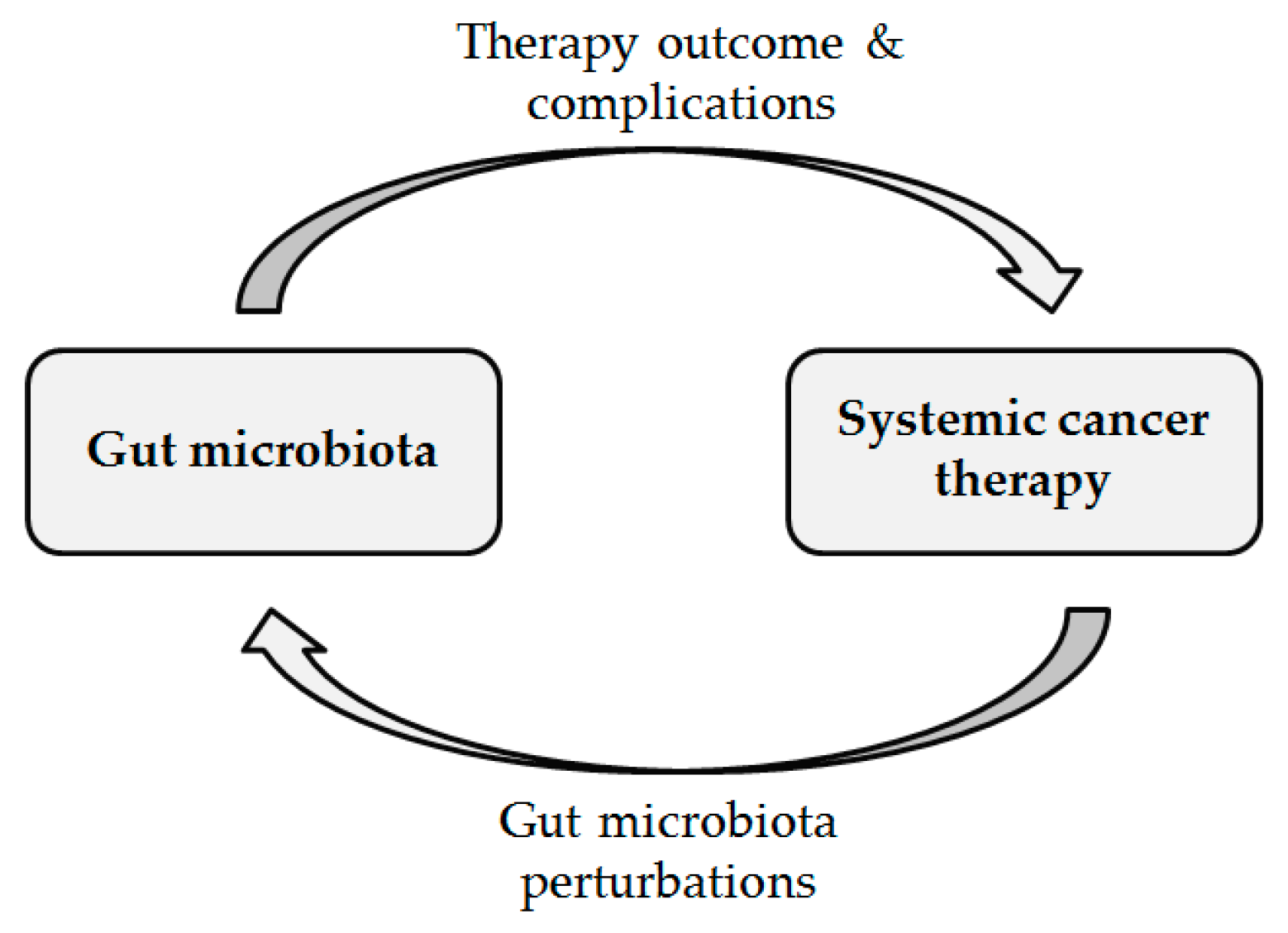
The Clinical Link between Human Intestinal Microbiota and Systemic Cancer Therapy

 , and
, and
Abstract
:1. Introduction
2. Baseline Human Intestinal Microbiota Characteristics Are Associated with the Development of Complications and Systemic Cancer Therapy Outcome
2.1. Chemotherapy
2.2. Immunotherapy
2.3. Hormonal Therapy
3. Human Intestinal Microbiota Changes during Systemic Cancer Therapy
3.1. Chemotherapy
3.2. Immunotherapy
3.3. Hormonal Therapy
4. Discussion
4.1. Baseline Human Intestinal Microbiota Is Associated with the Development of Complications and Systemic Cancer Therapy Outcome
4.2. Human Intestinal Microbiota Changes during Systemic Cancer Therapy
4.3. Strengths and Limitations
5. Materials and Methods
5.1. Review Questions
- Baseline human intestinal microbiota was associated with systemic cancer therapy outcome
- Human intestinal microbiota changed during systemic cancer therapy
5.2. Review Search
5.3. Eligibility Criteria
- Types of participants: human participants with any type of cancer.
- Types of interventions: systemic cancer therapy with chemotherapy, immunotherapy, or hormone therapy.
- Types of comparators: studies comparing baseline and or follow up intestinal microbiota composition in patients starting and/or receiving systemic cancer therapy with either healthy controls, no intervention, follow up samples, and/or therapy outcomes.
- Types of outcome measures: intestinal microbiota associated therapy outcomes and intestinal microbiota composition changes analyzed with any type of detection method.
- Types of study design: observational studies or intervention studies with a control and/or placebo group.
