Comprehensive Analysis of CRISPR/Cas9-Mediated Mutagenesis in Arabidopsis thaliana by Genome-Wide Sequencing

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Summary of On-Target Variations in CRISPR/Cas9-Edited Plants
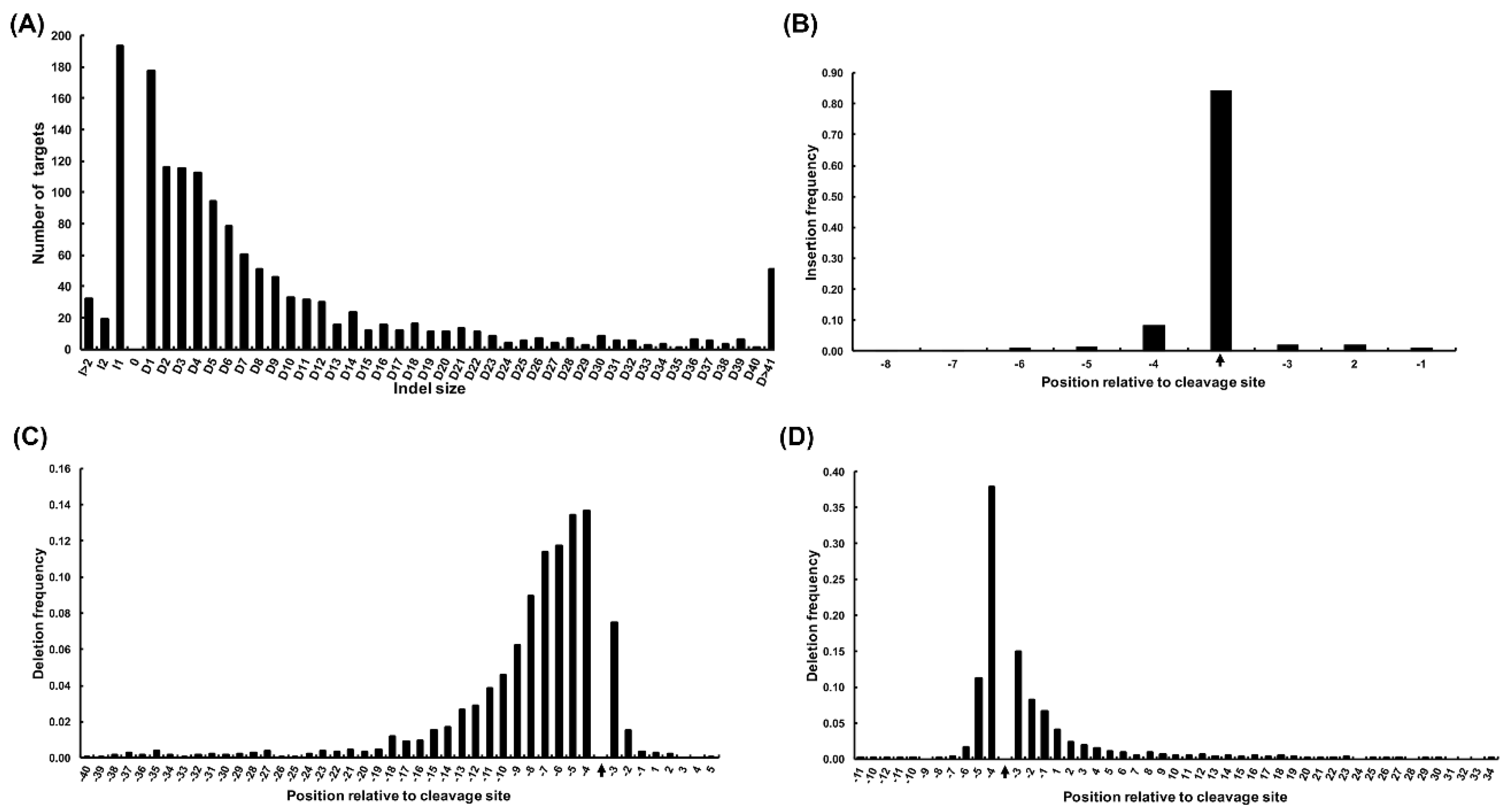
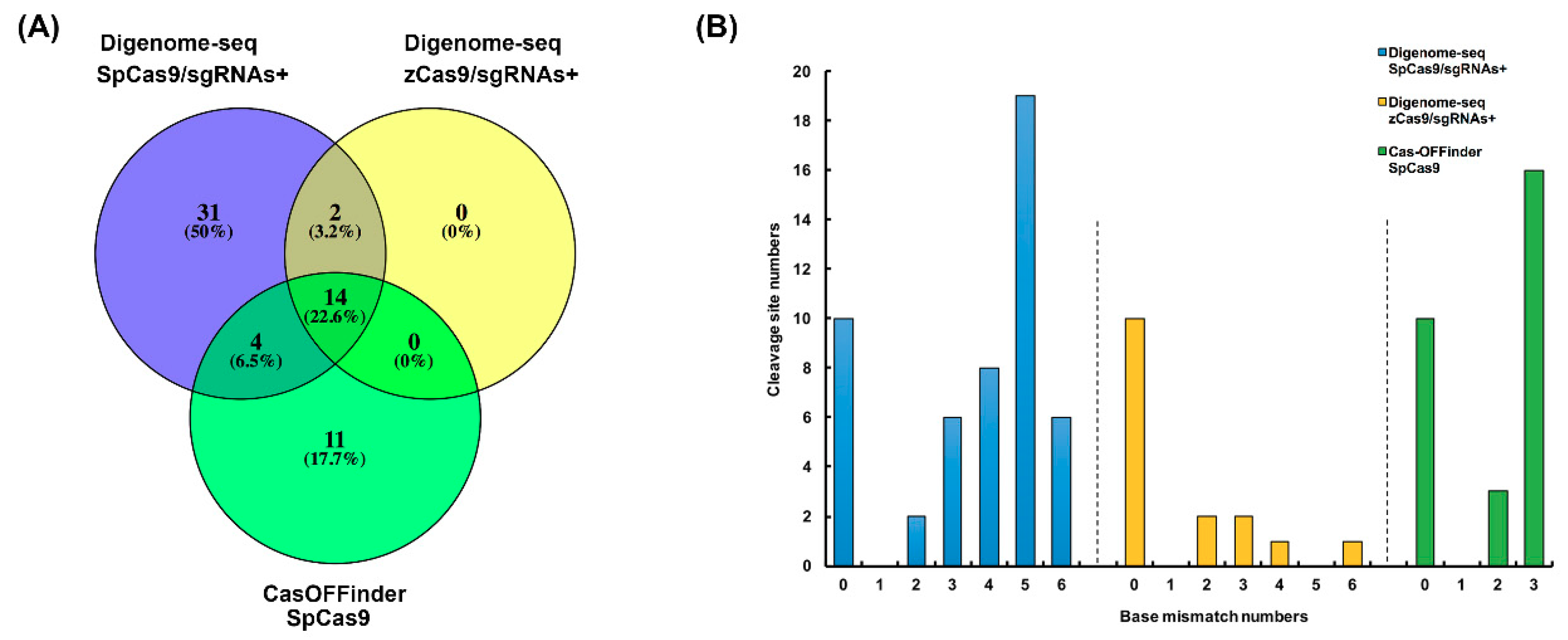
2.2. Off-target Sites Predicted by Digenome-seq
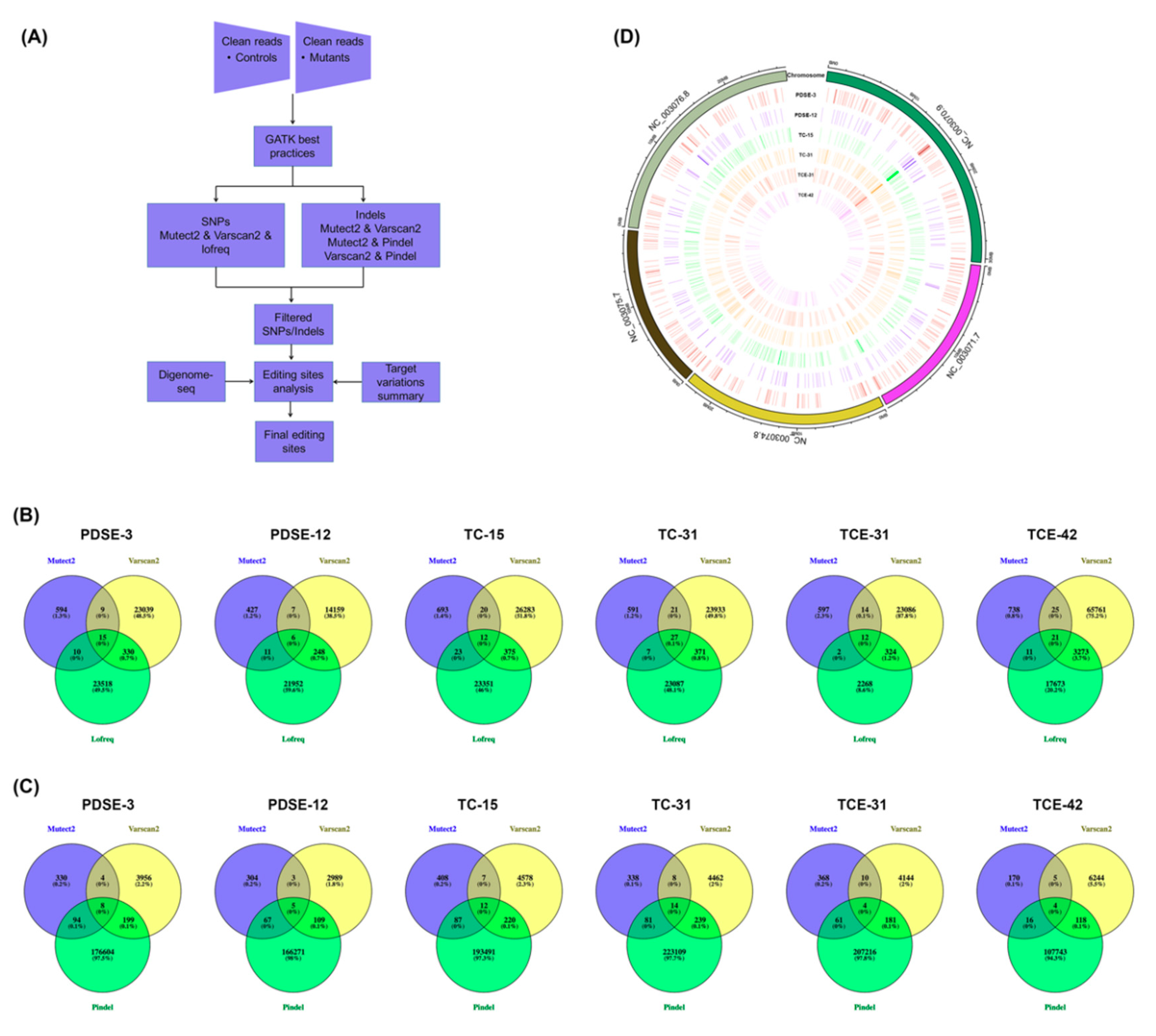
2.3. Detection of Genome-Wide Variations and Editing Sites
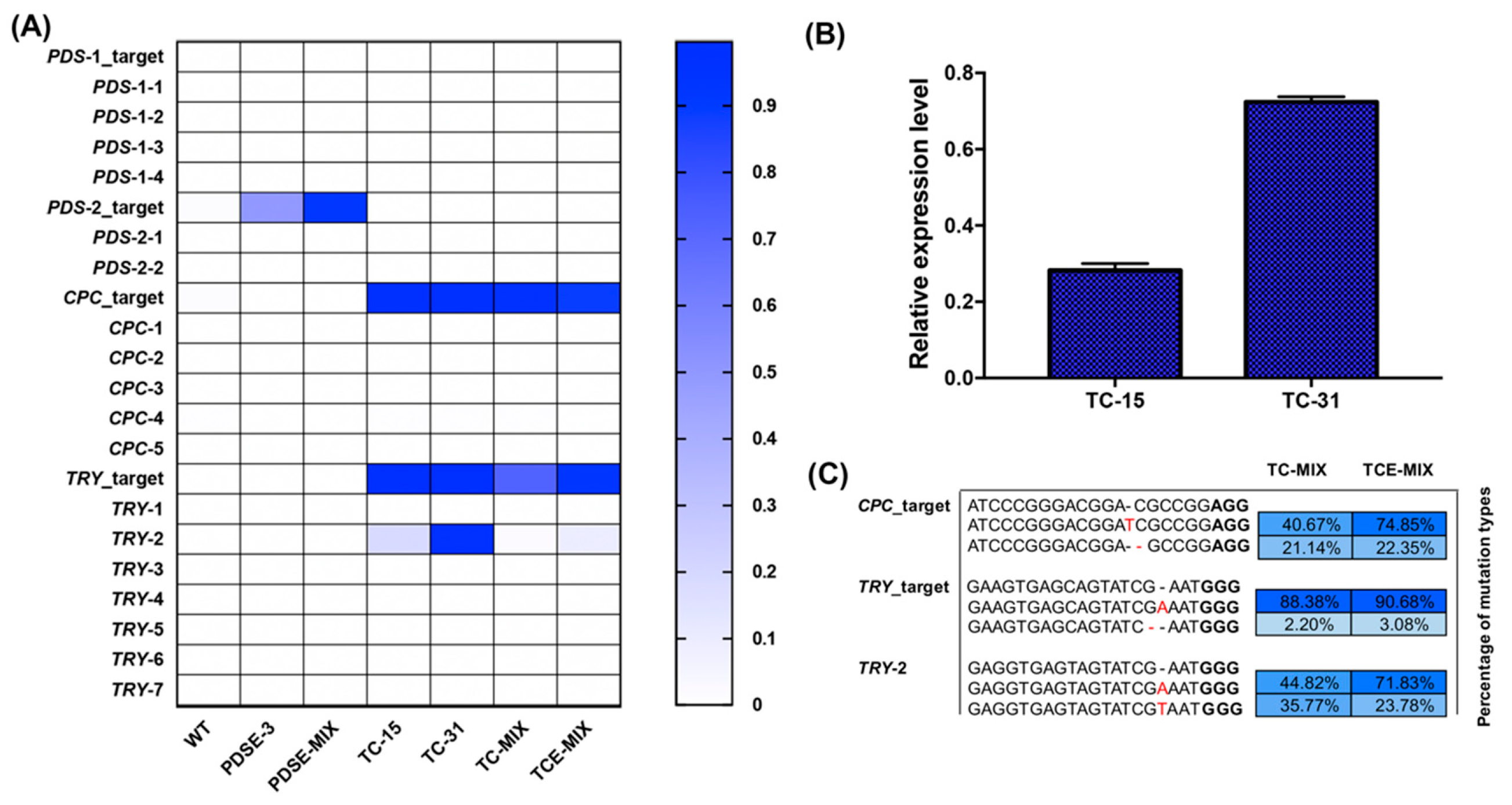
2.4. Analysis of Targeted Amplification Deep Sequencing
3. Discussion
4. Materials and Methods
4.1. Summary of Targeting Precision in Genome-Edited Plants
4.2. In Vitro Cleavage of Genomic DNA and Cleavage Site Analysis
4.3. Guide RNA Design and Vector Construction
4.4. Arabidopsis Transformation and Mutagenesis Analysis at On-Target Sites
4.5. Whole-Genome Sequencing and Variation Analysis
4.6. Targeted Amplification Deep Sequencing
4.7. Real-time Quantitative PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| Cas | CRISPR-associated protein |
| sgRNA | Single guide RNA |
| GMOs | Genetically modified organisms |
| PAM | Protospacer adjacent motif |
| indel | Insertion and deletion |
| SNV | Single nucleotide variation |
| WGS | Whole-genome sequencing |
| NHEJ | Non-homologous end joining |
| DSB | Double-strand break |
| HDR | Homology-directed repair |
References
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening Sequences of Regularly Spaced Prokaryotic Repeats Derive from Foreign Genetic Elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.; Benoit, Q.; Alexei, S.; Dusko, E. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [Green Version]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenyan, J.; David, B.; David, C.; Feng, Z.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar]
