GRP94 Is Involved in the Lipid Phenotype of Brain Metastatic Cells

 , , ,
, , ,  , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Brain Metastatic Cells Increase Total Fatty Acid (TFA) and Decrease Total Unsaturated Fatty Acid (TUFA) Content
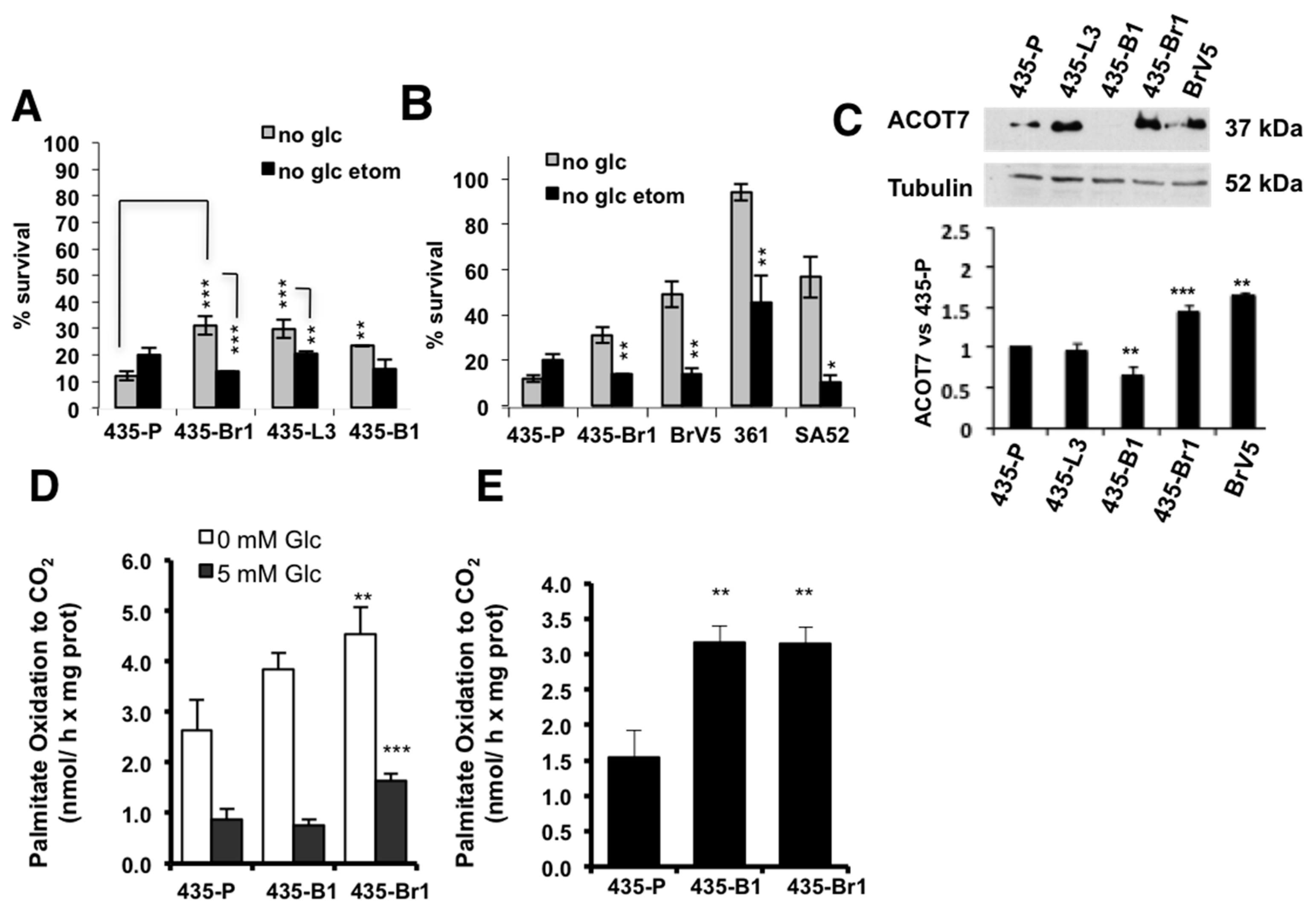
2.2. Inhibition of FA Import to the Mitochondria Impairs BrM Cell Survival after Metabolic Stress
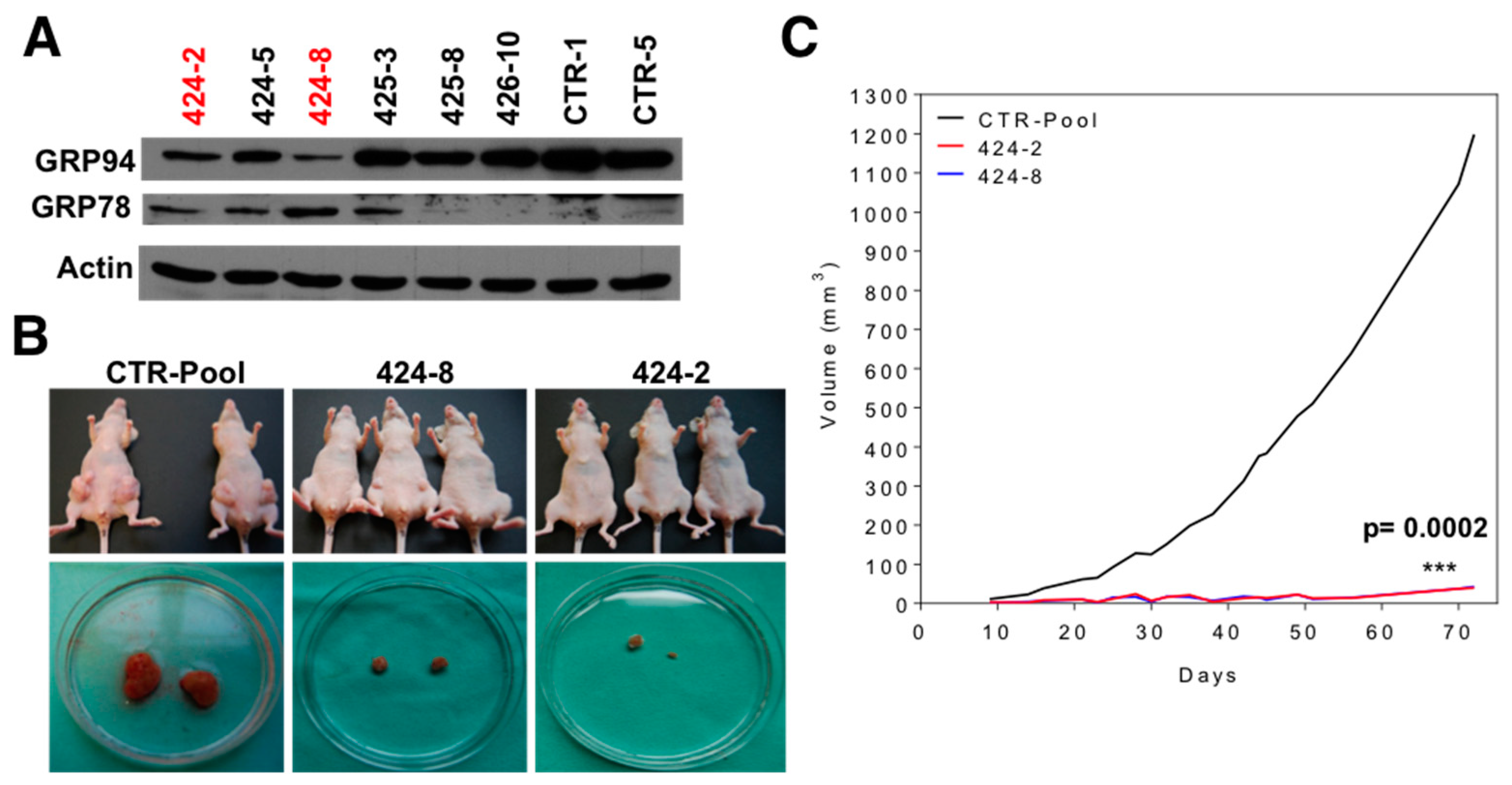
2.3. GRP94 Is Required for Tumor Growth In Vivo and It Modulates the Lipogenic Phenotype of Brain Metastatic Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Cell Viability
4.3. Fluorescent Stainings
4.4. Stable GRP94 Protein Knockdown
4.5. Protein Expression
4.6. 14C-Pamitate Oxidation
4.7. Animal Models
4.8. Raman Spectroscopy
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GRP94 | Glucose-Regulated Protein 94 |
| FA | Fatty Acids |
| ER | Endoplasmic Reticulum |
| UPR | Unfolded Protein Response |
| ERSRP | ER stress-resistance phenotype |
| IRE1 | Inositol-requiring enzyme 1 |
| CNS | Central Nervous system |
| TFA | Total Fatty Acid |
| TUFA | Total Unsaturated Fatty Acid |
| RS | Raman Spectroscopy |
| PCA | Principal Component Analysis |
| SREBP-1 | Sterol Regulatory Element-Binding Protein- 1 |
| LXR-α | Liver X Receptor |
| BrM | Brain metastasis |
| FAO | Fatty Acid Oxidation |
| Mfn | Mitofusin |
| KD | Knock-down |
References
- Maharaj, K.; Que, N.; Hong, F.; Huck, J.; Gill, S.; Wu, S.; Li, Z.; Gewirth, D. Exploring the functional complementation between Grp94 and Hsp90. PLoS ONE 2016, 11, e0166271. [Google Scholar] [CrossRef] [PubMed]
- Hoter, A.; El-Sabban, M.; Naim, H. The HSP90 family: Structure, regulation, function, and implications in health and disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Z. Roles of heat shock protein gp96 in the ER quality control: Redundant or unique function? Mol. Cells 2005, 20, 173–182. [Google Scholar] [PubMed]