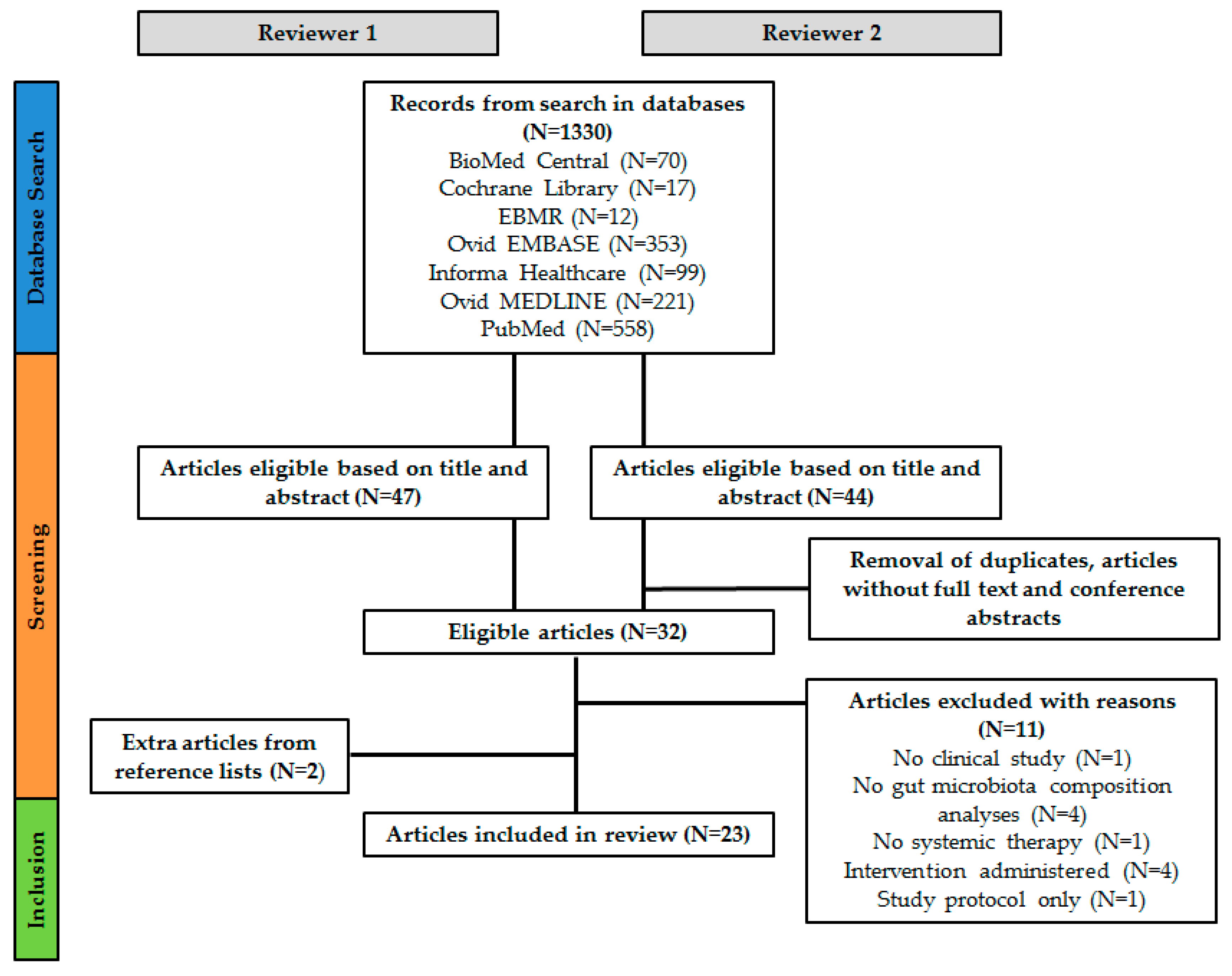
5.4. Study Selection
5.5. Data Collection Process
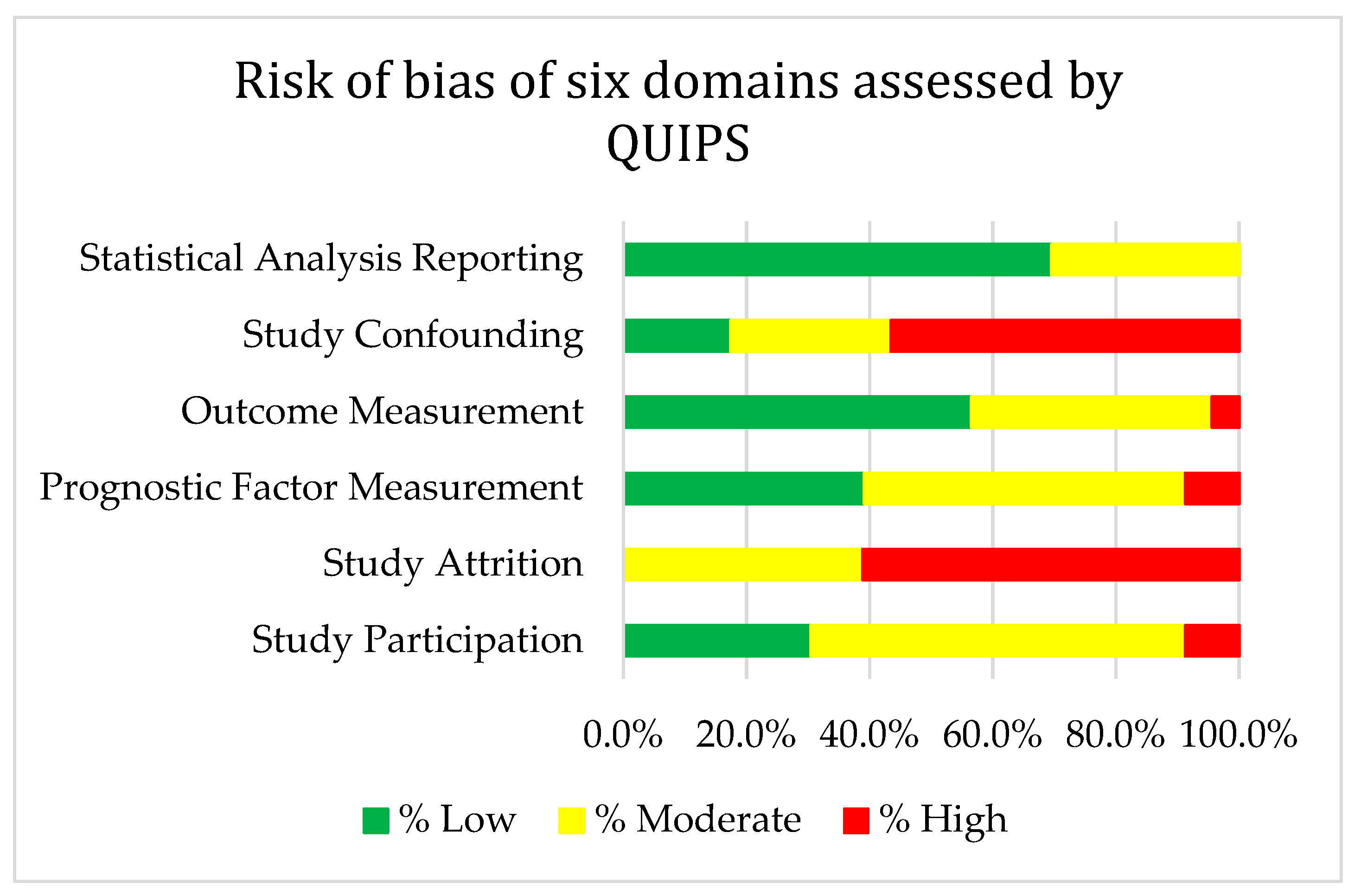
5.6. Risk of Bias Assessment
6. Future Directions
- The predictive ability of pre-treatment intestinal microbiota concerning development of complications and response to cancer treatment.
