The Complex Phosphorylation Patterns That Regulate the Activity of Hsp70 and Its Cochaperones


Abstract
1. Introduction
2. Protein Folding and Molecular Chaperones
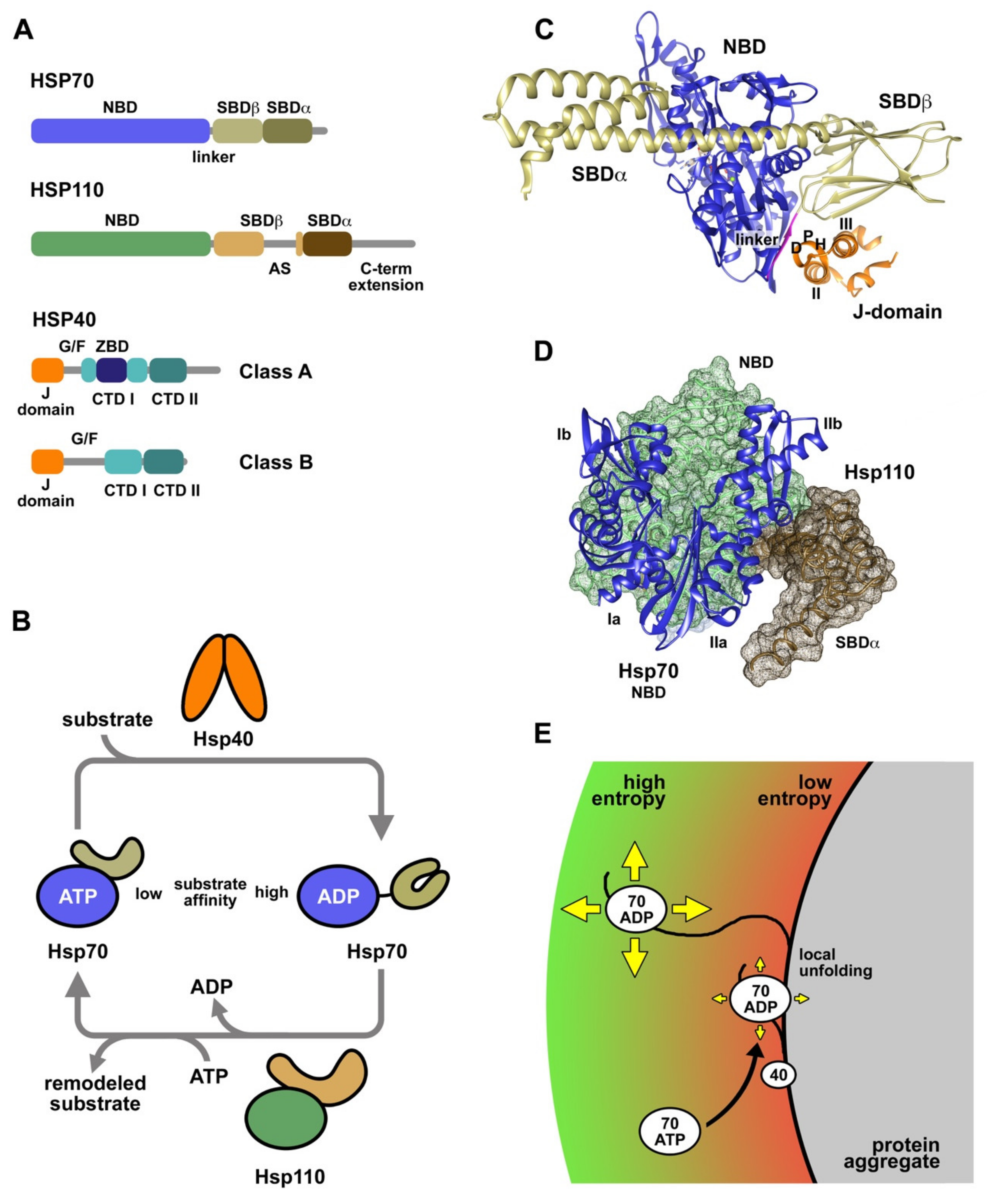
3. Molecular Chaperones Involved in Protein Folding and Protein Aggregate Reactivation
4. Interaction of Chaperones with Substrate Proteins and Protein Aggregates
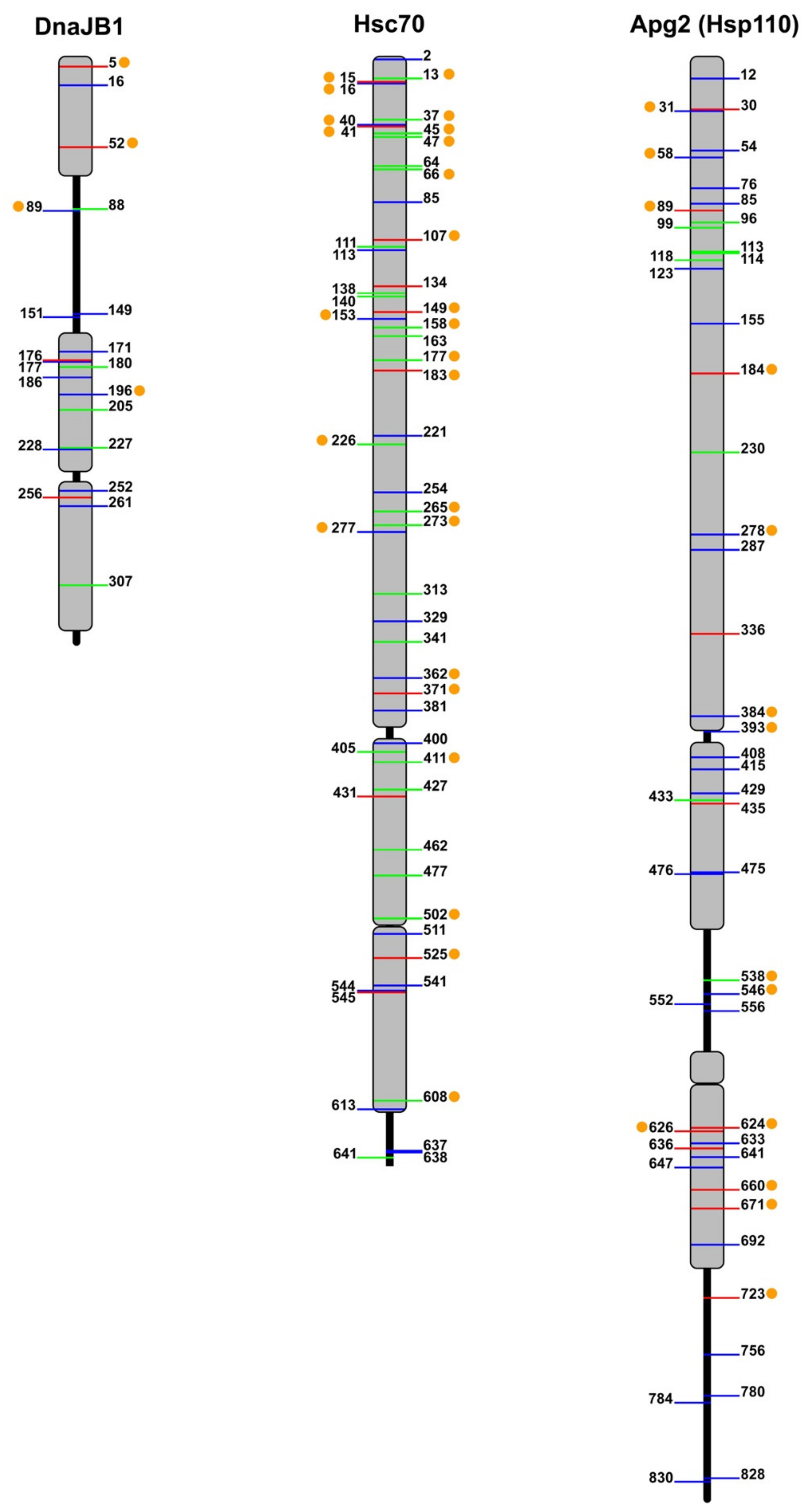
5. Regulation of Chaperone Activity by Phosphorylation
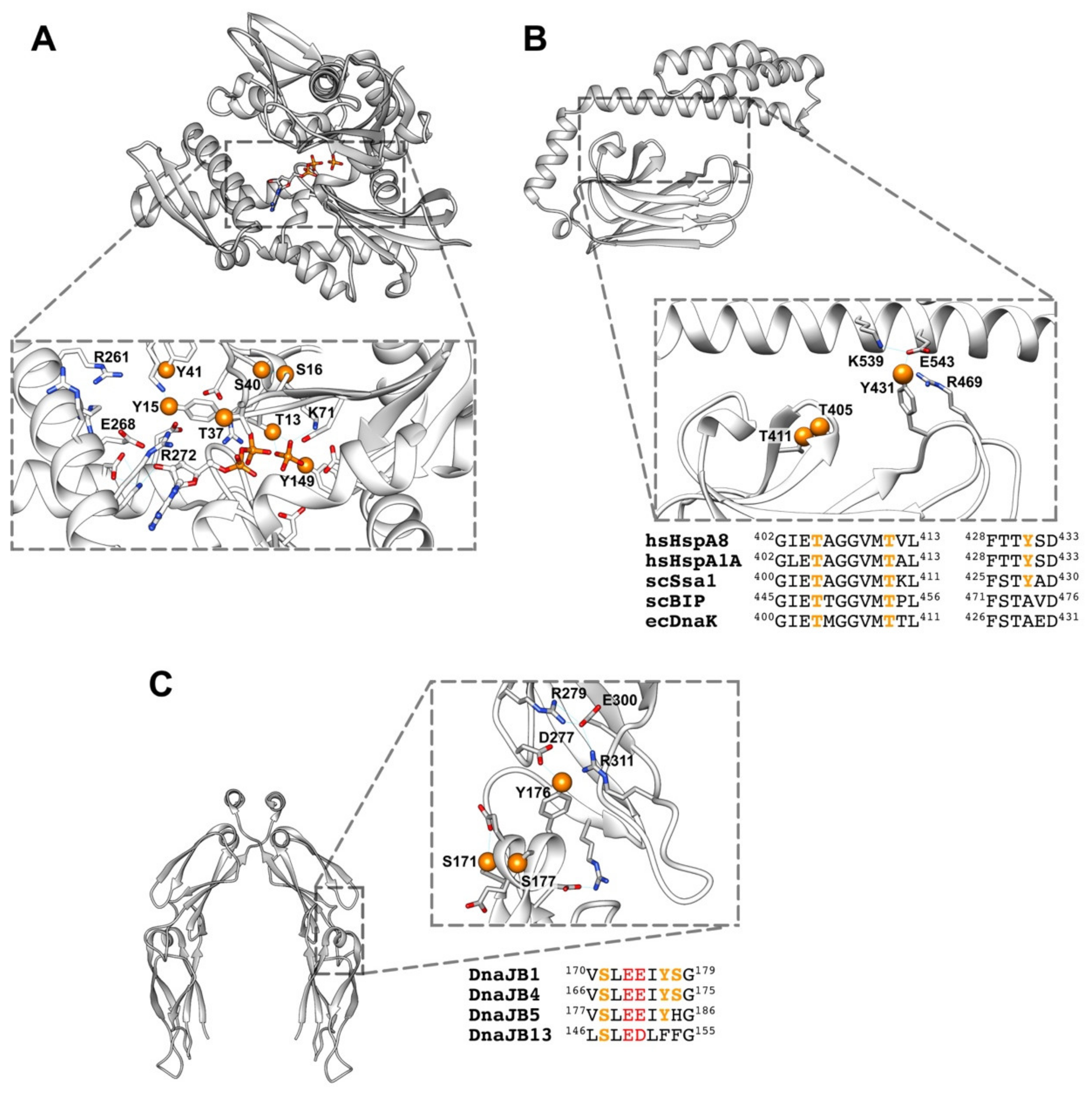
5.1. Hsp40
5.2. Hsp110
5.3. Hsp70
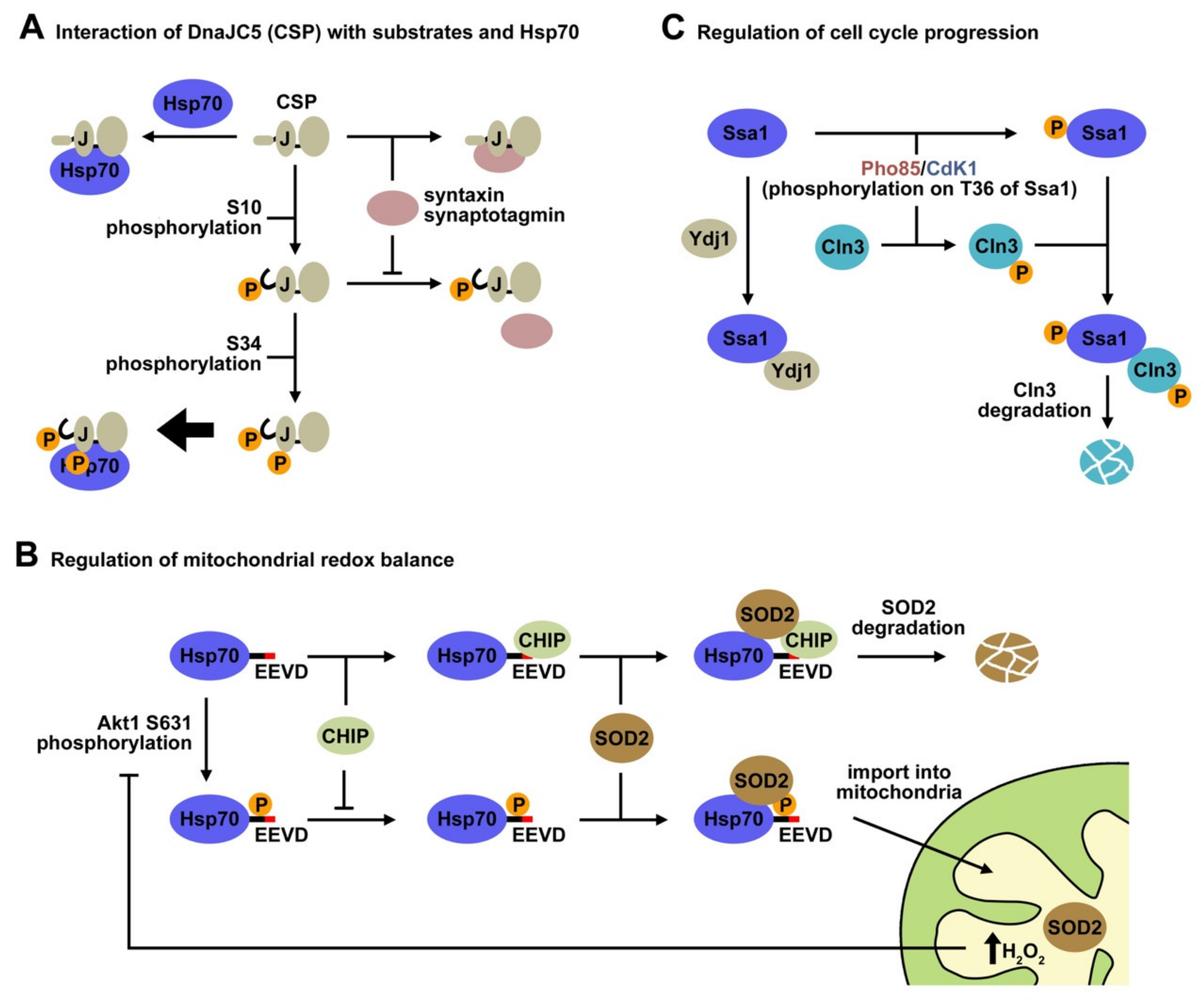
5.3.1. Regulation of the Mitochondrial Redox Balance
5.3.2. Host-Pathogen Interaction
5.3.3. Regulation of Hsp70 Dimerization by Phosphorylation
5.3.4. Regulation of the Balance between Protein Folding and Degradation
5.3.5. Regulation of the Cell Cycle Progression
5.3.6. Hsp70 Phosphorylation Regulates Drug Resistance in Cancer Cells
5.3.7. Regulation of Apoptosis
6. Phosphorylation as Part of the Chaperone Code
6.1. How Phosphorylation Regulates the Conformation and Activity of These Chaperones?
6.2. How Phosphorylation Modulates Intermolecular Interactions?
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PTM | Posttranslational modifications |
| Hsp | Heat shock protein |
| PQC | Protein quality control |
| NBD | Nucleotide binding domain |
| SBD | Substrate binding domain |
| CTD | C-terminal domain |
| TLA | Three letter acronym |
| ZBD | Zinc binding domain |
| Apg | ATP and peptide-binding protein in germ cells |
| EEVD | Glu-Glu-Val-Asp |
| sHsp | Small heat shock protein |
| αsyn | α-synuclein |
| Htt | Huntingtin |
| AS | Proline-rich acidic subdomain |
| NEF | Nucleotide exchange factor |
| TPR | Tetratricopeptide repeat |
| MK5 | Mitogen-activated protein kinase-activated protein kinase 5 |
| HSF1 | Heat shock factor 1 |
| CSP | Cysteine string protein |
| SNAP25 | Synaptosomal nerve-associated protein 25 |
| PKC | Protein kinase C |
| SOD2 | Superoxide dismutase-2 |
| CHIP | C-terminus of Hsc70 interacting protein |
| Akt1 | RAC-alpha serine/threonine-protein kinase |
| UPS | Ubiquitin-proteasome system |
| PP2C | Protein phosphatase 2C |
| HOP | Hsp70/Hsp90 organizing protein |
| CDK | Cyclin-dependent protein kinase |
| G6PDH | Glucose 6 phosphate dehydrogenase |
| Hsc | Heat shock cognate |
| NMR | Nuclear magnetic resonance |
References
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative Phosphoproteomics Reveals Widespread Full Phosphorylation Site Occupancy During Mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Gatto, G.J.; Garneau-Tsodikova, S.; Garneau-Tsodikova, S.; Garneau-Tsodikova, S. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P. The origins of protein phosphorylation. Nature 2002, 4, E127–E130. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M.; Šali, A.; Karplus, M. Protein Folding: A Perspective from Theory and Experiment. Angew. Chem. Int. Ed. 1998, 37, 868–893. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular Chaperone Functions in Protein Folding and Proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Wolynes, P.G. Evolution, energy landscapes and the paradoxes of protein folding. Biochimie 2015, 119, 218–230. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Zimmerman, S.B.; Trach, S.O. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 1991, 222, 599–620. [Google Scholar] [CrossRef]
- Christiansen, A.; Wang, Q.; Cheung, M.S.; Wittung-Stafshede, P. Effects of macromolecular crowding agents on protein folding in vitro and in silico. Biophys. Rev. 2013, 5, 137–145. [Google Scholar] [CrossRef]
- Ellis, R.J.; Minton, A.P. Protein aggregation in crowded environments. Biol. Chem. 2006, 387, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, A.I.; Radford, S.E. An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nat. Struct. Mol. Biol. 2009, 16, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.B.; Ho, C.-T.; Winkler, J.; Khokhrina, M.; Neuner, A.; Mohamed, M.Y.; Guilbride, D.L.; Richter, K.; Lisby, M.; Schiebel, E.; et al. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. EMBO J. 2015, 34, 778–797. [Google Scholar] [CrossRef] [PubMed]
- Lindner, A.B.; Madden, R.; Demarez, A.; Stewart, E.J.; Taddei, F. Asymmetric segregation of protein aggregates is associated with cellular aging and rejuvenation. Proc. Natl. Acad. Sci. USA 2008, 105, 3076–3081. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Desplats, P.; Sigurdson, C.; Tsigelny, I.; Masliah, E. Cell-to-cell transmission of non-prion protein aggregates. Nat. Rev. Neurol. 2010, 6, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Yue, Z. Neuronal aggregates: Formation, clearance and spreading. Dev. Cell 2015, 32, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Freilich, R.; Arhar, T.; Abrams, J.L.; Gestwicki, J.E. Protein–Protein Interactions in the Molecular Chaperone Network. Acc. Chem. Res. 2018, 51, 940–949. [Google Scholar] [CrossRef]