- Dali, L.; Zhongwei, Q.; Yanjiao, S.; Yuting, C.; Yuting, G.; Meizhen, L.; Yongmei, L.; Na, G.; Liren, W.; Xiaoling, L. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 681. [Google Scholar]
- Platt, R.; Chen, S.; Zhou, Y.; Yim, M.; Swiech, L.; Kempton, H.; Dahlman, J.; Parnas, O.; Eisenhaure, T.; Jovanovic, M. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [Green Version]
- Shan, Q.; Wang, Y.; Li, J.; Zhang, Y.; Chen, K.; Liang, Z.; Zhang, K.; Liu, J.; Xi, J.J.; Qiu, J.L. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Alptekin, B.; Budak, H. CRISPR/Cas9 genome editing in wheat. Funct. Integr. Genom. 2018, 18, 31–41. [Google Scholar] [CrossRef]
- Feng, C.; Yuan, J.; Wang, R.; Liu, Y.; Birchler, J.A.; Han, F. Efficient Targeted Genome Modification in Maize Using CRISPR/Cas9 System. J. Genet. Genom. 2015, 43, 37–43. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, B.; Ding, W.; Liu, X.; Yang, D.L.; Wei, P.; Cao, F.; Zhu, S.; Zhang, F.; Mao, Y. Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 2013, 23, 1229–1232. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganathan, D.; Ramasamy, K.; Sellamuthu, G.; Jayabalan, S.; Venkataraman, G. CRISPR for Crop Improvement: An Update Review. Front. Plant Sci. 2018, 9, 985. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Zhao, K. Genome editing technologies and their applications in crop improvement. Plant Biotechnol. Rep. 2018, 12, 57–68. [Google Scholar] [CrossRef]