- The potential use of microbiota-modulating strategies in order to improve cancer therapy outcome.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| AML | Acute myeloid leukemia |
| ALL | Acute lymphoblastic leukemia |
| ATT | Androgen receptor axis-targeted therapies |
| BSI | Bloodstream infections |
| CPIs | Checkpoint inhibitors |
| CRC | Colorectal cancer |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CV | Coefficient of variation |
| dHPLC | Denaturing high-performance liquid chromatography |
| FISH | Fluorescent in situ hybridization |
| FMT | Fecal microbiota transplantation |
| GNRH | Gonadotropin-releasing hormone |
| GPCR | G protein-coupled receptor |
| I | Ipilimumab |
| IN | Ipilimumab + nivolumab |
| LDA | Linear discriminant analysis |
| N | Nivolumab |
| NET | Neuroendocrine tumors |
| NSCLC | Non-small cell lung cancer |
| OTU | Operational taxonomic unit |
| OS | Overall survival |
| P | Pembrolizumab |
| PCA | Principal component analysis |
| PCoA | Principal coordinate analysis |
| PCR-DGGE | Polymerase chain reaction denaturing gradient gel electrophoresis |
| PD-1 | Programmed cell death protein 1 |
| PFS | Progression free survival |
| PICOS | Participants, interventions, comparators, outcome measures, study design |
| qPCR | Quantitative polymerase chain reaction |
| QUIPS | Quality In Prognosis Studies |
| RECIST | Response evaluation criteria in solid tumors |
| RCC | Renal cell carcinoma |
| SCFA | Short-chain fatty acids |
| WMGS | Whole metagenome sequencing |
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Article | Study Participation | Study Attrition | Prognostic Factor Measurement | Outcome Measurement | Study Confounding | Statistical Analysis and Reporting |
|---|---|---|---|---|---|---|
| Deng (2018), [36] | NA | |||||
| Gopalakrishnan (2018), [25] | NA | |||||
| Matson (2018), [24] | NA | |||||
| Routy (2018), [26] | ||||||
| Sfanos (2018), [42] | NA | |||||
| Youssef (2018), [35] | NA | |||||
| Chaput (2017), [27] | ||||||
| Frankel (2017), [28] | ||||||
| Galloway Pena (2017), [21] | ||||||
| Sze (2017), [34] | ||||||
| Dubin (2016), [29] | ||||||
| Rajagopala (2016), [15] | ||||||
| Galloway-Pena (2016), [20] | ||||||
| Montassier (2016), [22] | ||||||
| Vetizou (2015), [41] | ||||||
| Montassier (2015), [33] | ||||||
| Pal (2015), [23] | ||||||
| Montassier (2014), [6] | ||||||
| Stringer (2013), [37] | ||||||
| Zwielehner (2011), [32] | ||||||
| Dörffel (2011), [31] | ||||||
| Wada (2010), [38] | ||||||
| Van Vliet (2009), [39] |


Appendix B
| α-diversity | Number and evenness of distribution of taxa within a given sample |
| β-diversity | The difference in diversity of taxa from one sample to another, i.e., the number of taxa that are not the same (or not similarly distributed) in two different samples. |
| 16S rRNA gene | Marker gene for bacterial identification, containing evolutionary conserved universal as well as variable regions |
| Operational taxonomic unit (OTU) | Cluster of nearly-identical sequences (e.g., 97% similarity), often used in microbiota research instead of ‘species’ |
| 16S rRNA gene sequencing | Sequencing of the 16S rRNA marker gene |
| Metagenomic sequencing | Sequencing of the entire metagenome (all the genetic material in a sample), also allowing analysis of the functional capacity of the microbiome |
References
- Savage, D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepage, P.; Hasler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Dore, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Verdam, F.J.; Fuentes, S.; de Jonge, C.; Zoetendal, E.G.; Erbil, R.; Greve, J.W.; Buurman, W.A.; de Vos, W.M.; Rensen, S.S. Human intestinal microbiota composition is associated with local and systemic inflammation in obesity. Obesity 2013, 21, E607–E615. [Google Scholar] [CrossRef] [PubMed]
- Noce, A.; Marrone, G.; Di Daniele, F.; Ottaviani, E.; Wilson Jones, G.; Bernini, R.; Romani, A.; Rovella, V. Impact of Gut Microbiota Composition on Onset and Progression of Chronic Non-Communicable Diseases. Nutrients 2019, 11, 1073. [Google Scholar] [CrossRef] [PubMed]
- Montassier, E.; Batard, E.; Massart, S.; Gastinne, T.; Carton, T.; Caillon, J.; Le Fresne, S.; Caroff, N.; Hardouin, J.B.; Moreau, P.; et al. 16S rRNA gene pyrosequencing reveals shift in patient faecal microbiota during high-dose chemotherapy as conditioning regimen for bone marrow transplantation. Microb. Ecol. 2014, 67, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Huttenhower, C. Chapter 12: Human microbiome analysis. PLoS Comput. Biol. 2012, 8, e1002808. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The Intestinal Microbiome and Estrogen Receptor-Positive Female Breast Cancer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Liao, M.; Yao, Z.; Liang, W.; Li, Q.; Liu, J.; Yang, H.; Ji, Y.; Wei, W.; Tan, A.; et al. Breast cancer in postmenopausal women is associated with an altered gut metagenome. Microbiome 2018, 6, 136. [Google Scholar] [CrossRef] [PubMed]
