blood pressure in humans: Role for nitrite-derived NO. Hypertension 2010, 56, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Tlaskalova-Hogenova, H.; Stepankova, R.; Hudcovic, T.; Tuckova, L.; Cukrowska, B.; Lodinova-Zadnikova, R.; Kozakova, H.; Rossmann, P.; Bartova, J.; Sokol, D.; et al. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol. Lett. 2004, 93, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Rutger Persson, G. Rheumatoid arthritis and periodontitis-inflammatory and infectious connections. Review of the literature. J. Oral Microbiol. 2012, 4. [Google Scholar] [CrossRef]
- Temoin, S.; Chakaki, A.; Askari, A.; El-Halaby, A.; Fitzgerald, S.; Marcus, R.E.; Han, Y.W.; Bissada, N.F. Identification of oral bacterial DNA in synovial fluid of patients with arthritis with native and failed prosthetic joints. J. Clin. Rheumatol. 2012, 18, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Bright, R.; Proudman, S.M.; Bartold, P.M. Does periodontal treatment influence clinical and biochemical measures for rheumatoid arthritis? A systematic review and meta-analysis. Semin. Arthritis Rheum. 2014, 44, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.D.; Offenbacher, S. Systemic effects of periodontitis: Epidemiology of periodontal disease and cardiovascular disease. J. Periodontol. 2005, 76, 2089–2100. [Google Scholar] [CrossRef]
- Serra e Silva Filho, W.; Casarin, R.C.; Nicolela, E.L., Jr.; Passos, H.M.; Sallum, A.W.; Gonçalves, R.B. Microbial diversity similarities in periodontal pockets and atheromatous plaques of cardiovascular disease patients. PLoS ONE 2014, 9, e109761. [Google Scholar] [CrossRef]
- Heo, S.M.; Haase, E.M.; Lesse, A.J.; Gill, S.R.; Scannapieco, F.A. Genetic relationships between respiratory pathogens isolated from dental plaque and bronchoalveolar lavage fluid from patients in the intensive care unit undergoing mechanical ventilation. Clin. Infect. Dis. 2008, 47, 1562–1570. [Google Scholar] [CrossRef]
- Filkins, L.M.; Hampton, T.H.; Gifford, A.H.; Gross, M.J.; Hogan, D.A.; Sogin, M.L.; Morrison, H.G.; Paster, B.J.; O’Toole, G.A. Prevalence of streptococci and increased polymicrobial diversity associated with cystic fibrosis patient stability. J. Bacteriol. 2012, 194, 4709–4717. [Google Scholar] [CrossRef]
- Antunes, A.A.; de Santana Santos, T.; de Carvalho, R.W.; Avelar, R.L.; Pereira, C.U.; Pereira, J.C. Brain abscess of odontogenic origin. J. Craniofac. Surg. 2011, 22, 2363–2365. [Google Scholar] [CrossRef] [PubMed]
- Pierce, D.; Calkins, B.C.; Thornton, K. Infectious endocarditis: Diagnosis and treatment. Am. Fam. Physician 2012, 85, 981–986. [Google Scholar] [PubMed]
- Gil-Montoya, J.A.; Sanchez-Lara, I.; Carnero-Pardo, C.; Fornieles, F.; Montes, J.; Vilchez, R.; Burgos, J.S.; Gonzalez-Moles, M.A.; Barrios, R.; Bravo, M. Is periodontitis a risk factor for cognitive impairment and dementia? A case-control study. J. Periodontol. 2015, 86, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Backhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Keku, T.O.; Dulal, S.; Deveaux, A.; Jovov, B.; Han, X. The gastrointestinal microbiota and colorectal cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G351–G363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candela, M.; Turroni, S.; Biagi, E.; Carbonero, F.; Rampelli, S.; Fiorentini, C.; Brigidi, P. Inflammation and colorectal cancer, when microbiota-host mutualism breaks. World J. Gastroenterol. 2014, 20, 908–922. [Google Scholar] [CrossRef]
- Bien, J.; Palagani, V.; Bozko, P. The intestinal microbiota dysbiosis and Clostridium difficile infection: Is there a relationship with inflammatory bowel disease? Therap. Adv. Gastroenterol. 2013, 6, 53–68. [Google Scholar] [CrossRef]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel. Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Abreu, M.T.; Peek, R.M., Jr. Gastrointestinal malignancy and the microbiome. Gastroenterology 2014, 146, 1534–1546.e1533. [Google Scholar] [CrossRef]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z.; et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Proctor, D.M.; Relman, D.A. The Landscape Ecology and Microbiota of the Human Nose, Mouth, and Throat. Cell Host Microbe 2017, 21, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Lynch, D.B.; Brown, J.M.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef]