- Wang, F.; Wang, C.; Liu, P.; Lei, C.; Hao, W.; Gao, Y.; Liu, Y.G.; Zhao, K. Enhanced Rice Blast Resistance by CRISPR/Cas9-Targeted Mutagenesis of the ERF Transcription Factor Gene OsERF922. Plos ONE 2016, 11, e0154027. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, H.; Wang, H.; Lafitte, H.R.; Archibald, R.L.; Yang, M.; Hakimi, S.M.; Mo, H.; Habben, J.E. ARGOS8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotechnol. J. 2017, 15, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Waltz, E. Gene-edited CRISPR mushroom escapes US regulation. Nature 2016, 532, 293. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Zhang, Y.; Ge, X.; Yang, F.; Zhang, L.; Zheng, J.; Tan, X.; Jin, Z.B.; Qu, J.; Gu, F. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 2014, 4, 5405. [Google Scholar] [CrossRef]
- Meng, X.; Hu, X.; Liu, Q.; Song, X.; Gao, C.; Li, J.; Wang, K. Robust genome editing of CRISPR-Cas9 at NAG PAMs in rice. Sci. China Life Sci. 2018, 61, 122–125. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef]
- Yang, L.; Grishin, D.; Wang, G.; Aach, J.; Zhang, C.Z.; Chari, R.; Homsy, J.; Cai, X.; Zhao, Y.; Fan, J.B.; et al. Targeted and genome-wide sequencing reveal single nucleotide variations impacting specificity of Cas9 in human stem cells. Nat. Commun. 2014, 5, 5507. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target specificity of the CRISPR-Cas9 system. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Xing, H.L.; Wang, Z.P.; Zhang, H.Y.; Yang, F.; Wang, X.C.; Chen, Q.J. Potential high-frequency off-target mutagenesis induced by CRISPR/Cas9 in Arabidopsis and its prevention. Plant Mol. Biol. 2018, 96, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wolt, J.D. Risk associated with off-target