- Rajagopala, S.V.; Yooseph, S.; Harkins, D.M.; Moncera, K.J.; Zabokrtsky, K.B.; Torralba, M.G.; Tovchigrechko, A.; Highlander, S.K.; Pieper, R.; Sender, L.; et al. Gastrointestinal microbial populations can distinguish pediatric and adolescent Acute Lymphoblastic Leukemia (ALL) at the time of disease diagnosis. BMC Genom. 2016, 17, 635. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Ha, E.M. Combination Therapy of Lactobacillus plantarum Supernatant and 5-Fluouracil Increases Chemosensitivity in Colorectal Cancer Cells. J. Microbiol. Biotechnol. 2016, 26, 1490–1503. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillere, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef]
- Florez, A.B.; Sierra, M.; Ruas-Madiedo, P.; Mayo, B. Susceptibility of lactic acid bacteria, bifidobacteria and other bacteria of intestinal origin to chemotherapeutic agents. Int. J. Antimicrob. Agents 2016, 48, 547–550. [Google Scholar] [CrossRef] [Green Version]
- Attman, E.; Aittoniemi, J.; Sinisalo, M.; Vuento, R.; Lyytikainen, O.; Karki, T.; Syrjanen, J.; Huttunen, R. Etiology, clinical course and outcome of healthcare-associated bloodstream infections in patients with hematological malignancies: A retrospective study of 350 patients in a Finnish tertiary care hospital. Leuk. Lymphoma 2015, 56, 3370–3377. [Google Scholar] [CrossRef]
- Galloway-Pena, J.R.; Smith, D.P.; Sahasrabhojane, P.; Ajami, N.J.; Wadsworth, W.D.; Daver, N.G.; Chemaly, R.F.; Marsh, L.; Ghantoji, S.S.; Pemmaraju, N.; et al. The role of the gastrointestinal microbiome in infectious complications during induction chemotherapy for acute myeloid leukemia. Cancer 2016, 122, 2186–2196. [Google Scholar] [CrossRef]
- Galloway-Pena, J.R.; Smith, D.P.; Sahasrabhojane, P.; Wadsworth, W.D.; Fellman, B.M.; Ajami, N.J.; Shpall, E.J.; Daver, N.; Guindani, M.; Petrosino, J.F.; et al. Characterization of oral and gut microbiome temporal variability in hospitalized cancer patients. Genome Med. 2017, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montassier, E.; Al-Ghalith, G.A.; Ward, T.; Corvec, S.; Gastinne, T.; Potel, G.; Moreau, P.; de la Cochetiere, M.F.; Batard, E.; Knights, D. Pretreatment gut microbiome predicts chemotherapy-related bloodstream infection. Genome Med. 2016, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Li, S.M.; Wu, X.; Qin, H.; Kortylewski, M.; Hsu, J.; Carmichael, C.; Frankel, P. Stool Bacteriomic Profiling in Patients with Metastatic Renal Cell Carcinoma Receiving Vascular Endothelial Growth Factor-Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2015, 21, 5286–5293. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2017, 28, 1368–1379. [Google Scholar] [CrossRef]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef]
- Dubin, K.; Callahan, M.K.; Ren, B.; Khanin, R.; Viale, A.; Ling, L.; No, D.; Gobourne, A.; Littmann, E.; Huttenhower, C.; et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun. 2016, 7, 10391. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Dörffel, Y.; Swidsinski, A.; Loening-Baucke, V.; Wiedenmann, B.; Pavel, M. Common biostructure of the colonic microbiota in neuroendocrine tumors and Crohn’s disease and the effect of therapy. Inflamm. Bowel Dis. 2012, 18, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Zwielehner, J.; Lassl, C.; Hippe, B.; Pointner, A.; Switzeny, O.J.; Remely, M.; Kitzweger, E.; Ruckser, R.; Haslberger, A.G. Changes in human fecal microbiota due to chemotherapy analyzed by TaqMan-PCR, 454 sequencing and PCR-DGGE fingerprinting. PLoS ONE 2011, 6, e28654. [Google Scholar] [CrossRef] [PubMed]
- Montassier, E.; Gastinne, T.; Vangay, P.; Al-Ghalith, G.A.; Bruley des Varannes, S.; Massart, S.; Moreau, P.; Potel, G.; de La Cochetiere, M.F.; Batard, E.; et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment. Pharmacol. Ther. 2015, 42, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Sze, M.A.; Baxter, N.T.; Ruffin, M.T.T.; Rogers, M.A.M.; Schloss, P.D. Normalization of the microbiota in patients after treatment for colonic lesions. Microbiome 2017, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Youssef, O.; Lahti, L.; Kokkola, A.; Karla, T.; Tikkanen, M.; Ehsan, H.; Carpelan-Holmstrom, M.; Koskensalo, S.; Bohling, T.; Rautelin, H.; et al. Stool Microbiota Composition Differs in Patients with Stomach, Colon, and Rectal Neoplasms. Dig. Dis. Sci. 2018, 63, 2950–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Li, Z.; Li, G.; Li, B.; Jin, X.; Lyu, G. Comparison of Microbiota in Patients Treated by Surgery or Chemotherapy by 16S rRNA Sequencing Reveals Potential Biomarkers for Colorectal Cancer Therapy. Front. Microbiol. 2018, 9, 1607. [Google Scholar] [CrossRef] [PubMed]
- Stringer, A.M.; Al-Dasooqi, N.; Bowen, J.M.; Tan, T.H.; Radzuan, M.; Logan, R.M.; Mayo, B.; Keefe, D.M.; Gibson, R.J. Biomarkers of chemotherapy-induced diarrhoea: A clinical study of intestinal microbiome alterations, inflammation and circulating matrix metalloproteinases. Support. Care Cancer 2013, 21, 1843–1852. [Google Scholar] [CrossRef]
- Wada, M.; Nagata, S.; Saito, M.; Shimizu, T.; Yamashiro, Y.; Matsuki, T.; Asahara, T.; Nomoto, K. Effects of the enteral administration of Bifidobacterium breve on patients undergoing chemotherapy for pediatric malignancies. Support. Care Cancer 2010, 18, 751–759. [Google Scholar] [CrossRef]
- Van Vliet, M.J.; Tissing, W.J.; Dun, C.A.; Meessen, N.E.; Kamps, W.A.; de Bont, E.S.; Harmsen, H.J. Chemotherapy treatment in pediatric patients with acute myeloid leukemia receiving antimicrobial prophylaxis leads to a relative increase of colonization with potentially pathogenic bacteria in the gut. Clin. Infect. Dis. 2009, 49, 262–270. [Google Scholar] [CrossRef]