- Huck, J.; Que, N.; Hong, F.; Gewirth, D. Structural and functional analysis of GRP94 in the closed state reveals an essential role for the pre-N domain and a potential client-binding site. Cell Rep. 2017, 20, 2800–2809. [Google Scholar] [CrossRef] [PubMed]
- Ansa-Addo, E.; Thaxton, J.; Hong, F.; Wu, B.; Zhang, Y.; Fugle, C.; Metelli, A.; Riesenberg, B.; Williams, K.; Gewirth, D.; et al. Clients and oncogenic roles of molecular chaperone gp96/grp94. Curr. Top. Med. Chem. 2016, 16, 2765–2778. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ashok, B.; Liu, X.; Garikapaty, V.; Mittelman, A.; Tiwari, R. Induction of heat shock protein gp96 by immune cytokines. Cell Stress Chaperones 2003, 8, 242–248. [Google Scholar] [CrossRef]
- Zhu, G.; Lee, A. Role of the unfolded protein response, GRP78 and GRP94 in organ homeostasis. J. Cell Phisiol. 2015, 230, 1413–1420. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Weissman, A.M. The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Clarke, R.; Cook, K.L.; Hu, R.; Facey, C.O.; Tavassoly, I.; Schwartz, J.L.; Baumann, W.T.; Tyson, J.J.; Xuan, J.; Wang, Y.; et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012, 72, 1321–1331. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [Google Scholar] [CrossRef]
- Ghosh, S.; Shinogle, H.; Galeva, N.; Dobrowsky, R.; Blagg, B. Endoplasmic reticulum-resident heat shock protein 90 (HSP90) isoform glucose-regulated protein 94 (GRP94) regulates cell polarity and cancer cell migration by affecting intracellular transport. J. Biol. Chem. 2016, 291, 8309–8323. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, C.; Zhang, K. Role of unfolded protein response in lipogenesis. World J. Hepatol. 2010, 2, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Yang, Y.; Qiu, Z.; Staron, M.; Hong, F.; Li, Y.; Wu, S.; Li, Y.; Hao, B.; Bona, R.; et al. Folding of toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat. Commun. 2010, 1, 79. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, T.; Simmen, T. Endoplasmic reticulum chaperones and oxidoreductases: Critical regulators of tumor cell survival and immunorecognition. Front. Oncol. 2014, 4. [Google Scholar]
- Han, J.; Kaufman, R. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Noto, A.; Pisanu, M.; De Vitis, C.; Maugeri-Saccà, M.; Ciliberto, G. Metabolic features of cancer stem cells: The emerging role of lipid metabolism. Oncogene 2018, 37, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Sharma, N.; Gupta, V.; Durden, B.; Hadad, R.; Banerjee, S.; Dudeja, V.; Saluja, A.; Banerjee, S. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Lin, X.; DeAngelis, L. Treatment of brain metastasis. J. Clin. Oncol. 2015, 33, 3475–3484. [Google Scholar] [CrossRef]
- Martin, B.; Aragues, R.; Sanz, R.; Oliva, B.; Boluda, S.; Martinez, A.; Sierra, A. Biological pathways contributing to organ-specific phenotype of brain metastatic cells. J. Proteome Res. 2008, 7, 908–920. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Pamplona, R.; Aragues, R.; Driouch, K.; Martin, B.; Oliva, B.; Gil, M.; Boluda, S.; Fernandez, P.L.; Martinez, A.; Moreno, V.; et al. Expression of endoplasmic reticulum stress proteins is a candidate marker of brain metastasis in both ErbB-2+ and ErbB-2− primary breast tumors. Am. J. Pathol. 