plant genome editing and methods for its limitation. Emerg. Top. Life Sci. 2017, 1, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Kadam, U.S.; Shelake, R.M.; Chavhan, R.L.; Suprasanna, P. Concerns regarding ‘off-target’ activity of genome editing endonucleases. Plant Physiol. Biochem. 2018, 131, 22–30. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Veres, A.; Gosis, B.S.; Ding, Q.; Collins, R.; Ragavendran, A.; Brand, H.; Erdin, S.; Cowan, C.A.; Talkowski, M.E.; Musunuru, K. Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing. Cell Stem Cell 2014, 15, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Mao, Y.; Xu, N.; Zhang, B.; Wei, P.; Yang, D.L.; Wang, Z.; Zhang, Z.; Zheng, R.; Yang, L.; et al. Multigeneration analysis reveals the inheritance, specificity, and patterns of CRISPR/Cas-induced gene modifications in Arabidopsis. Proc. Natl. Acad. Sci. 2014, 111, 4632–4637. [Google Scholar] [CrossRef]
- Peterson, B.A.; Haak, D.C.; Nishimura, M.T.; Teixeira, P.J.; James, S.R.; Dangl, J.L.; Nimchuk, Z.L. Genome-Wide Assessment of Efficiency and Specificity in CRISPR/Cas9 Mediated Multiple Site Targeting in Arabidopsis. PLoS ONE 2016, 11, e0162169. [Google Scholar] [CrossRef]
- Tang, X.; Liu, G.; Zhou, J.; Ren, Q.; You, Q.; Tian, L.; Xin, X.; Zhong, Z.; Liu, B.; Zheng, X.; et al. A large-scale whole-genome sequencing analysis reveals highly specific genome editing by both Cas9 and Cpf1 (Cas12a) nucleases in rice. Genome Biol. 2018, 19, 84. [Google Scholar] [CrossRef]