- Zhang, X.L.; Komada, Y.; Chipeta, J.; Li, Q.S.; Inaba, H.; Azuma, E.; Yamamoto, H.; Sakurai, M. Intracellular cytokine profile of T cells from children with acute lymphoblastic leukemia. Cancer Immunol. Immunother. 2000, 49, 165–172. [Google Scholar] [CrossRef]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sfanos, K.S.; Markowski, M.C.; Peiffer, L.B.; Ernst, S.E.; White, J.R.; Pienta, K.J.; Antonarakis, E.S.; Ross, A.E. Compositional differences in gastrointestinal microbiota in prostate cancer patients treated with androgen axis-targeted therapies. Prostate Cancer Prostatic Dis. 2018, 21, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Zhang, S.; Li, H.; Yang, F.; Mushtaq, N.; Ullah, S.; Shi, Y.; An, C.; Xu, J. The influence of gut microbiota dysbiosis to the efficacy of 5-Fluorouracil treatment on colorectal cancer. Biomed. Pharmacother. 2018, 108, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Buschmann, M.M.; Gilbert, J.A. Pharmacomicrobiomics: The Holy Grail to Variability in Drug Response? Clin. Pharmacol. Ther. 2019, 106, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.L.; Wilson, I.D.; Teare, J.; Marchesi, J.R.; Nicholson, J.K.; Kinross, J.M. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 2019, 570, 462–467. [Google Scholar] [CrossRef]
- Bronckaers, A.; Balzarini, J.; Liekens, S. The cytostatic activity of pyrimidine nucleosides is strongly modulated by Mycoplasma hyorhinis infection: Implications for cancer therapy. Biochem. Pharmacol. 2008, 76, 188–197. [Google Scholar] [CrossRef]
- Wallace, B.D.; Wang, H.; Lane, K.T.; Scott, J.E.; Orans, J.; Koo, J.S.; Venkatesh, M.; Jobin, C.; Yeh, L.A.; Mani, S.; et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 2010, 330, 831–835. [Google Scholar] [CrossRef]
- Roberts, A.B.; Wallace, B.D.; Venkatesh, M.K.; Mani, S.; Redinbo, M.R. Molecular insights into microbial beta-glucuronidase inhibition to abrogate CPT-11 toxicity. Mol. Pharmacol. 2013, 84, 208–217. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef] [PubMed]
- Van der Beek, C.M.; Dejong, C.H.C.; Troost, F.J.; Masclee, A.A.M.; Lenaerts, K. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr. Rev. 2017, 75, 286–305. [Google Scholar] [CrossRef] [PubMed]
- Miko, E.; Kovacs, T.; Sebo, E.; Toth, J.; Csonka, T.; Ujlaki, G.; Sipos, A.; Szabo, J.; Mehes, G.; Bai, P. Microbiome-Microbial Metabolome-Cancer Cell Interactions in Breast Cancer-Familiar, but Unexplored. Cells 2019, 8, 293. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Pérez, O.; Cruz-Ramón, V.; Chinchilla-López, P.; Méndez-Sánchez, N. The role of the gut microbiota in bile acid metabolism. Ann. Hepatol. 2018, 16, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Miko, E.; Vida, A.; Kovacs, T.; Ujlaki, G.; Trencsenyi, G.; Marton, J.; Sari, Z.; Kovacs, P.; Boratko, A.; Hujber, Z.; et al. Lithocholic acid, a bacterial metabolite reduces breast cancer cell proliferation and aggressiveness. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 958–974. [Google Scholar] [CrossRef] [PubMed]
- Colosimo, D.A.; Kohn, J.A.; Luo, P.M.; Piscotta, F.J.; Han, S.M.; Pickard, A.J.; Rao, A.; Cross, J.R.; Cohen, L.J.; Brady, S.F. Mapping Interactions of Microbial Metabolites with Human G-Protein-Coupled Receptors. Cell Host Microbe 2019, 26, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Yu, S.; Qin, S.; Liu, Q.; Xu, H.; Zhao, W.; Chu, Q.; Wu, K. Gut microbiome modulates efficacy of immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 47. [Google Scholar] [CrossRef] [PubMed]
- Asmar, N.; Ibrahim, T.; Rey, J.F. Checkpoint Inhibitors: Conquering Cancer with a Little (T)-Help from Our Microbial Friends. Dig. Dis. Sci. 2018, 63, 2177–2179. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.H.; Yin, X.; Welander, P.V. Sterol Synthesis in Diverse Bacteria. Front. Microbiol. 2016, 7, 990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridlon, J.M.; Ikegawa, S.; Alves, J.M.; Zhou, B.; Kobayashi, A.; Iida, T.; Mitamura, K.; Tanabe, G.; Serrano, M.; De Guzman, A.; et al. Clostridium scindens: A human gut microbe with a high potential to convert glucocorticoids into androgens. J. Lipid Res. 2013, 54, 2437–2449. [Google Scholar] [CrossRef]
- Fuhrman, B.J.; Feigelson, H.S.; Flores, R.; Gail, M.H.; Xu, X.; Ravel, J.; Goedert, J.J. Associations of the fecal microbiome with urinary estrogens and estrogen metabolites in postmenopausal women. J. Clin. Endocrinol. Metab. 2014, 99, 4632–4640. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Velicer, C.M.; Heckbert, S.R.; Lampe, J.W.; Potter, J.D.; Robertson, C.A.; Taplin, S.H. Antibiotic use in relation to the risk of breast cancer. JAMA 2004, 291, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Kirkup, B.; McKee, A.; Makin, K.; Paveley, J.; Caim, S.; Alcon-Giner, C.; Leclaire, C.; Dalby, M.; Le Gall, G.; Andrusaite, A. Perturbation of the gut microbiota by antibiotics results in accelerated breast tumour growth and metabolic dysregulation. BioRxiv 2019, 553602. [Google Scholar] [CrossRef]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.P.T.; Green, S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0. Available online: https://www.handbook.cochrane.org (accessed on 6 May 2019).