- Aguado, A.; Fernández-Higuero, J.Á.; Moro, F.; Muga, A. Chaperone-assisted protein aggregate reactivation: Different solutions for the same problem. Arch. Biochem. Biophys. 2015, 580, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 2018, 69, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.F.J.; Katrukha, E.A.; Van Straaten, W.; Verlhac, P.; Reggiori, F.; Kapitein, L.C. Probing aggrephagy using chemically-induced protein aggregates. Nat. Commun. 2018, 9, 4245. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J. The Mammalian Disaggregase Machinery: Hsp110 Synergizes with Hsp70 and Hsp40 to Catalyze Protein Disaggregation and Reactivation in a Cell-Free System. PLoS ONE 2011, 6, e26319. [Google Scholar] [CrossRef]
- Rampelt, H.; Kirstein-Miles, J.; Nillegoda, N.B.; Chi, K.; Scholz, S.R.; Morimoto, R.I.; Bukau, B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012, 31, 4221–4235. [Google Scholar] [CrossRef]
- Gao, X.; Carroni, M.; Nussbaum-Krammer, C.; Mogk, A.; Nillegoda, N.B.; Szlachcic, A.; Guilbride, D.L.; Saibil, H.R.; Mayer, M.P.; Bukau, B. Human Hsp70 Disaggregase Reverses Parkinson’s-Linked alpha-Synuclein Amyloid Fibrils. Mol. Cell 2015, 59, 781–793. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The Hsp70 chaperone machinery: J-proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Bracher, A.; Verghese, J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2015, 2, 10. [Google Scholar] [CrossRef]
- Vogel, M.; Mayer, M.P.; Bukau, B. Allosteric Regulation of Hsp70 Chaperones Involves a Conserved Interdomain Linker. J. Biol. Chem. 2006, 281, 38705–38711. [Google Scholar] [CrossRef]
- Kityk, R.; Kopp, J.; Mayer, M.P. Molecular Mechanism of J-Domain-Triggered ATP Hydrolysis by Hsp70 Chaperones. Mol. Cell 2018, 69, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Polier, S.; Dragovic, Z.; Hartl, F.U.; Bracher, A. Structural Basis for the Cooperation of Hsp70 and Hsp110 Chaperones in Protein Folding. Cell 2008, 133, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Swain, J.F.; Dinler, G.; Sivendran, R.; Montgomery, D.L.; Stotz, M.; Gierasch, L.M. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell 2007, 26, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Kityk, R.; Kopp, J.; Sinning, I.; Mayer, M.P. Structure and Dynamics of the ATP-Bound Open Conformation of Hsp70 Chaperones. Mol. Cell 2012, 48, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Schlecht, R.; Erbse, A.H.; Bukau, B.; Mayer, M.P. Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat. Struct. Mol. Biol. 2011, 18, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Rios, P.D.L.; Christen, P.; Lustig, A.; Goloubinoff, P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat. Methods 2010, 6, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.J.; Hageman, J.; Carra, S.; Kampinga, H.H. Structural and Functional Diversities between Members of the Human HSPB, HSPH, HSPA, and DNAJ Chaperone Families. Biochemistry 2008, 47, 7001–7011. [Google Scholar] [CrossRef] [PubMed]
- Wall, D.; Zylicz, M.; Georgopoulos, C. The NH2-terminal 108 amino acids of the Escherichia coli DnaJ protein stimulate the ATPase activity of DnaK and are sufficient for lambda replication. J. Biol. Chem. 1994, 269, 5446–5451. [Google Scholar] [PubMed]
- Douglas, M.G. A Conserved HPD Sequence of the J-domain Is Necessary for YDJ1 Stimulation of Hsp70 ATPase Activity at a Site Distinct from Substrate Binding. J. Biol. Chem. 1996, 271, 9347–9354. [Google Scholar]
- Li, J.; Qian, X.; Sha, B. The Crystal Structure of the Yeast Hsp40 Ydj1 Complexed with Its Peptide Substrate. Structure 2003, 11, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Nillegoda, N.B.; Kirstein, J.; Szlachcic, A.; Berynskyy, M.; Stank, A.; Stengel, F.; Arnsburg, K.; Gao, X.; Scior, A.; Aebersold, R.; et al. Crucial HSP70 co–chaperone complex unlocks metazoan protein disaggregation. Nature 2015, 524, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Korszun, R.; Hartl, F.U.; Flanagan, J. A zinc finger-like domain of the molecular chaperone DnaJ is involved in binding to denatured protein substrates. EMBO J. 1996, 15, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Arsene-Ploetze, F.; Rist, W.; Rudiger, S.; Schneider-Mergener, J.; Mayer, M.P.; Bukau, B. Molecular basis for regulation of the heat shock transcription factor sigma32 by the DnaK and DnaJ chaperones. Mol. Cell 2008, 32, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, J.; Perales-Calvo, J.; Muga, A.; Valpuesta, J.M.; Moro, F. Structural insights into the chaperone activity of the 40-kDa heat shock protein DnaJ: Binding and remodeling of a native substrate. J. Biol. Chem. 2013, 288, 15065–15074. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Cyr, D.M. Protein Folding Activity of Hsp70 Is Modified Differentially by the Hsp40 CochaperoneCochaperones Sis1 and Ydj1. J. Biol. Chem. 1998, 273, 27824–27830. [Google Scholar] [CrossRef] [PubMed]