- Li, J.; Manghwar, H.; Sun, L.; Wang, P.; Wang, G.; Sheng, H.; Zhang, J.; Liu, H.; Qin, L.; Rui, H.; et al. Whole genome sequencing reveals rare off-target mutations and considerable inherent genetic or/and somaclonal variations in CRISPR/Cas9-edited cotton plants. Plant Biotechnol. J. 2018, 17, 858–868. [Google Scholar] [CrossRef]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef]
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef]
- Aidan, O.B.; Bailey, T.L. GT-Scan: Identifying unique genomic targets. Bioinformatics 2014, 30, 2673–2675. [Google Scholar]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.I.; Kim, J.S. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef]
- Akcakaya, P.; Bobbin, M.L.; Guo, J.A.; Malagon-Lopez, J.; Clement, K.; Garcia, S.P.; Fellows, M.D.; Porritt, M.J.; Firth, M.A.; Carreras, A.; et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 2018, 561, 416–419. [Google Scholar] [CrossRef]
- Xing, H.L.; Dong, L.; Wang, Z.P.; Zhang, H.Y.; Han, C.Y.; Liu, B.; Wang, X.C.; Chen, Q.J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014, 14, 327. [Google Scholar] [CrossRef]
- Qin, G.; Gu, H.; Ma, L.; Peng, Y.; Deng, X.W.; Chen, Z.; Qu, L.J. Disruption of phytoene desaturase gene results in albino and dwarf phenotypes in Arabidopsis by impairing chlorophyll, carotenoid, and gibberellin biosynthesis. Cell Res. 2007, 17, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Zhang, J.H.; Pandey, M.; Kahler, J.F.; Loshakov, A.; Harris, B.; Dagur, P.K.; Mo, Y.Y.; Simonds, W.F. Improving the specificity and efficacy of CRISPR/CAS9 and gRNA through target specific DNA reporter. J. Biotechnol. 2014, 189, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, S.; Kim, S.; Park, J.; Kim, J.S. Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 2016, 26, 406–415. [Google Scholar] [CrossRef] [Green Version]