- Aarnoutse, R.; de Vos-Geelen, J.; Penders, J.; Boerma, E.G.; Warmerdam, F.; Goorts, B.; Olde Damink, S.W.M.; Soons, Z.; Rensen, S.S.M.; Smidt, M.L. Study protocol on the role of intestinal microbiota in colorectal cancer treatment: A pathway to personalized medicine 2.0. Int. J. Colorectal Dis. 2017, 32, 1077–1084. [Google Scholar] [CrossRef] [PubMed]


| Study Design | Main Findings | |||||
|---|---|---|---|---|---|---|
| Study | Cancer Type | n | Type of Therapy | Analysis Method | Therapy Outcome | Microbial Outcomes Found to Be Different |
| Chemotherapy | ||||||
| Galloway-Peña et al. (2017), [21] | AML | n = 35 | Induction chemotherapy | 16S rRNA gene sequencing | Increased risk for infections | ↑ intra-patient temporal variability of α-diversity (CV of Shannon index) ↑ Stenotrophomonas |
| Galloway-Peña et al. (2016), [20] | AML | n = 34 | Induction chemotherapy | 16S rRNA gene sequencing | Increased risk for infections | ↓ baseline α-diversity (Shannon index) |
| Pal et al. (2015), [23] | Metastatic RCC | n = 20 | VEGF-TKI | 16S rRNA gene sequencing | Increased risk to develop diarrhea | ↑ Bacteroides ↓ Prevotella |
| Immunotherapy | ||||||
| Matson et al. (2018), [24] | Metastatic melanoma | n = 42 | Anti-PD-1 (n = 38) Anti-CTLA-4 (n = 4) | 16S rRNA gene sequencing Metagenomic shotgun sequencing qPCR | Response (n = 16) | ↑ Bifidobacteriaceae ↑ Enterococcus faecium ↑ Collinsella aerofaciens ↑ Bifidobacterium adolescentis ↑ Klebsiella pneumoniae ↑ Veillonella parvula ↑ Parabacteroides merdae ↑ Lactobacillus sp. ↑ Bifidobacterium longum ↓ Ruminococcus obeum ↓ Roseburia intestinalis |
| Gopalakrishnan et al. (2018), [25] | Metastatic melanoma | n = 43 | Anti-PD-1 | 16S rRNA gene sequencing | Response (n = 30) | ↑ α-diversity (inverse Simpson score) ↑ between-group β-diversity ↑ Clostridiales ↑ Ruminococcaceae ↑ Faecalibacterium ↓ Bacteroidales |
| n = 25 | Anti-PD-1 | Metagenomic whole-genome shotgun sequencing | Response (n = 14) | ↑ Faecalibacterium sp. ↑ Clostridium sp. ↑ Clostridiales ↑ Eubacterium sp. ↑ Oscillibacter sp. ↑ Ruminococcaceae ↓ Bacteroides thetaiotaomicron ↓ Escherichia coli ↓ Oxalobacter formigenes ↓ Anaerotruncus colihominis ↓ Klebsiella variicola | ||
| n = 39 | Anti-PD-1 | Metagenomic whole-genome shotgun sequencing | Prolonged PFS (n = 19) | ↑ Faecalibacterium ↓ Bacteroidales | ||
| Routy et al. (2018), [26] | NSCLC (n = 60) RCC (n = 40) | n = 100 | Anti-PD-1 | Metagenomic shotgun sequencing | Response | ↑ α-diversity (richness) ↑ Akkermansia muciniphila ↑ Firmicutes and 4x unclassified ↑ Eubacterium sp. ↑ Lachnospiraceae ↑ Erysipelotrichaceae bacterium ↑ Cloacibacillus porcorum ↑ Enterococcus faecium ↑ Intestinimonas ↑ 2x unclassified Clostridialis ↑ Alistipes ↑ Bacteroides sp. ↑ Alistipes indistinctus ↑ Firmicutes bacterium ↑ Prevotella ↓ Prevotella ↓ Clostridium sp. ↓ unclassified Firmicutes ↓ Prevotella sp. ↓ Clostridiales ↓ Clostridium bolteae ↓ Firmicutes bacterium ↓Closteridiales bacterium ↓ Blautia ↓ Bacteroides clanus ↓ Proteobacteria ↓ Bacteroides nordii ↓ Parabacteroides distasonis |
| NSCLC + RCC | n = 78 | Anti-PD-1 | Metagenomic shotgun sequencing | PFS > 3 months | ↑ unclassified Firmicutes 6x ↑ Eubacterium sp. ↑ Alistipes 2x ↑ Akkermansia muciniphila ↑ Intestinimonas ↑ Bacteroides nordii ↑ Bacteroides xylanisolvens ↑ Blautia ↑ Lachnospiraceae ↑ Firmicutes bacterium ↑ Firmicutes ↑ unclassified Clostridiales 2x ↑ Clostridialis ↑ Ruminococcaceae ↑ Clostridium sp. ↑ Flavonifractor ↑ Bacteroides caccae ↑ unclassified Ruminococcaceae ↑ Ruminococcus sp. ↓ unclassified Clostridialis ↓ Parabacteroides distasonis ↓ Firmicutes bacterium ↓ Clostridiales ↓ Clostridialis VE202-14 ↓ Anaerotruncus colihominis ↓ Lachnospiraceae ↓ Erysipelotrichaceae | |
| Chaput et al. (2017), [27] | Metastatic melanoma | n = 26 | Ipilimumab | 16S rRNA gene sequencing | Colitis and good response | ↑ Firmicutes ↓ Bacteroidetes |
| ↑ PFS ↑ OS ↑ % clinical benefit | ↑ Faecalibacterium ↑ Firmicutes ↑ unclassified Ruminococcaceae ↑ Clostridium XIVa ↑ Blautia | |||||
| Frankel et al. (2017), [28] | Metastatic/ unresectable melanoma | n = 39 | Ipilimumab, Nivolumab Ipilimumab + Nivolumab Pembrolizumab | Metagenomic shotgun sequencing | Response (n = 24) | ↑ Bacteroides caccae ↑ Streptococcus parasanguinis |
| n = 24 | Ipilimumab + Nivolumab | Metagenomic shotgun sequencing | Response (n = 16) | ↑ Faecalibacterium prausnitzii ↑ Holdemania filiformis ↑ Bacteroides thetaiotamicron | ||
| n = 13 | Pembrolizumab | Metagenomic shotgun sequencing | Response (n = 6) | ↑ Dorea formicigenerans | ||
| Dubin et al. (2016), [29] | Metastatic Melanoma | n = 34 | Ipilimumab | 16S rRNA gene sequencing | Colitis free | ↑ Bacteroidaceae ↑ Bacteroides ↑ Barnesiellaceae ↑ Barnesiellaceae unclassified ↑ Rikenellaceae ↑ Rikenellaceae unclassified ↑ Bacteroidetes ↑ Bacteroidia ↑ Bacteroidales ↑ Bacteroidetes |