- Perales-Calvo, J.; Muga, A.; Moro, F. Role of DnaJ G/F-rich Domain in Conformational Recognition and Binding of Protein Substrates. J. Biol. Chem. 2010, 285, 34231–34239. [Google Scholar] [CrossRef]
- Laufen, T.; Mayer, M.; Beisel, C.; Klostermeier, D.; Mogk, A.; Reinstein, J.; Bukau, B. Mechanism of regulation of Hsp70 chaperones by DnaJ cochaperones. Proc. Natl. Acad. Sci. USA 1999, 96, 5452–5457. [Google Scholar] [CrossRef]
- Huang, K.; Flanagan, J.M.; Prestegard, J.H. The influence of C-terminal extension on the structure of the “J-domain” in E. coli DnaJ. Protein Sci. 1999, 8, 203–214. [Google Scholar] [CrossRef]
- Qian, Y.Q.; Patel, D.; Hartl, F.-U.; McColl, D.J. Nuclear Magnetic Resonance Solution Structure of the Human Hsp40 (HDJ-1) J-domain. J. Mol. Biol. 1996, 260, 224–235. [Google Scholar] [CrossRef]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in Hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef] [PubMed]
- Suh, W.-C.; Burkholder, W.F.; Lu, C.Z.; Zhao, X.; Gottesman, M.E.; Gross, C.A. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc. Natl. Acad. Sci. USA 1998, 95, 15223–15228. [Google Scholar] [CrossRef] [PubMed]
- Aron, R.; Lopez, N.; Walter, W.; Craig, E.A.; Johnson, J. In Vivo Bipartite Interaction between the Hsp40 Sis1 and Hsp70 in Saccharomyces cerevisiae. Genetics 2005, 169, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Y.; Ziegelhoffer, T.; Craig, E.A. Functionality of Class A and Class B J-protein Cochaperones with Hsp70. FEBS Lett. 2015, 589, 2825–2830. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-C.; Zhou, C.-J.; Zhou, Z.-R.; Wu, M.; Cao, C.-Y.; Hu, H.-Y. The C-terminal Helices of Heat Shock Protein 70 Are Essential for J-domain Binding and ATPase Activation. J. Biol. Chem. 2012, 287, 6044–6052. [Google Scholar] [CrossRef] [PubMed]
- Scheufler, C.; Brinker, A.; Bourenkov, G.; Pegoraro, S.; Moroder, L.; Bartunik, H.; Hartl, F.U.; Moarefi, I. Structure of TPR domain-peptide complexes: Critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 2000, 101, 199–210. [Google Scholar] [CrossRef]
- Connell, P.; Ballinger, C.A.; Jiang, J.; Wu, Y.; Thompson, L.J.; Hohfeld, J.; Patterson, C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001, 3, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, N.; Yokota, M.; Yasuda, K.; Saito, Y.; Nagata, K.; Hatayama, T. Characterization of stress sensitivity and chaperone activity of Hsp105 in mammalian cells. Biochem. Biophys. Res. Commun. 2011, 409, 90–95. [Google Scholar] [CrossRef]
- Liu, Q.; Hendrickson, W.A. Insights into Hsp70 Chaperone Activity from a Crystal Structure of the Yeast Hsp110 Sse1. Cell 2007, 131, 106–120. [Google Scholar] [CrossRef]
- Cabrera, Y.; Dublang, L.; Fernández-Higuero, J.A.; Albesa-Jové, D.; Lucas, M.; Viguera, A.R.; Guerin, M.E.; Vilar, J.M.; Muga, A.; Moro, F. Regulation of Human Hsc70 ATPase and Chaperone Activities by Apg2: Role of the Acidic Subdomain. J. Mol. Biol. 2019, 431, 444–461. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Yamagishi, N.; Hatayama, T. Different localization of Hsp105 family proteins in mammalian cells. Exp. Cell Res. 2007, 313, 3707–3717. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Yamagishi, N.; Hatayama, T. Nuclear localization mechanism of Hsp105beta and its possible function in mammalian cells. J. Biochem. 2009, 145, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Andréasson, C.; Fiaux, J.; Rampelt, H.; Druffel-Augustin, S.; Bukau, B. Insights into the structural dynamics of the Hsp110–Hsp70 interaction reveal the mechanism for nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2008, 105, 16519–16524. [Google Scholar] [CrossRef] [PubMed]
- Raviol, H.; Bukau, B.; Mayer, M.P. Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett. 2006, 580, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Goeckeler, J.L.; Petruso, A.P.; Aguirre, J.; Clement, C.C.; Chiosis, G.; Brodsky, J.L. The Yeast Hsp110, Sse1p, Exhibits High Affinity Peptide Binding. FEBS Lett. 2008, 582, 2393–2396. [Google Scholar] [CrossRef]
- Oh, H.J.; Chen, X.; Subjeck, J.R. hsp110 Protects Heat-denatured Proteins and Confers Cellular Thermoresistance. J. Biol. Chem. 1997, 272, 31636–31640. [Google Scholar] [CrossRef]
- Oh, H.J.; Easton, D.; Murawski, M.; Kaneko, Y.; Subjeck, J.R. The Chaperoning Activity of hsp110: Identification of Functional Domains by Use of Targeted Deletions. J. Biol. Chem. 1999, 274, 15712–15718. [Google Scholar] [CrossRef]
- Dragovic, Z.; Broadley, S.A.; Shomura, Y.; Bracher, A.; Hartl, F.U. Molecular chaperones of the Hsp110 family act as nucleotide exchange factors of Hsp70s. EMBO J. 2006, 25, 2519–2528. [Google Scholar] [CrossRef]