- Peters, B.A.; Wu, J.; Hayes, R.B.; Ahn, J. The oral fungal mycobiome: Characteristics and relation to periodontitis in a pilot study. BMC Microbiol 2017, 17, 157. [Google Scholar] [CrossRef] [PubMed]
- Sanapareddy, N.; Legge, R.M.; Jovov, B.; McCoy, A.; Burcal, L.; Araujo-Perez, F.; Randall, T.A.; Galanko, J.; Benson, A.; Sandler, R.S.; et al. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. Isme. J. 2012, 6, 1858–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, M.; Komiyama, Y.; Mitsuyama, K.; Andoh, A.; Aoyama, T.; Matsumoto, Y.; Kanauchi, O. Prebiotic treatment reduced preneoplastic lesions through the downregulation of toll like receptor 4 in a chemo-induced carcinogenic model. J. Clin. Biochem. Nutr. 2011, 49, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Zheng, J.; Hu, G.; Wang, J.; Huang, C.; Lou, L.; Wang, X.; Zeng, Y. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci. Rep. 2016, 6, 26337. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillere, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Daillere, R.; Vetizou, M.; Waldschmitt, N.; Yamazaki, T.; Isnard, C.; Poirier-Colame, V.; Duong, C.P.M.; Flament, C.; Lepage, P.; Roberti, M.P.; et al. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity 2016, 45, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Koliarakis, I.; Psaroulaki, A.; Nikolouzakis, T.K.; Kokkinakis, M.; Sgantzos, M.N.; Goulielmos, G.; Androutsopoulos, V.P.; Tsatsakis, A.; Tsiaoussis, J. Intestinal microbiota and colorectal cancer: A new aspect of research. JBUON. 2018, 23, 1216–1234. [Google Scholar] [PubMed]
- Hibberd, A.A.; Lyra, A.; Ouwehand, A.C.; Rolny, P.; Lindegren, H.; Cedgard, L.; Wettergren, Y. Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ. Open Gastroenterol. 2017, 4, e000145. [Google Scholar] [CrossRef]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 2014, 15, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Zenobia, C.; Hajishengallis, G. Porphyromonas gingivalis virulence factors involved in subversion of leukocytes and microbial dysbiosis. Virulence 2015, 6, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Arimatsu, K.; Yamada, H.; Miyazawa, H.; Minagawa, T.; Nakajima, M.; Ryder, M.I.; Gotoh, K.; Motooka, D.; Nakamura, S.; Iida, T.; et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci. Rep. 2014, 4, 4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, M.; Arimatsu, K.; Kato, T.; Matsuda, Y.; Minagawa, T.; Takahashi, N.; Ohno, H.; Yamazaki, K. Oral Administration of P. gingivalis Induces Dysbiosis of Gut Microbiota and Impaired Barrier Function Leading to Dissemination of Enterobacteria to the Liver. PLoS ONE 2015, 10, e0134234. [Google Scholar] [CrossRef] [PubMed]
- Palm, N.W.; de Zoete, M.R.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014, 158, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Liang, S.; Payne, M.A.; Hashim, A.; Jotwani, R.; Eskan, M.A.; McIntosh, M.L.; Alsam, A.; Kirkwood, K.L.; Lambris, J.D.; et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 2011, 10, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takahashi, N.; Kato, T.; Matsuda, Y.; Yokoji, M.; Yamada, M.; Nakajima, T.; Kondo, N.; Endo, N.; Yamamoto, R.; et al. Aggravation of collagen-induced arthritis by orally administered Porphyromonas gingivalis through modulation of the gut microbiota and gut immune system. Sci. Rep. 2017, 7, 6955. [Google Scholar] [CrossRef]
- Kato, T.; Yamazaki, K.; Nakajima, M.; Date, Y.; Kikuchi, J.; Hase, K.; Ohno, H.; Yamazaki, K. Oral Administration of Porphyromonas gingivalis Alters the Gut Microbiome and Serum Metabolome. mSphere 2018, 3, e00460-18. [Google Scholar] [CrossRef] [Green Version]
- Ottosson, F.; Brunkwall, L.; Ericson, U.; Nilsson, P.M.; Almgren, P.; Fernandez, C.; Melander, O.; Orho-Melander, M. Connection Between BMI-Related Plasma Metabolite Profile and Gut Microbiota. J. Clin. Endocrinol. Metab. 2018, 103, 1491–1501. [Google Scholar] [CrossRef] [Green Version]
- Konturek, P.C.; Harsch, I.A.; Konturek, K.; Schink, M.; Konturek, T.; Neurath, M.F.; Zopf, Y. Gut(-)Liver Axis: How Do Gut Bacteria Influence the Liver? Med. Sci. 2018, 6, 79. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Merritt, M.E.; Donaldson, J.R. Effect of bile salts on the DNA and membrane integrity of enteric bacteria. J. Med. Microbiol. 2009, 58, 1533–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, M.; Naka, S.; Nakano, K.; Wada, K.; Endo, H.; Mawatari, H.; Imajo, K.; Nomura, R.; Hokamura, K.; Ono, M.; et al. Involvement of a periodontal pathogen, Porphyromonas gingivalis on the pathogenesis of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef] [PubMed]