| Other | ||||||
| Montassier et al. (2016), [22] | Non-Hodgkin lymphoma | n = 28 | HSCT | 16S rRNA high-throughput DNA sequencing | Increased risk to develop bloodstream infections | ↑ Erysipelotrichaceae ↑ Veillonella ↓ α-diversity (phylogenetic diversity, observed species, Chao1 & Shannon indices) ↓ Barnesiellaceae ↑ Butyricimonas ↓ Christensenellaceae ↓ Faecalibacterium ↓ Oscillospira ↓ Christensenella ↓ Dehalobacterium ↓ Desulfovibrio ↓ Sutterella ↓ Oxalobacter ↓ Coriobacteriaceae |
| Study Design | Main Findings | |||||
|---|---|---|---|---|---|---|
| Study | Type of Cancer | n | Type of Therapy | Sampling Time Points | Method Used for Microbiota Analysis | Effects of Therapy on Microbiota |
| Chemotherapy | ||||||
| Galloway-Peña et al. (2017), [21] | AML | n = 35 | Induction chemotherapy | Baseline: before or within first 24h of chemotherapy; Follow-up: every 96h until neutrophil recovery | 16S rRNA gene sequencing | ↑ intra-patient temporal variability of α-diversity (CV of Shannon) ↑ Staphylococcus ↑ Streptococcus ↑ Akkermansia ↑ Subdilogranulum ↑ Pseudobutyrivibrio |
| Sze et al. (2017), [34] | CRC | n = 26 | 12 surgery 9 surgery + chemotherapy 5 surgery + chemotherapy + radiation | Before and after treatment | 16S rRNA gene sequencing | Change in community structure Shift towards healthy microbiota |
| Galloway-Peña et al. (2016), [20] | AML | n = 34 | Induction chemotherapy | Baseline: before therapy; Follow-up: every 96 h until neutrophil recovery | 16S rRNA gene sequencing | ↑ Lactobacillus ↓ α-diversity (Shannon index) ↓ Blautia |
| Rajagopala et al. (2016), [15] | ALL | n = 28 | Chemotherapy | (1) Before therapy, (2) during induction chemotherapy (3) during consolidation chemotherapy (4) during maintenance chemotherapy | 16S rRNA gene sequencing | ↑ α-diversity (Shannon index) |
| Montassier et al. (2015), [33] | Non-Hodgkin’s lymphoma | n = 28 | Chemotherapy | Baseline: before chemotherapy; Follow-up: 7 days later | 16S rRNA gene sequencing | ↑ Proteobacteria ↑ Citrobacter ↑ Klebsiella ↑ Enterococcus ↑ Megasphaera ↑ Parabacreroides ↓ α-diversity (Faith’s phylogenetic diversity, observed species) ↓ Firmicutes ↓ Actinobacteria ↓ Ruminococcus ↓ Oscillospira ↓ Blautia ↓ Lachnospira ↓ Roseburia ↓ Dorea ↓ Coprococcus ↓ Anaerostipes ↓ Clostridium ↓ Collinsella ↓ Adlercreutzia ↓ Bifidobacterium |
| Montassier et al. (2014), [6] | Non-Hodgkin’s lymphoma | n = 8 | Chemotherapy | Baseline: before chemotherapy; Follow-up: 1 week after chemotherapy | 16S rRNA gene pyrosequencing/dHPLC | ↑ Bacteroidetes ↑ Proteobacteria ↑ Bacteroides ↑ Escherichia ↓ α-diversity (OTUs, Chao index, Shannon index) ↓ Firmicutes ↓ Actinobacteria ↓ Blautia ↓ Faecalibacterium ↓ Roseburia ↓ Bifidobacterium |
| Stringer et al. (2013), [37] | Breast cancer, gastrointestinal cancer | n = 10 | Chemotherapy (FOLFOX4, FOLFOX6, FOLFIRI, capecitabine) | (1) Before chemotherapy (2) Day 2 of chemotherapy (3) Day 5 (4) Day 10 | Bacterial growth tests with selective media, real-time PCR | ↑ E.coli ↑ Lactobacillus spp. (until day 5, then decrease) |
| Dörffel et al. (2012), [31] | NET | n = 13 | Chemotherapy | Before and during therapy | FISH | ↑ Faecalibacterium prausnitzii (midgut NET only) |
| Zwielehner et al. (2011), [32] | Different types of cancer | n = 17 | Chemotherapy | (1) Before chemotherapy (2) Day 1–4 after chemotherapy (5) Day 5–9 after chemotherapy | qPCR/PCR-DGGE | ↓ Bacteroides ↓ Bifidobacteria ↓ Clostridium cluster IV ↓ Clostridium cluster XIVa |
| n = 2 | Chemotherapy | (1) Before chemotherapy (2) Day 1–4 after chemotherapy | High throughput sequencing | ↑ Enterococcus faecium ↑ Clostridium difficile ↑ Peptostreptococcaceae ↓ Faecalibacterium prausnitzii ↓ Lactobacilli ↓ Veillonella spp. ↓ Bifidobacteria ↓ E.coli/Shigella | ||
| Wada et al. (2010), [38] | Different types of cancer | n = 23 | Chemotherapy | (1) Before chemotherapy (2) Within 24 h after initiation (3) Once weekly | Bacterial cultures (n = 3) | ↑ Enterobacteriaceae |
| Van Vliet et al. (2009), [39] | Pediatric AML | n = 9 | Chemotherapy | (1) Day 2 of chemotherapy (2) Day 11 of chemotherapy (3) ≥6 weeks after treatment | PCR-DGGE | ↓ α-diversity |
| Immunotherapy | ||||||
| Routy et al. (2018), [26] | NSCLC (n = 15) RCC (n = 17) | n = 32 | Anti-PD-1 | (1) Before treatment (2) After 2nd injection (1 month) (3) After 4th injection (2 months) (4) After 12th injection (6 months | Metagenomic shotgun sequencing | ↑ α-diversity (Richness) ↑ Candidatus Alistipes marseilloanorexicus ↑ Clostridium scindens ↑ Eubacterium sp. ↑ Clostridium sp. ↑ Streptococcus salivarius ↑ Clostridiales ↑ Eubacterium eligens |