- Raviol, H.; Sadlish, H.; Rodriguez, F.; Mayer, M.P.; Bukau, B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006, 25, 2510–2518. [Google Scholar] [CrossRef]
- Schuermann, J.P.; Jiang, J.; Cuellar, J.; Llorca, O.; Wang, L.; Gimenez, L.E.; Jin, S.; Taylor, A.B.; Demeler, B.; Morano, K.A.; et al. Structure of the Hsp110:Hsc70 Nucleotide Exchange Machine. Mol. Cell 2008, 31, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular Chaperones and Protein Quality Control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, M.R.; Valpuesta, J.M. Hsp70 chaperone: A master player in protein homeostasis. F1000Research 2018, 7, 1497. [Google Scholar] [CrossRef] [PubMed]
- Wentink, A.; Nussbaum-Krammer, C.; Bukau, B. Modulation of Amyloid States by Molecular Chaperones. Cold Spring Harb. Perspect. Biol. 2019, 11, a033969. [Google Scholar] [CrossRef]
- Mannini, B.; Cascella, R.; Zampagni, M.; Van Waarde-Verhagen, M.; Meehan, S.; Roodveldt, C.; Campioni, S.; Boninsegna, M.; Penco, A.; Relini, A.; et al. Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA 2012, 109, 12479–12484. [Google Scholar] [CrossRef]
- Ojha, J.; Masilamoni, G.; Dunlap, D.; Udoff, R.A.; Cashikar, A.G. Sequestration of Toxic Oligomers by HspB1 as a Cytoprotective Mechanism. Mol. Cell. Biol. 2011, 31, 3146–3157. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Echeverria, A.; Shorter, J. Small Heat Shock Proteins Potentiate Amyloid Dissolution by Protein Disaggregases from Yeast and Humans. PLoS Biol. 2012, 10, e1001346. [Google Scholar] [CrossRef]
- Scior, A.; Buntru, A.; Arnsburg, K.; Ast, A.; Iburg, M.; Juenemann, K.; Pigazzini, M.L.; Mlody, B.; Puchkov, D.; Priller, J.; et al. Complete suppression of Htt fibrilization and disaggregation of Htt fibrils by a trimeric chaperone complex. EMBO J. 2018, 37, 282–299. [Google Scholar] [CrossRef]
- Schlieker, C.; Tews, I.; Bukau, B.; Mogk, A. Solubilization of aggregated proteins by ClpB/DnaK relies on the continuous extraction of unfolded polypeptides. FEBS Lett. 2004, 578, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Acebron, S.P.; Martin, I.; Del Castillo, U.; Moro, F.; Muga, A. DnaK-mediated association of ClpB to protein aggregates. A bichaperone network at the aggregate surface. FEBS Lett. 2009, 583, 2991–2996. [Google Scholar] [CrossRef] [PubMed]
- Acebron, S.P.; Fernandez-Saiz, V.; Taneva, S.G.; Moro, F.; Muga, A. DnaJ recruits DnaK to protein aggregates. J. Biol. Chem. 2008, 283, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Rios, P.D.L.; Ben-Zvi, A.; Slutsky, O.; Azem, A.; Goloubinoff, P. Hsp70 chaperones accelerate protein translocation and the unfolding of stable protein aggregates by entropic pulling. Proc. Natl. Acad. Sci. USA 2006, 103, 6166–6171. [Google Scholar] [CrossRef] [PubMed]
- Sousa, R.; Lafer, E.M. The Physics of Entropic Pulling: A Novel Model for the Hsp70 Motor Mechanism. Int. J. Mol. Sci. 2019, 20, 2334. [Google Scholar] [CrossRef] [PubMed]
- Kostenko, S.; Jensen, K.L.; Moens, U. Phosphorylation of heat shock protein 40 (Hsp40/DnaJB1) by mitogen-activated protein kinase-activated protein kinase 5 (MK5/PRAK). Int. J. Biochem. Cell Biol. 2014, 47, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Prescott, G.R.; Burgoyne, R.D.; Lian, L.-Y.; Morgan, A. Phosphorylation of Cysteine String Protein Triggers a Major Conformational Switch. Structure 2016, 24, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Shirafuji, T.; Ueyama, T.; Adachi, N.; Yoshino, K.I.; Sotomaru, Y.; Uwada, J.; Kaneoka, A.; Ueda, T.; Tanaka, S.; Hide, I.; et al. The Role of Cysteine String Protein alpha Phosphorylation at Serine 10 and 34 by Protein Kinase Cgamma for Presynaptic Maintenance. J. Neurosci. 2018, 38, 278–290. [Google Scholar] [CrossRef]
- Ishihara, K.; Yamagishi, N.; Hatayama, T. Protein kinase CK2 phosphorylates Hsp105 alpha at Ser509 and modulates its function. Biochem. J. 2003, 371, 917–925. [Google Scholar] [CrossRef]
- Zemanovic, S.; Ivanov, M.V.; Ivanova, L.V.; Bhatnagar, A.; Michalkiewicz, T.; Teng, R.-J.; Kumar, S.; Rathore, R.; Pritchard, K.A.; Konduri, G.G.; et al. Dynamic Phosphorylation of the C Terminus of Hsp70 Regulates the Mitochondrial Import of SOD2 and Redox Balance. Cell Rep. 2018, 25, 2605–2616. [Google Scholar] [CrossRef]