- Komazaki, R.; Katagiri, S.; Takahashi, H.; Maekawa, S.; Shiba, T.; Takeuchi, Y.; Kitajima, Y.; Ohtsu, A.; Udagawa, S.; Sasaki, N.; et al. Periodontal pathogenic bacteria, Aggregatibacter actinomycetemcomitans affect non-alcoholic fatty liver disease by altering gut microbiota and glucose metabolism. Sci. Rep. 2017, 7, 13950. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Nyman, M.; Fak, F. Modulation of gut microbiota in rats fed high-fat diets by processing whole-grain barley to barley malt. Mol. Nutr. Food Res. 2015, 59, 2066–2076. [Google Scholar] [CrossRef] [PubMed]
- Lourenςo, T.G.B.; Spencer, S.J.; Alm, E.J.; Colombo, A.P.V. Defining the gut microbiota in individuals with periodontal diseases: An exploratory study. J. Oral Microbiol. 2018, 10, 1487741. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Matin, P.; White, M.B.; Fagan, A.; Deeb, J.G.; Acharya, C.; Dalmet, S.S.; Sikaroodi, M.; Gillevet, P.M.; Sahingur, S.E. Periodontal therapy favorably modulates the oral-gut-hepatic axis in cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G824–G837. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Chen, C.Y.; Hayes, R.B. Oral microbiome and oral and gastrointestinal cancer risk. Cancer Causes Control. 2012, 23, 399–404. [Google Scholar] [CrossRef]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef]
- Hale, V.L.; Chen, J.; Johnson, S.; Harrington, S.C.; Yab, T.C.; Smyrk, T.C.; Nelson, H.; Boardman, L.A.; Druliner, B.R.; Levin, T.R.; et al. Shifts in the Fecal Microbiota Associated with Adenomatous Polyps. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chiu, J.; Chen, Y.; Huang, Y.; Higashimori, A.; Fang, J.; Brim, H.; Ashktorab, H.; Ng, S.C.; Ng, S.S.M.; et al. Fecal Bacteria Act as Novel Biomarkers for Noninvasive Diagnosis of Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Warren, R.D.; Barrett, M.P.; Cisek, K.; Das, A.; Jeffery, I.B.; Hurley, E.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. The oral microbiota in colorectal cancer is distinctive and predictive. Gut 2018, 67, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Babic, A.; Tworoger, S.S.; Zhang, L.; Wu, K.; Smith-Warner, S.A.; Ogino, S.; Chan, A.T.; Meyerhardt, J.; Giovannucci, E.; et al. Periodontal disease, tooth loss and colorectal cancer risk: Results from the Nurses’ Health Study. Int. J. Cancer 2017, 140, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Cai, Q.; Shu, X.O.; Steinwandel, M.D.; Blot, W.J.; Zheng, W.; Long, J. Prospective study of oral microbiome and colorectal cancer risk in low-income and African American populations. Int. J. Cancer 2019, 144, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Flanagan, L.; Schmid, J.; Ebert, M.; Soucek, P.; Kunicka, T.; Liska, V.; Bruha, J.; Neary, P.; Dezeeuw, N.; Tommasino, M.; et al. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur. J. Clin. Microbiol. Infect. Dis 2014, 33, 1381–1390. [Google Scholar] [CrossRef]
- Signat, B.; Roques, C.; Poulet, P.; Duffaut, D. Fusobacterium nucleatum in periodontal health and disease. Curr. Issues Mol. Biol. 2011, 13, 25–36. [Google Scholar]
- Ahn, J.; Sinha, R.; Pei, Z.; Dominianni, C.; Wu, J.; Shi, J.; Goedert, J.J.; Hayes, R.B.; Yang, L. Human gut microbiome and risk for colorectal cancer. J. Natl. Cancer Inst. 2013, 105, 1907–1911. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Cao, Y.; Chan, A.T.; Qian, Z.R.; Nowak, J.A.; Masugi, Y.; Shi, Y.; Song, M.; da Silva, A.; Gu, M.; et al. Fusobacterium nucleatum in Colorectal Carcinoma Tissue According to Tumor Location. Clin. Transl. Gastroenterol. 2016, 7, e200. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Yamamoto, E.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.O.; Sugai, T.; An, B.; et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014, 74, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Tran Van Nhieu, J.; Furet, J.P. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kanno, S.; Nosho, K.; Sukawa, Y.; Mitsuhashi, K.; Kurihara, H.; Igarashi, H.; Takahashi, T.; Tachibana, M.; Takahashi, H.; et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int. J. Cancer 2015, 137, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Phipps, A.I.; Chan, A.T.; Ogino, S. Anatomic subsite of primary colorectal cancer and subsequent risk and distribution of second cancers. Cancer 2013, 119, 3140–3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, M.; Morikawa, T.; Kuchiba, A.; Imamura, Y.; Qian, Z.R.; Nishihara, R.; Liao, X.; Waldron, L.; Hoshida, Y.; Huttenhower, C.; et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012, 61, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Suehiro, Y.; Hashimoto, S.; Hoshida, T.; Fujimoto, M.; Watanabe, M.; Imanaga, D.; Sakai, K.; Matsumoto, T.; Nishioka, M.; et al. Fusobacterium nucleatum as a prognostic marker of colorectal cancer in a Japanese population. J. Gastroenterol. 2018, 53, 517–524. [Google Scholar] [CrossRef]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e516. [Google Scholar] [CrossRef]
- Komiya, Y.; Shimomura, Y.; Higurashi, T.; Sugi, Y.; Arimoto, J.; Umezawa, S.; Uchiyama, S.; Matsumoto, M.; Nakajima, A. Patients with colorectal cancer have identical strains of Fusobacterium nucleatum in their colorectal cancer and oral cavity. Gut 2019, 68, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012, 13, R42. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ge, Y.; Cheng, L.; Zeng, B.; Yu, J.; Peng, X.; Zhao, J.; Li, W.; Ren, B.; Li, M.; et al. Oral bacteria colonize and compete with gut microbiota in gnotobiotic mice. Int. J. Oral Sci. 2019, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecher, B.; Hardt, W.D. Mechanisms controlling pathogen colonization of the gut. Curr. Opin. Microbiol. 2011, 14, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Seedorf, H.; Griffin, N.W.; Ridaura, V.K.; Reyes, A.; Cheng, J.; Rey, F.E.; Smith, M.I.; Simon, G.M.; Scheffrahn, R.H.; Woebken, D.; et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 2014, 159, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Suda, W.; Chen, K.; Gevers, D.; Hattori, M.; Honda, K.; Johnson, R.C.; Kiguchi, Y.; Kolls, J.K.; Elinav, E.; et al. Ectopic colonization of oral bacteria in the intestine drives T(H)1 cell induction and inflammation. Science 2017, 358, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.S.; Hayward, M.R.; Coelho, L.P.; Li, S.S.; Costea, P.I.; Voigt, A.Y.; Wirbel, J.; Maistrenko, O.M.; Alves, R.J.; Bergsten, E.; et al. Extensive transmission of microbes along the gastrointestinal tract. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Bohm, J.; Brunetti, F.; Habermann, N.; et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Saygun, I.; Nizam, N.; Keskiner, I.; Bal, V.; Kubar, A.; Acikel, C.; Serdar, M.; Slots, J. Salivary infectious agents and periodontal disease status. J. Periodontal. Res. 2011, 46, 235–239. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Martinsen, T.C.; Bergh, K.; Waldum, H.L. Gastric juice: A barrier against infectious diseases. Basic Clin. Pharmacol. Toxicol. 2005, 96, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.Y.; Pratap, S.; Southerland, J.H.; Farmer-Dixon, C.M.; Lakshmyya, K.; Gangula, P.R. Role of oral and gut microbiome in nitric oxide-mediated colon motility. Nitric. Oxide 2018, 73, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Imhann, F.; Bonder, M.J.; Vich Vila, A.; Fu, J.; Mujagic, Z.; Vork, L.; Tigchelaar, E.F.; Jankipersadsing, S.A.; Cenit, M.C.; Harmsen, H.J.; et al. Proton pump inhibitors affect the gut microbiome. Gut 2016, 65, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Geva-Zatorsky, N.; Sefik, E.; Kua, L.; Pasman, L.; Tan, T.G.; Ortiz-Lopez, A.; Yanortsang, T.B.; Yang, L.; Jupp, R.; Mathis, D.; et al. Mining the Human Gut Microbiota for Immunomodulatory Organisms. Cell 2017, 168, 928–943.e911. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Herrero, E.; Boon, N.; Pauwels, M.; Bernaerts, K.; Slomka, V.; Quirynen, M.; Teughels, W. Necrotrophic growth of periodontopathogens is a novel virulence factor in oral biofilms. Sci. Rep. 2017, 7, 1107. [Google Scholar] [CrossRef] [PubMed]
- Parahitiyawa, N.B.; Jin, L.J.; Leung, W.K.; Yam, W.C.; Samaranayake, L.P. Microbiology of odontogenic bacteremia: Beyond endocarditis. Clin. Microbiol. Rev. 2009, 22, 46–64. [Google Scholar] [CrossRef]
- Hajishengallis, G. Periodontitis. From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host defense against oral microbiota by bone-damaging T cells. Nat. Commun. 2018, 9, 701. [Google Scholar] [CrossRef]
- Carrion, J.; Scisci, E.; Miles, B.; Sabino, G.J.; Zeituni, A.E.; Gu, Y.; Bear, A.; Genco, C.A.; Brown, D.L.; Cutler, C.W. Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. J. Immunol. 2012, 189, 3178–3187. [Google Scholar] [CrossRef]