- Oliveros, J.C.; Franch, M.; Tabas-Madrid, D.; San-Leon, D.; Montoliu, L.; Cubas, P.; Pazos, F. Breaking-Cas-interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBL genomes. Nucleic Acids Res. 2016, 44, W267–W271. [Google Scholar] [CrossRef]
- Pawluk, A.; Amrani, N.; Zhang, Y.; Garcia, B.; Hidalgo-Reyes, Y.; Lee, J.; Edraki, A.; Shah, M.; Sontheimer, E.J.; Maxwell, K.L.; et al. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell 2016, 167, 1829–1838. [Google Scholar] [CrossRef]
- Oakes, B.L.; Fellmann, C.; Rishi, H.; Arkin, A.P.; Doudna, J.A.; Savage, D.F. CRISPR-Cas9 Circular Permutants as Programmable Scaffolds for Genome Modification. Cell 2019, 176, 256–267. [Google Scholar] [CrossRef]
- Shou, J.; Li, J.; Liu, Y.; Wu, Q. Precise and Predictable CRISPR Chromosomal Rearrangements Reveal Principles of Cas9-Mediated Nucleotide Insertion. Mol. Cell 2018, 71, 498–509. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, D.; Kim, S.; Kim, J.S. Genotyping with CRISPR-Cas-derived RNA-guided endonucleases. Nat. Commun. 2014, 5, 3157. [Google Scholar] [CrossRef]
- Wang, Z.P.; Xing, H.L.; Dong, L.; Zhang, H.Y.; Han, C.Y.; Wang, X.C.; Chen, Q.J. Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol. 2015, 16, 144. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Mismatch | SgRNAs | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| GLV 1-1 | GLV 6-1 | GLV 7-2 | GLV 8-2 | GLV 10-1 | GLV 10-2 | PDS-1 | PDS-2 | CPC | TRY | |
| 0 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 |
| 1 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 |
| 2 | 0/0 | 0/0 | 0/0 | 0/0 | 0/1 | 0/0 | 0/0 | 0/0 | 0/0 | 2/2 |