- Knowlton, A.A.; Grenier, M.; Kirchhoff, S.R.; Salfity, M. Phosphorylation at tyrosine-524 influences nuclear accumulation of HSP72 with heat stress. Am. J. Physiol. Circ. Physiol. 2000, 278, 2143–2149. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.M.; Taylor, I.R.; Ruggero, D.; Gestwicki, J.E.; Shokat, K.M.; Mukherjee, S. A Legionella pneumophila Kinase Phosphorylates the Hsp70 Chaperone Family to Inhibit Eukaryotic Protein Synthesis. Cell Host Microbe 2019, 25, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Morgner, N.; Schmidt, C.; Beilsten-Edmands, V.; Ebong, I.-O.; Patel, N.A.; Clérico, E.M.; Kirschke, E.; Daturpalli, S.; Jackson, S.E.; Agard, D.; et al. Hsp70 Forms Antiparallel Dimers Stabilized by Post-translational Modifications to Position Clients for Transfer to Hsp90. Cell Rep. 2015, 11, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Ruckova, E.; Halada, P.; Coates, P.J.; Hrstka, R.; Lane, D.P.; Vojtesek, B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to cochaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene 2013, 32, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Lai, K.-C.; Kuo, H.-H.; Chow, L.-P.; Yih, L.-H.; Lee, T.-C. HSP70 colocalizes with PLK1 at the centrosome and disturbs spindle dynamics in cells arrested in mitosis by arsenic trioxide. Arch. Toxicol. 2014, 88, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Truman, A.W.; Kristjansdottir, K.; Wolfgeher, D.; Hasin, N.; Polier, S.; Zhang, H.; Perrett, S.; Prodromou, C.; Jones, G.W.; Kron, S.J. CDK-Dependent Hsp70 Phosphorylation Controls G1 Cyclin Abundance and Cell-Cycle Progression. Cell 2012, 151, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Dean, A.; Ashwini, S.; Sheridan, P.P.; Bhushan, A.; Lai, J.C.; Cao, S.; Daniels, C.K. Identification and characterization of a 66-68-kDa protein as a methotrexate-binding protein in murine leukemia L1210 cells. Cell Stress Chaperones 2013, 18, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.L.; Xu, G.; Tang, H.L.; Zhu, S.Y.; Zhao, L.J.; Ren, H.; Zhao, P.; Qi, Z.T.; Wang, W. Anchoring of both PKA-RIIalpha and 14-3-3theta regulates retinoic acid induced 16 mediated phosphorylation of heat shock protein 70. Oncotarget 2015, 6, 15540–15550. [Google Scholar] [CrossRef]
- O’Regan, L.; Sampson, J.; Richards, M.W.; Knebel, A.; Roth, D.; Hood, F.E.; Straube, A.; Royle, S.J.; Bayliss, R.; Fry, A.M. Hsp72 is targeted to the mitotic spindle by Nek6 to promote K-fiber assembly and mitotic progression. J. Cell Biol. 2015, 209, 349–358. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Andreasson, C.; Barducci, A.; Cheetham, M.E.; Cyr, D.; Emanuelsson, C.; Genevaux, P.; Gestwicki, J.E.; Goloubinoff, P.; Huerta-Cepas, J.; et al. Function, evolution, and structure of J-domain proteins. Cell Stress Chaperones 2019, 24, 7–15. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Mosser, D.D.; Morimoto, R.I. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998, 12, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D.; Morgan, A. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin. Cell Dev. Biol. 2015, 40, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, F.; Nicoll, W.S.; Zimmermann, R.; Cheetham, M.E.; Blatch, G.L. Not all J domains are created equal: Implications for the specificity of Hsp40–Hsp70 interactions. Protein Sci. 2005, 14, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, C.B.; Mastrogiacomo, A.; Faull, K.; Umbach, J.A. Extensive lipidation of a Torpedo cysteine string protein. J. Biol. Chem. 1994, 269, 19197–19199. [Google Scholar] [PubMed]
- Hilger, M.; Bonaldi, T.; Gnad, F.; Mann, M. Systems-wide Analysis of a Phosphatase Knock-down by Quantitative Proteomics and Phosphoproteomics. Mol. Cell. Proteom. 2009, 8, 1908–1920. [Google Scholar] [CrossRef]
- Evans, G.J.O.; Wilkinson, M.C.; Graham, M.E.; Turner, K.M.; Chamberlain, L.H.; Burgoyne, R.D.; Morgan, A. Phosphorylation of Cysteine String Protein by Protein Kinase A. J. Biol. Chem. 2001, 276, 47877–47885. [Google Scholar] [CrossRef]
- Collins, M.O.; Yu, L.; Coba, M.P.; Husi, H.; Campuzano, I.; Blackstock, W.P.; Choudhary, J.S.; Grant, S.G. Proteomic analysis of in vivo phosphorylated synaptic proteins. J. Biol. Chem. 2005, 280, 5972–5982. [Google Scholar] [CrossRef]
- Evans, G.J.; Morgan, A. Phosphorylation-dependent interaction of the synaptic vesicle proteins cysteine string protein and synaptotagmin I. Biochem. J. 2002, 364, 343–347. [Google Scholar] [CrossRef]
- Chiang, N.; Hsiao, Y.T.; Yang, H.J.; Lin, Y.C.; Lu, J.C.; Wang, C.T. Phosphomimetic mutation of cysteine string protein-alpha increases the rate of regulated exocytosis by modulating fusion pore dynamics in PC12 cells. PLoS ONE 2014, 9, e99180. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A Proteome-wide, Quantitative Survey of In Vivo Ubiquitylation Sites Reveals Widespread Regulatory Roles. Mol. Cell. Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef] [PubMed]
- Boal, F.; Zhang, H.; Tessier, C.; Scotti, P.; Lang, J. The Variable C-Terminus of Cysteine String Proteins Modulates Exocytosis and Protein—Protein Interactions†. Biochemistry 2004, 43, 16212–16223. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, P.; Coulombe, B. Regulation of molecular chaperones through post-translational modifications: Decrypting the chaperone code. Biochim. Biophys. Acta 2013, 1829, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Nitika, T.A.W. Cracking the chaperone code: Cellular roles for Hsp70 phosphorylation. Trends Biochem. Sci. 2017, 42, 932–935. [Google Scholar] [CrossRef] [PubMed]