- Kolenbrander, P.E.; Palmer, R.J., Jr.; Periasamy, S.; Jakubovics, N.S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 2010, 8, 471–480. [Google Scholar] [CrossRef]
- Chenicheri, S.; Usha, R.; Ramachandran, R.; Thomas, V.; Wood, A. Insight into Oral Biofilm: Primary, Secondary and Residual Caries and Phyto-Challenged Solutions. Open Dent. J. 2017, 11, 312–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Konstantinov, S.R.; Smits, R.; Peppelenbosch, M.P. Bacterial Biofilms in Colorectal Cancer Initiation and Progression. Trends Mol. Med. 2017, 23, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.L.; Freeman, D.J.; Pleasance, S.; Watson, P.; Moore, R.A.; Cochrane, K.; Allen-Vercoe, E.; Holt, R.A. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome 2013, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Sears, C.L. Do biofilms confer a pro-carcinogenic state? Gut Microbes 2016, 7, 54–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes 2017, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.; Hansson, G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Sicard, J.F.; Le Bihan, G.; Vogeleer, P.; Jacques, M.; Harel, J. Interactions of Intestinal Bacteria with Components of the Intestinal Mucus. Front. Cell Infect. Microbiol. 2017, 7, 387. [Google Scholar] [CrossRef]
- Jorth, P.; Turner, K.H.; Gumus, P.; Nizam, N.; Buduneli, N.; Whiteley, M. Metatranscriptomics of the human oral microbiome during health and disease. MBio 2014, 5, e01012-14. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 2012, 27, 409–419. [Google Scholar] [CrossRef]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Amitay, E.L.; Werner, S.; Vital, M.; Pieper, D.H.; Hofler, D.; Gierse, I.J.; Butt, J.; Balavarca, Y.; Cuk, K.; Brenner, H. Fusobacterium and colorectal cancer: Causal factor or passenger? Results from a large colorectal cancer screening study. Carcinogenesis 2017, 38, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Tomkovich, S.; Dejea, C.M.; Winglee, K.; Drewes, J.L.; Chung, L.; Housseau, F.; Pope, J.L.; Gauthier, J.; Sun, X.; Muhlbauer, M.; et al. Human colon mucosal biofilms from healthy or colon cancer hosts are carcinogenic. J. Clin. Investig. 2019, 130, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Tomkovich, S.; Yang, Y.; Winglee, K.; Gauthier, J.; Muhlbauer, M.; Sun, X.; Mohamadzadeh, M.; Liu, X.; Martin, P.; Wang, G.P.; et al. Locoregional Effects of Microbiota in a Preclinical Model of Colon Carcinogenesis. Cancer Res. 2017, 77, 2620–2632. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N. Microbial ecosystem in the oral cavity: Metabolic diversity in an ecological niche and its relationship with oral diseases. ICS 2005, 1284, 103–112. [Google Scholar] [CrossRef]
- Milella, L. The Negative Effects of Volatile Sulphur Compounds. J. Vet. Dent. 2015, 32, 99–102. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Eley, B.M.; Cox, S.W. Proteolytic and hydrolytic enzymes from putative periodontal pathogens: Characterization, molecular genetics, effects on host defenses and tissues and detection in gingival crevice fluid. Periodontology 2000 2003, 31, 105–124. [Google Scholar] [CrossRef]
- Linden, S.K.; Florin, T.H.; McGuckin, M.A. Mucin dynamics in intestinal bacterial infection. PLoS ONE 2008, 3, e3952. [Google Scholar] [CrossRef]
- Potempa, J.; Sroka, A.; Imamura, T.; Travis, J. Gingipains, the major cysteine proteinases and virulence factors of Porphyromonas gingivalis: Structure, function and assembly of multidomain protein complexes. Curr. Protein Pept. Sci. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Kilian, M.; Reinholdt, J.; Nyvad, B.; Frandsen, E.V.; Mikkelsen, L. IgA1 proteases of oral streptococci: Ecological aspects. Immunol. Invest. 1989, 18, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Mariggio, M.A.; Vinella, A.; Pasquetto, N.; Curci, E.; Cassano, A.; Fumarulo, R. In vitro effects of polyamines on polymorphonuclear cell apoptosis and implications in the pathogenesis of periodontal disease. Immunopharmacol. Immunotoxicol. 2004, 26, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.N.; Wortham, B.W.; Lines, J.L.; Fetherston, J.D.; Perry, R.D.; Oliveira, M.A. Polyamines are essential for the formation of plague biofilm. J. Bacteriol 2006, 188, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Attene-Ramos, M.S.; Wagner, E.D.; Plewa, M.J.; Gaskins, H.R. Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. 2006, 4, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, M.R.; Szabo, C. Hydrogen Sulfide and Cancer. Handb. Exp. Pharmacol. 2015, 230, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, S.I.; Jin, L.; Gasparovich, S.R.; Tao, L. Multiple alcohol dehydrogenases but no functional acetaldehyde dehydrogenase causing excessive acetaldehyde production from ethanol by oral streptococci. Microbiology 2013, 159, 1437–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meurman, J.H.; Uittamo, J. Oral micro-organisms in the etiology of cancer. Acta. Odontol. Scand. 2008, 66, 321–326. [Google Scholar] [CrossRef]
- Muto, M.; Hitomi, Y.; Ohtsu, A.; Shimada, H.; Kashiwase, Y.; Sasaki, H.; Yoshida, S.; Esumi, H. Acetaldehyde production by non-pathogenic Neisseria in human oral microflora: Implications for carcinogenesis in upper aerodigestive tract. Int. J. Cancer 2000, 88, 342–350. [Google Scholar] [CrossRef]
- Yang, L.; Ganly, I.; Morris, L.; Palmer, F.; Deng, H.; Ahn, J.; Hayes, R.B.; Wang, B.; Pei, Z. Relevance of Microbiome to Cigarette Smoking and Oral Cancer. J. Dent. Res. 2011, 90, 120. [Google Scholar]
- Verna, L.; Whysner, J.; Williams, G.M. N-nitrosodiethylamine mechanistic data and risk assessment: Bioactivation, DNA-adduct formation, mutagenicity, and tumor initiation. Pharmacol. Ther. 1996, 71, 57–81. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Ostaff, M.J.; Stange, E.F.; Wehkamp, J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol. Med. 2013, 5, 1465–1483. [Google Scholar] [CrossRef]
- Rescigno, T.; Micolucci, L.; Tecce, M.F.; Capasso, A. Bioactive Nutrients and Nutrigenomics in Age-Related Diseases. Molecules 2017, 22, 105. [Google Scholar] [CrossRef]
- Mehta, R.S.; Nishihara, R.; Cao, Y.; Song, M.; Mima, K.; Qian, Z.R.; Nowak, J.A.; Kosumi, K.; Hamada, T.; Masugi, Y.; et al. Association of Dietary Patterns with Risk of Colorectal Cancer Subtypes Classified by Fusobacterium nucleatum in Tumor Tissue. JAMA Oncol. 2017, 3, 921–927. [Google Scholar] [CrossRef]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, S.E.; Lamont, R.J. Oral bacteria and cancer. PLoS Pathog. 2014, 10, e1003933. [Google Scholar] [CrossRef] [PubMed]
- Coppenhagen-Glazer, S.; Sol, A.; Abed, J.; Naor, R.; Zhang, X.; Han, Y.W.; Bachrach, G. Fap2 of Fusobacterium nucleatum is a galactose-inhibitable adhesin involved in coaggregation, cell adhesion, and preterm birth. Infect. Immun. 2015, 83, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Abed, J.; Emgard, J.E.; Zamir, G.; Faroja, M.; Almogy, G.; Grenov, A.; Sol, A.; Naor, R.; Pikarsky, E.; Atlan, K.A.; et al. Fap2 Mediates Fusobacterium nucleatum Colorectal Adenocarcinoma Enrichment by Binding to Tumor-Expressed Gal-GalNAc. Cell Host Microbe 2016, 20, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.W.; Ikegami, A.; Rajanna, C.; Kawsar, H.I.; Zhou, Y.; Li, M.; Sojar, H.T.; Genco, R.J.; Kuramitsu, H.K.; Deng, C.X. Identification and characterization of a novel adhesin unique to oral fusobacteria. J. Bacteriol. 2005, 187, 5330–5340. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, O.; Yao, L.; Maeda, K.; Rose, T.M.; Lewis, E.L.; Duman, M.; Lamont, R.J.; Ojcius, D.M. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cell Microbiol. 2008, 10, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, S.; Park, Y.; Hasegawa, Y.; Tribble, G.D.; James, C.E.; Handfield, M.; Stavropoulos, M.F.; Yilmaz, O.; Lamont, R.J. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 2007, 9, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Jermanus, C.; Barbetta, B.; Choi, C.; Verbeke, P.; Ojcius, D.M.; Yilmaz, O. Porphyromonas gingivalis infection sequesters pro-apoptotic Bad through Akt in primary gingival epithelial cells. Mol. Oral Microbiol. 2010, 25, 89–101. [Google Scholar] [CrossRef]