| 3 | 0/1 | 0/2 | 0/3 | 0/0 | 1/3 | 1/5 | 0/0 | 0/0 | 0/0 | 0/2 |
| 4 | 0/24 | 0/12 | 0/52 | 0/52 | 0/32 | 0/33 | 1/2 | 0/6 | 0/37 | 0/5 |
| 5 | 0/161 | 0/180 | 0/350 | 0/350 | 0/273 | 0/273 | 0/36 | 0/55 | 0/27 | 0/94 |
| Mutant | SNV | Indel | ||
|---|---|---|---|---|
| WT− | WT−/DT- | WT− | WT−/DT- | |
| PDSE-3 | 608 | 15 | 3120 | 305 |
| PDSE-12 | 10 | 6 | 1968 | 184 |
| TC-15 | 612 | 12 | 2525 | 326 |
| TC-31 | 31 | 27 | 2332 | 342 |
| TCE-31 | 14 | 12 | 2586 | 256 |
| TCE-42 | 24 | 21 | 690 | 143 |
| Mutant | PDS-2 (%) | CPC (%) | TRY (%) | TRY Off Target (%) |
|---|---|---|---|---|
| PDSE-3 | 38.1 | - | - | - |
| PDSE-12 | 46.1 | - | - | - |
| TC-15 | - | 84.1 | 83.5 | 20.9 |
| TC-31 | - | 85.7 | 88.1 | 97.3 |
| TCE-31 | - | 87.6 | 95.3 | 9.8 |
| TCE-42 | - | 100 | 84.2 | 19 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, W.; Fu, W.; Zhu, P.; Li, Z.; Wang, C.; Wang, C.; Zhang, Y.; Zhu, S. Comprehensive Analysis of CRISPR/Cas9-Mediated Mutagenesis in Arabidopsis thaliana by Genome-Wide Sequencing. Int. J. Mol. Sci. 2019, 20, 4125. https://doi.org/10.3390/ijms20174125
Xu W, Fu W, Zhu P, Li Z, Wang C, Wang C, Zhang Y, Zhu S. Comprehensive Analysis of CRISPR/Cas9-Mediated Mutagenesis in Arabidopsis thaliana by Genome-Wide Sequencing. International Journal of Molecular Sciences. 2019; 20(17):4125. https://doi.org/10.3390/ijms20174125
Chicago/Turabian StyleXu, Wenjie, Wei Fu, Pengyu Zhu, Zhihong Li, Chenguang Wang, Chaonan Wang, Yongjiang Zhang, and Shuifang Zhu. 2019. "Comprehensive Analysis of CRISPR/Cas9-Mediated Mutagenesis in Arabidopsis thaliana by Genome-Wide Sequencing" International Journal of Molecular Sciences 20, no. 17: 4125. https://doi.org/10.3390/ijms20174125