- Beltrao, P.; Albanèse, V.; Kenner, L.R.; Swaney, D.L.; Burlingame, A.; Villén, J.; Lim, W.A.; Fraser, J.S.; Frydman, J.; Krogan, N.J. Systematic Functional Prioritization of Protein Post-translational Modifications. Cell 2012, 150, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Fernandes, J.; Go, Y.-M.; Jones, D.P. Redox dynamics of manganese as a mitochondrial life-death switch. Biochem. Biophys. Res. Commun. 2017, 482, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Culotta, V.C.; Yang, M.; O’Halloran, T.V. Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta 2006, 1763, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Karnati, S.; Lüers, G.; Pfreimer, S.; Baumgart-Vogt, E. Mammalian SOD2 is exclusively located in mitochondria and not present in peroxisomes. Histochem. Cell Biol. 2013, 140, 105–117. [Google Scholar] [CrossRef]
- Afolayan, A.J.; Teng, R.J.; Eis, A.; Rana, U.; Broniowska, K.A.; Corbett, J.A.; Pritchard, K.; Konduri, G.G. Inducible HSP70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L351–L360. [Google Scholar] [CrossRef]
- Afolayan, A.J.; Alexander, M.; Holme, R.L.; Michalkiewicz, T.; Rana, U.; Teng, R.J.; Zemanovic, S.; Sahoo, D.; Pritchard, K.A., Jr.; Konduri, G.G. Domain Mapping of Heat Shock Protein 70 Reveals That Glutamic Acid 446 and Arginine 447 Are Critical for Regulating Superoxide Dismutase 2 Function. J. Biol. Chem. 2017, 292, 2369–2378. [Google Scholar] [CrossRef]
- VanPelt, J.; Page, R.C. Unraveling the CHIP:Hsp70 complex as an information processor for protein quality control. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Finsel, I.; Hilbi, H. Formation of a pathogen vacuole according to Legionella pneumophila: How to kill one bird with many stones. Cell. Microbiol. 2015, 17, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Flayhan, A.; Bergé, C.; Baïlo, N.; Doublet, P.; Bayliss, R.; Terradot, L. The structure of Legionella pneumophila LegK4 type four secretion system (T4SS) effector reveals a novel dimeric eukaryotic-like kinase. Sci. Rep. 2015, 5, 14602. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Richter, K.; Buchner, J. Mixed Hsp90-cochaperone complexes are important for the progression of the reaction cycle. Nat. Struct. Mol. Biol. 2011, 18, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Ebong, I.-O.; Morgner, N.; Zhou, M.; Saraiva, M.A.; Daturpalli, S.; Jackson, S.E.; Robinson, C.V. Heterogeneity and dynamics in the assembly of the Heat Shock Protein 90 chaperone complexes. Proc. Natl. Acad. Sci. USA 2011, 108, 17939–17944. [Google Scholar] [CrossRef]
- Qi, R.; Sarbeng, E.B.; Liu, Q.; Le, K.Q.; Xu, X.; Xu, H.; Yang, J.; Wong, J.L.; Vorvis, C.; Hendrickson, W.A.; et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat. Struct. Mol. Biol. 2013, 20, 900–907. [Google Scholar] [CrossRef]
- Sarbeng, E.B.; Liu, Q.; Tian, X.; Yang, J.; Li, H.; Wong, J.L.; Zhou, L.; Liu, Q. A Functional DnaK Dimer Is Essential for the Efficient Interaction with Hsp40 Heat Shock Protein. J. Biol. Chem. 2015, 290, 8849–8862. [Google Scholar] [CrossRef]
- Hernandez, H.; Robinson, C.V. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat. Protoc. 2007, 2, 715–726. [Google Scholar] [CrossRef]
- Southworth, D.R.; Agard, D.A. Client-Loading Conformation of the Hsp90 Molecular Chaperone Revealed in the Cryo-EM Structure of the Human Hsp90:Hop Complex. Mol. Cell 2011, 42, 771–781. [Google Scholar] [CrossRef]
- Alvira, S.; Cuellar, J.; Röhl, A.; Yamamoto, S.; Itoh, H.; Alfonso, C.; Rivas, G.; Buchner, J.; Valpuesta, J.M. Structural characterization of the substrate transfer mechanism in Hsp70/Hsp90 folding machinery mediated by Hop. Nat. Commun. 2014, 5, 5484. [Google Scholar] [CrossRef]
- Kundrat, L.; Regan, L. Balance between Folding and Degradation for Hsp90-Dependent Client Proteins: A Key Role for CHIP. Biochemistry 2010, 49, 7428–7438. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Girotra, M.; Merchant, N.; Nair, P.; Dutta, S.K. Evidence of Multimeric Forms of HSP70 with Phosphorylation on Serine and Tyrosine Residues—Implications for Roles of HSP70 in Detection of GI Cancers. Asian Pac. J. Cancer Prev. 2013, 14, 5741–5745. [Google Scholar] [CrossRef] [PubMed]
- Saeki, Y.; Tanaka, K. Assembly and Function of the Proteasome. Methods Mol. Biol. 2012, 832, 315–337. [Google Scholar] [PubMed]