| Chaput et al. (2017), [27] | Metastatic melanoma with colitis | n = 7 | Ipilimumab | At baseline and at the time of colitis occurrence | 16S rRNA gene sequencing | ↓ α-diversity (Shannon index) ↓ Ruminococcus ↓ Lachnospiracea incertae sedis ↓ Blautia ↓ Clostridium IV ↓ Eubacterium ↓ unclassified Lachnospiraceae ↓ Pseudoflavonifracto ↓ Butyrate producing bacterium L2-21 ↓ Ruminococcus bromii ↓ Blautia obeum 1-33 ↓ Eubacterium coprostanoligenes HL ↓ Clostridium clostridioforme LCR24 ↓ Alistipes spe 627 ↓ Blautia obeum ↓ Butyrate producing bacterium PH08AY04 ↓ Clostridium leptum DSM 753T ↓ Bacterium ASF500 ↓ Clostridium sp JC3 ↓ Rumen bacterium 2-293-25 ↓ Bacterium ic ↓ Butyrate producing bacterium M21-2 ↓ Unidentified bacterium CCCM23 ↓ Unidentified bacterium CCCM41 ↓ Ruminococcus bromii L2-63 ↓ Clostridiales bacterium JN18-V41 |
| Vetizou et al. (2015), [41] | Metastatic melanoma | n = 18 | Ipilimumab | See Chaput et al. (2017) | 16S rRNA gene sequencing | ↑ Bacteroides salyersiae ↑ Bacteroides acidifaciens ↑ Bacteroides uniformis ↓ Prevotella copri ↓ Bacteroides sp. ↓ Barnesiella intestinohominis ↓ Parabacteroides distasonis |
| Dörffel et al. (2012), [31] | Midgut NET | n = 11 | Interferon alpha-2b | Before and during therapy | FISH | ↑ Faecalibacterium prausnitzii |
| Study Design | Main Findings | |||||
|---|---|---|---|---|---|---|
| Study | Type of Cancer | Type of Therapy | n Cases | n Controls | Method Used for Microbiota Analysis | Effects of Therapy on Microbiota |
| Chemotherapy | ||||||
| Youssef et al. (2018), [35] | Gastrointestinal cancer | Chemotherapy and/or radiotherapy | n = 20 (treated patients) | Non-treated patients: n = 43 | 16S rRNA gene sequencing | ↑ Lactobacillaceae ↑ Lactobacillus |
| Healthy controls: n = 13 | 16S rRNA gene sequencing | ↓ Bifidobacteriaceae ↓ Ruminiclostridium ↓ Lachnoclosteridium ↓ Oscillibacter | ||||
| Deng et al. (2018), [36] | CRC | Oxaliplatin + tegafur | n = 14 | n = 33 | 16S rRNA gene sequencing | ↑ Veillonella ↑ Veillonella dispar ↑ Prevotella copri ↑ Bacteroides plebeius |
| Stringer et al. (2013), [37] | CRC, breast cancer, laryngeal cancer, esophageal cancer, melanoma | Chemotherapy | n = 16 | n = 2 | Bacterial growth tests with selective media, real-time PCR | ↑ Escherichia coli ↑ Staphylococcus spp. ↓ Lactobacillus spp. ↓ Bacteroides spp. ↓ Bifidobacterium spp. ↓ Enterococcus spp. |
| Van Vliet et al. (2009), [39] | Pediatric AML | Chemotherapy | n = 9 | n = 11 | FISH | ↑ Enterococci ↓ total number of bacteria ↓ Bacteroides ↓ Clostridium cluster XIVa ↓ Faecalibacterium prausnitzii ↓ Bifidobacterium ↓ Streptococci |
| Hormonal Therapy | ||||||
| Sfanos et al. (2018), [42] | Prostate cancer | ATT/GNRH | ATT: n = 9 GNRH: n = 5 | n = 16 (no medication) | 16S rDNA sequencing | Smallest β-diversity within ATT compared to GNRH and controls Greatest β-diversity between ATT and no medication |
| ATT | n = 9 | n = 16 (no medication) | 16S rDNA sequencing | ↑ Akkermansia muciniphila ↑ Ruminococcaceae ↑ Blautia wexlerae ↑ Clostridium oroticum ↑ Lachnospiraceae_Clostridium_XlVa ↑ Robinsoniella peoriensis ↑ Anaerococcus tetradius ↑ Bacteroides stercoris ↑ Verrucomicrobiaceae ↑ Lachnospiraceae ↑ Clostridiales insertae sedis XIII ↑ Staphylococcaceae ↑ Bacillales ↑ Aerococcaceae ↑ Selenomonadales ↓ Clostridiales ↓ Brevibacteriaceae ↓ Erysipelotrichaceae ↓ Streptococcaceae ↓ Clostridiales_unassigned ↓ Prevotellaceae | ||
| ATT | n = 9 | n = 16 (no medication) n = 5 (GNRH) | qPCR | ↑ Akkermansia muciniphila | ||
| GNRH | n = 5 | n = 16 (no medication) | 16S rDNA sequencing | ↑ Blautia wexlerae ↑ Clostridium oroticum ↑ Anaerococcus tetradius ↑ Lachnospiraceae ↑ Staphylococcaceae ↑ Aerococcaceae ↑ Selenomonadales | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aarnoutse, R.; Ziemons, J.; Penders, J.; Rensen, S.S.; de Vos-Geelen, J.; Smidt, M.L. The Clinical Link between Human Intestinal Microbiota and Systemic Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 4145. https://doi.org/10.3390/ijms20174145
Aarnoutse R, Ziemons J, Penders J, Rensen SS, de Vos-Geelen J, Smidt ML. The Clinical Link between Human Intestinal Microbiota and Systemic Cancer Therapy. International Journal of Molecular Sciences. 2019; 20(17):4145. https://doi.org/10.3390/ijms20174145
Chicago/Turabian StyleAarnoutse, Romy, Janine Ziemons, John Penders, Sander S. Rensen, Judith de Vos-Geelen, and Marjolein L. Smidt. 2019. "The Clinical Link between Human Intestinal Microbiota and Systemic Cancer Therapy" International Journal of Molecular Sciences 20, no. 17: 4145. https://doi.org/10.3390/ijms20174145
APA StyleAarnoutse, R., Ziemons, J., Penders, J., Rensen, S. S., de Vos-Geelen, J., & Smidt, M. L. (2019). The Clinical Link between Human Intestinal Microbiota and Systemic Cancer Therapy. International Journal of Molecular Sciences, 20(17), 4145. https://doi.org/10.3390/ijms20174145