- Nakhjiri, S.F.; Park, Y.; Yilmaz, O.; Chung, W.O.; Watanabe, K.; El-Sabaeny, A.; Park, K.; Lamont, R.J. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol. Lett. 2001, 200, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Sugita, H.; Kuboniwa, M.; Iwai, S.; Hamada, M.; Noda, T.; Morisaki, I.; Lamont, R.J.; Amano, A. Porphyromonas gingivalis promotes invasion of oral squamous cell carcinoma through induction of proMMP9 and its activation. Cell Microbiol. 2014, 16, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Levy, M.; Korem, T.; Dohnalova, L.; Shapiro, H.; Jaitin, D.A.; David, E.; Winter, D.R.; Gury-BenAri, M.; Tatirovsky, E.; et al. Microbiota Diurnal Rhythmicity Programs Host Transcriptome Oscillations. Cell 2016, 167, 1495–1510.e1412. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Iliev, I.D. The mycobiota: Interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 2014, 14, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Santaolalla, R.; Sussman, D.A.; Ruiz, J.R.; Davies, J.M.; Pastorini, C.; Espana, C.L.; Sotolongo, J.; Burlingame, O.; Bejarano, P.A.; Philip, S.; et al. TLR4 activates the beta-catenin pathway to cause intestinal neoplasia. PLoS ONE 2013, 8, e63298. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Peng, Y.; Yu, J.; Chen, T.; Wu, Y.; Shi, L.; Li, Q.; Wu, J.; Fu, X. Invasive Fusobacterium nucleatum activates beta-catenin signaling in colorectal cancer via a TLR4/P-PAK1 cascade. Oncotarget 2017, 8, 31802–31814. [Google Scholar] [CrossRef]
- Dharmani, P.; Strauss, J.; Ambrose, C.; Allen-Vercoe, E.; Chadee, K. Fusobacterium nucleatum infection of colonic cells stimulates MUC2 mucin and tumor necrosis factor alpha. Infect. Immun. 2011, 79, 2597–2607. [Google Scholar] [CrossRef]
- Ye, X.; Wang, R.; Bhattacharya, R.; Boulbes, D.R.; Fan, F.; Xia, L.; Adoni, H.; Ajami, N.J.; Wong, M.C.; Smith, D.P.; et al. Fusobacterium Nucleatum Subspecies Animalis Influences Proinflammatory Cytokine Expression and Monocyte Activation in Human Colorectal Tumors. Cancer Prev. Res. (Phila.) 2017, 10, 398–409. [Google Scholar] [CrossRef]
- Park, H.E.; Kim, J.H.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Intratumoral Fusobacterium nucleatum abundance correlates with macrophage infiltration and CDKN2A methylation in microsatellite-unstable colorectal carcinoma. Virchows. Arch. 2017, 471, 329–336. [Google Scholar] [CrossRef]
- Mima, K.; Sukawa, Y.; Nishihara, R.; Qian, Z.R.; Yamauchi, M.; Inamura, K.; Kim, S.A.; Masuda, A.; Nowak, J.A.; Nosho, K.; et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol. 2015, 1, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Mahlaoui, N.; Aguilar, C.; Bach, P.; Join-Lambert, O.; Garraffo, A.; Seksik, P.; Danion, F.; Jegou, S.; Straube, M.; et al. Intestinal dysbiosis in inflammatory bowel disease associated with primary immunodeficiency. J. Allergy Clin. Immunol. 2019, 143, 775–778.e776. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygard, S.; Vesterhus, M.; Hoivik, M.L.; Troseid, M.; Marschall, H.U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Spooner, R.; Yilmaz, O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int. J. Mol. Sci. 2011, 12, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, O. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol 2013, 15, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Szkaradkiewicz, A.K.; KarpiĹski, T.M. Microbiology of chronic periodontitis. J. Biol. Earth Sci. 2013, 3, 14–20. [Google Scholar]
- Ivanov, K.; Kolev, N.; Tonev, A.; Nikolova, G.; Krasnaliev, I.; Softova, E.; Tonchev, A. Comparative analysis of prognostic significance of molecular markers of apoptosis with clinical stage and tumor differentiation in patients with colorectal cancer: A single institute experience. Hepatogastroenterology 2009, 56, 94–98. [Google Scholar]
- Guarner, F. Enteric flora in health and disease. Digestion 2006, 73, 5–12. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Oral Bacteria. | Sampling/Size | Methods | Main Findings | References |
|---|---|---|---|---|
| Fusobacterium, Gemella, Peptostreptococcus and Parvimonas | —Colonic mucosa/ control (n = 61), colonic adenoma-normal adjacent pair (n = 47), tumor tissue-normal adjacent pair (n = 52) | 16S rRNA gene sequencing | —Increased abundance of presented bacteria in CRC —Mucosal microbiota demonstrates distinct changes across stages of CRC tumorigenesis. | Nakatsu et al. 2015 [80] |
| Actinomyces, Corynebacterium, Haemophilus, Mogibacterium, and Porphyromonas | —Feces/colonic adenoma patients (n = 233), control (n = 547) | 16S rRNA gene sequencing | —Increased abundance of presented bacteria in colonic adenomas | Hale et al. 2017 [81] |
| Fusobacterium, Oscillibacter, Peptostreptococcus, Porphyromonas, Roseburia, and Ruminococcus | —Colonic mucosa/tumor tissue (n = 59), colonic adenoma (n = 21), control (n = 56) | 16S rRNA gene sequencing, real-time qPCR | —Increased abundance of presented bacteria in CRC | Flemer et al. 2017 [45] |