- Assimon, V.A.; Southworth, D.R.; Gestwicki, J.E. Specific binding of tetratricopeptide repeat (TPR) proteins to heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) is regulated by affinity and phosphorylation. Biochemistry 2015, 54, 7120–7131. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Matherly, L.H.; Goldman, I.D. Membrane transporters and folate homeostasis: Intestinal absorption and transport into systemic compartments and tissues. Expert Rev. Mol. Med. 2009, 11, e4. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, Y.; Drori, S.; Cole, P.D.; Kamen, B.A.; Sirota, J.; Ifergan, I.; Ben Arush, M.W.; Elhasid, R.; Sahar, D.; Kaspers, G.J.L.; et al. Reduced folate carrier mutations are not the mechanism underlying methotrexate resistance in childhood acute lymphoblastic leukemia. Cancer 2004, 100, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Manola, J.B.; Oh, W.K.; Parslow, D.L.; George, D.J.; Austin, C.L.; Kantoff, P.W. Plasma Levels of Heat Shock Protein 70 in Patients with Prostate Cancer: A Potential Biomarker for Prostate Cancer. Clin. Prostate Cancer 2004, 3, 49–53. [Google Scholar] [CrossRef]
- Chuma, M.; Sakamoto, M.; Yamazaki, K.; Ohta, T.; Ohki, M.; Asaka, M.; Hirohashi, S. Expression profiling in multistage hepatocarcinogenesis: Identification of HSP70 as a molecular marker of early hepatocellular carcinoma. Hepatology 2003, 37, 198–207. [Google Scholar] [CrossRef]
- Kumar, P.; Jha, N.K.; Jha, S.K.; Ramani, K.; Ambasta, R.K. Tau phosphorylation, molecular chaperones, and ubiquitin E3 ligase: Clinical relevance in Alzheimer’s disease. J. Alzheimers Dis. 2015, 43, 341–361. [Google Scholar] [CrossRef]
- Nishi, H.; Hashimoto, K.; Panchenko, A.R. Phosphorylation in protein-protein binding: Effect on stability and function. Structure 2011, 19, 1807–1815. [Google Scholar] [CrossRef]
- Johnson, L.N. The regulation of protein phosphorylation. Biochem. Soc. Trans. 2009, 37, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Presler, M.; Van Itallie, E.; Klein, A.M.; Kunz, R.; Coughlin, M.L.; Peshkin, L.; Gygi, S.P.; Wühr, M.; Kirschner, M.W. Proteomics of phosphorylation and protein dynamics during fertilization and meiotic exit in the Xenopus egg. Proc. Natl. Acad. Sci. USA 2017, 114, E10838–E10847. [Google Scholar] [CrossRef] [PubMed]
- Wieteska, L.; Shahidi, S.; Zhuravleva, A. Allosteric fine-tuning of the conformational equilibrium poises the chaperone BiP for post-translational regulation. eLife 2017, 6, e29430. [Google Scholar] [CrossRef] [PubMed]
- Zanzoni, A.; Carbajo, D.; Diella, F.; Gherardini, P.F.; Tramontano, A.; Helmer-Citterich, M.; Via, A. Phospho3D 2.0: An enhanced database of three-dimensional structures of phosphorylation sites. Nucleic Acids Res. 2011, 39, D268–D271. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lai, A.L.; Clerico, E.M.; Blackburn, M.E.; Patel, N.A.; Robinson, C.V.; Borbat, P.P.; Freed, J.H.; Gierasch, L.M. Key features of an Hsp70 chaperone allosteric landscape revealed by ion-mobility native mass spectrometry and double electron-electron resonance. J. Biol. Chem. 2017, 292, 8773–8785. [Google Scholar] [CrossRef] [PubMed]
- Sikor, M.; Mapa, K.; Von Voithenberg, L.V.; Mokranjac, D.; Lamb, D.C. Real-time observation of the conformational dynamics of mitochondrial Hsp70 by spFRET. EMBO J. 2013, 32, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, A.; Gierasch, L.M. Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc. Natl. Acad. Sci. USA 2015, 112, E2865–E2873. [Google Scholar] [CrossRef] [PubMed]
- Zhuravleva, A.; Clerico, E.M.; Gierasch, L.M. An interdomain energetic tug-of-war creates the allosterically active state in Hsp70 molecular chaperones. Cell 2012, 151, 1296–1307. [Google Scholar] [CrossRef] [PubMed]
- Salazar, C.; Höfer, T. Multisite protein phosphorylation—From molecular mechanisms to kinetic models. FEBS J. 2009, 276, 3177–3198. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Chimenti, M.S.; Pemble, H.; Schönichen, A.; Thompson, O.; Jacobson, M.P.; Wittmann, T. Multisite Phosphorylation Disrupts Arginine-Glutamate Salt Bridge Networks Required for Binding of Cytoplasmic Linker-associated Protein 2 (CLASP2) to End-binding Protein 1 (EB1). J. Biol. Chem. 2012, 287, 17050–17064. [Google Scholar] [CrossRef]
- Ferrell, J.E., Jr.; Ha, S.H. Ultrasensitivity part II: Multisite phosphorylation, stoichiometric inhibitors, and positive feedback. Trends Biochem. Sci. 2014, 39, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. USA 2009, 106, 6591–6596. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, R.; Linial, M. Cooperativity within proximal phosphorylation sites is revealed from large-scale proteomics data. Biol. Direct 2010, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Freschi, L.; Osseni, M.; Landry, C.R. Functional Divergence and Evolutionary Turnover in Mammalian Phosphoproteomes. PLoS Genet. 2014, 10, e1004062. [Google Scholar] [CrossRef] [PubMed]
- Rubin, S.M. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 2013, 38, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Deshong, A.J.; Pelton, J.G.; Rubin, S.M. Phosphorylation-induced Conformational Changes in the Retinoblastoma Protein Inhibit E2F Transactivation Domain Binding. J. Biol. Chem. 2010, 285, 16286–16293. [Google Scholar] [CrossRef]
- Flaherty, K.M.; O’Brien, M.C.; McKay, D.B. Lysine 71 of the Chaperone Protein Hsc70 Is Essential for ATP Hydrolysis. J. Biol. Chem. 1996, 271, 15874–15878. [Google Scholar]
- Xu, X.; Sarbeng, E.B.; Vorvis, C.; Kumar, D.P.; Zhou, L.; Liu, Q. Unique peptide substrate binding properties of 110-kDa heat-shock protein (Hsp110) determine its distinct chaperone activity. J. Biol. Chem. 2012, 287, 5661–5672. [Google Scholar] [CrossRef]
- Kabani, M.; Kelley, S.S.; Morrow, M.W.; Montgomery, D.L.; Sivendran, R.; Rose, M.D.; Gierasch, L.M.; Brodsky, J.L. Dependence of Endoplasmic Reticulum-associated Degradation on the Peptide Binding Domain and Concentration of BiP. Mol. Biol. Cell 2003, 14, 3437–3448. [Google Scholar] [CrossRef]
- Mayer, M.; Schröder, H.; Rüdiger, S.; Paal, K.; Laufen, T.; Bukau, B. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat. Genet. 2000, 7, 586–593. [Google Scholar]
- Rüdiger, S.; Mayer, M.P.; Schneider-Mergener, J.; Bukau, B. Modulation of substrate specificity of the DnaK chaperone by alteration of a hydrophobic arch. J. Mol. Biol. 2000, 304, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Clerico, E.M.; McArthur, N.; Gierasch, L.M. Allosteric landscapes of eukaryotic cytoplasmic Hsp70s are shaped by evolutionary tuning of key interfaces. Proc. Natl. Acad. Sci. USA 2018, 115, 11970–11975. [Google Scholar] [CrossRef] [PubMed]
- Ehrenberger, T.; Cantley, L.C.; Yaffe, M.B. Computational Prediction of Protein-Protein Interactions. Methods Mol. Biol. 2015, 1278, 57–75. [Google Scholar] [PubMed]
- Quintavalle, C.; Di Costanzo, S.; Zanca, C.; Tasset, I.; Fraldi, A.; Incoronato, M.; Mirabelli, P.; Monti, M.; Ballabio, A.; Pucci, P.; et al. Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J. Cell. Physiol. 2014, 229, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Stetz, G.; Tse, A.; Verkhivker, G.M. Dissecting Structure-Encoded Determinants of Allosteric Cross-Talk between Post-Translational Modification Sites in the Hsp90 Chaperones. Sci. Rep. 2018, 8, 6899. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villen, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chaperone | Phosphorylation site(s) | Structural/Functional consequence(s) | Reference |
|---|---|---|---|
| hsDnaJB1 | Ser149, Ser151 and Ser171 | Inhibition of HSF1-mediated transcription | [86] |
| hsCSP (DnaJC5) | Ser10 | Order-to-disorder transition. Modulation of neurotransmitter release by inhibiting binding to syntaxin and synaptotagmin | [87] |
| hsCSP (DnaJC5) | Ser10 and Ser34 | Protection of the presynaptic terminal by promoting HSP70 chaperone activity | [88] |
| hsHsp105α (HSPH1) | Ser509 | Inhibition of the Hsp105-induced suppression of Hsc70-mediated refolding | [89] |
| hsHsp70 (HSPA1A) | Ser631 | Regulation of SOD2 import into the mitochondria and redox balance | [90] |
| hsHsp70 (HSPA1A) | Tyr524 | Enhanced nuclear accumulation and heat-shock injury resistance | [91] |
| hsHsc70 (HSPA8) | Thr495 | Inhibition of Hsp70 ATPase and refolding activities | [92] |
| ecHsp70 (DnaK) | Thr504 | Stabilization of Hsp70 antiparallel dimers to position the client for transfer to Hsp90 | [93] |
| hsHsc70 (HSPA8) | Thr636 | Enhanced interaction with HOP | [94] |
| hsHsc70 (HSPA8) | Ser631 and Ser633 | Recruitment of Hsc70 to the centrosomes leading to mitotic spindle elongation and prevention of apoptosis | [95] |
| scHsp70 (Ssa1) | Thr36 | Regulation of the cell cycle progression | [96] |
| mmHsc70 (Hspa8) | Tyr288 | Cell uptake of methotrexate | [97] |
| hsHsp70 (HSPA1A) | Ser486 | Inhibition of apoptosis | [98] |
| hsHsp70 (HSPA1A) | Thr66 | Promotion of K-fiber assembly and mitotic progression | [99] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velasco, L.; Dublang, L.; Moro, F.; Muga, A. The Complex Phosphorylation Patterns That Regulate the Activity of Hsp70 and Its Cochaperones. Int. J. Mol. Sci. 2019, 20, 4122. https://doi.org/10.3390/ijms20174122
Velasco L, Dublang L, Moro F, Muga A. The Complex Phosphorylation Patterns That Regulate the Activity of Hsp70 and Its Cochaperones. International Journal of Molecular Sciences. 2019; 20(17):4122. https://doi.org/10.3390/ijms20174122
Chicago/Turabian StyleVelasco, Lorea, Leire Dublang, Fernando Moro, and Arturo Muga. 2019. "The Complex Phosphorylation Patterns That Regulate the Activity of Hsp70 and Its Cochaperones" International Journal of Molecular Sciences 20, no. 17: 4122. https://doi.org/10.3390/ijms20174122
APA StyleVelasco, L., Dublang, L., Moro, F., & Muga, A. (2019). The Complex Phosphorylation Patterns That Regulate the Activity of Hsp70 and Its Cochaperones. International Journal of Molecular Sciences, 20(17), 4122. https://doi.org/10.3390/ijms20174122