| Fusobacterium nucleatum, Peptostreptococcus stomatitis, and Parvimonas micra | —Oral swabs, feces, colonic mucosa/CRC patients (n = 99), colonic adenoma patients (n = 32), Controls (n = 103) | 16S rRNA gene sequencing | —Increased abundance of presented bacteria in CRC —Oral microbiota is distinctive and predictive in CRC | Flemer et al. 2018 [83] |
| Treponema denticola, Bifidobacteriaceae, and Prevotella Carnobacteriaceae, Erysipelotrichaceae, Prevotella melaninogenica, Streptococcus, and Solobacterium | —Mouth rinse/CRC patients (n = 231), Control (n = 462) | 16S rRNA gene sequencing | —The former group of bacteria was associated with increased risk of CRC —The latter group of bacteria was associated with reduced risk of CRC | Yang et al. 2018 [86] |
| Fusobacterium spp. (F. nucleatum, F. mortiferum, and F. necrophorum) | —Colonic mucosa/tumor tissue-normal adjacent pair (n = 95) | qPCR, 16S rRNA gene sequencing, FISH | —Bacteria belonging to Fusobacterium were abundant in CRC | Kostic et al. 2012 [87] |
| Fusobacterium (F. nucleatum) | —Colonic mucosa/ tumor tissue-matched normal tissue (n = 99) | qPCR, 16S rRNA gene sequencing | —Increased abundance of Fusobacterium in CRC was positively associated with lymph node metastasis | Castellarin et al. 2012 [49] |
| Fusobacterium spp., Porphyromonas spp. | —Feces/CRC patients (n = 47), control (n = 94) | 16S rRNA gene sequencing | —Increased abundance of presented bacteria in CRC patients —Decreased abundance of Clostridium spp. was simultaneously detected | Ahn et al. 2013 [90] |
| Fusobacterium nucleatum | —Colonic mucosa/tumor tissue-matched normal tissue (n = 122), colonic adenoma-matched normal tissue (n = 52) —Feces/CRC patients (n = 7), colonic adenoma patients (n = 24), controls (n = 25) | qPCR | —Patients with high levels of F. nucleatum presented a significantly shorter survival time that patients with low levels of this species | Flanagan et al. 2014 [88] |
| Fusobacterium spp. | Colonic mucosa/tumor tissue (n = 149), normal adjacent tissue (n = 89), control (n = 72) | qPCR | —Fusobacterium enhancement is associated with specific molecular subsets of CRC | Tahara et al. 2014 [93] |
| Fusobacterium spp. and Lactococcus spp. | —Colonic mucosa/ tumor tissue (n = 31), normal adjacent tissue (n = 20) | 16S rRNA gene sequencing | —Increased abundance of presented bacteria in CRC —Pseudomonas and Escherichia-Shigella were decreased | Gao et al. 2015 [94] |
| Fusobacterium nucleatum | —Colonic mucosa/tumor tissue (n = 1102) | qPCR | —Increased abundance of this species in proximal CRC | Mima et al. 2016 [92] |
| Fusobacterium nucleatum | —Colonic mucosa/tumor tissue (n = 100), normal tissue (n = 72) | Droplet digital PCR | —Overabundance of this species correlated with KRAS mutation, tumor size, and shorter survival time | Yamaoka et al. 2018 [99] |
| Fusobacterium nucleatum | —Colonic mucosa/tumor tissue (n = 296) | HT RNA sequencing, real time qPCR | —Fusobacterium nucleatum promotes chemoresistance through modulation of autophagy in CRC | Yu et al. 2017 [100] |
| Fusobacterium nucleatum | —Colonic mucosa, saliva/CRC patients (n = 14) | AP-PCR, 16S rRNA gene sequencing | —Similar strains of Fusobacterium nucleatum are presented between oral cavity and colon in CRC patients | Komiya et al. 2019 [101] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koliarakis, I.; Messaritakis, I.; Nikolouzakis, T.K.; Hamilos, G.; Souglakos, J.; Tsiaoussis, J. Oral Bacteria and Intestinal Dysbiosis in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 4146. https://doi.org/10.3390/ijms20174146
Koliarakis I, Messaritakis I, Nikolouzakis TK, Hamilos G, Souglakos J, Tsiaoussis J. Oral Bacteria and Intestinal Dysbiosis in Colorectal Cancer. International Journal of Molecular Sciences. 2019; 20(17):4146. https://doi.org/10.3390/ijms20174146
Chicago/Turabian StyleKoliarakis, Ioannis, Ippokratis Messaritakis, Taxiarchis Konstantinos Nikolouzakis, George Hamilos, John Souglakos, and John Tsiaoussis. 2019. "Oral Bacteria and Intestinal Dysbiosis in Colorectal Cancer" International Journal of Molecular Sciences 20, no. 17: 4146. https://doi.org/10.3390/ijms20174146
APA StyleKoliarakis, I., Messaritakis, I., Nikolouzakis, T. K., Hamilos, G., Souglakos, J., & Tsiaoussis, J. (2019). Oral Bacteria and Intestinal Dysbiosis in Colorectal Cancer. International Journal of Molecular Sciences, 20(17), 4146. https://doi.org/10.3390/ijms20174146