2011, 179, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Aranda, A.; Hernández, V.; Guney, E.; Muixí, L.; Foj, R.; Baixeras, N.; Cuadras, D.; Moreno, V.; Urruticoechea, A.; Gil, M.; et al. FN14 and GRP94 expression are prognostic/predictive biomarkers of brain metastasis outcome that open up new therapeutic strategies. Oncotarget 2015, 6, 44254–44273. [Google Scholar] [CrossRef] [PubMed]
- Landemaine, T.; Jackson, A.; Bellahcene, A.; Rucci, N.; Sin, S.; Abad, B.M.; Sierra, A.; Boudinet, A.; Guinebretiere, J.M.; Ricevuto, E.; et al. A six-gene signature predicting breast cancer lung metastasis. Cancer Res. 2008, 68, 6092–6099. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.; Soto-Pantoja, D.; Clarke, P.; Cruz, M.; Zwart, A.; Wärri, A.; Hilakivi-Clarke, L.; Roberts, D.; Clarke, R. Endoplasmic reticulum stress protein GRP78 modulates lipid metabolism to control drug sensitivity and antitumor immunity in breast cancer. Cancer Res. 2016, 76, 5657–5670. [Google Scholar] [CrossRef]
- Freudiger, C.W.; Min, W.; Saar, B.G.; Lu, S.; Holtom, G.R.; He, C.; Tsai, J.C.; Kang, J.X.; Xie, X.S. Label-free biomedical imaging with high sensitivity by stimulated Raman scattering microscopy. Science 2008, 322, 1857–1861. [Google Scholar] [CrossRef]
- Greenspan, P.; Fowler, S.D. Spectrofluorometric studies of the lipid probe, nile red. J. Lipid Res. 1985, 26, 781–789. [Google Scholar]
- Pineau, L.; Colas, J.; Dupont, S.; Beney, L.; Fleurat-Lessard, P.; Berjeaud, J.M.; Berges, T.; Ferreira, T. Lipid-induced ER stress: Synergistic effects of sterols and saturated fatty acids. Traffic 2009, 10, 673–690. [Google Scholar] [CrossRef]
- Raghu, H.; Sodadasu, P.K.; Malla, R.R.; Gondi, C.S.; Estes, N.; Rao, J.S. Localization of uPAR and MMP-9 in lipid rafts is critical for migration, invasion and angiogenesis in human breast cancer cells. BMC Cancer 2010, 10, 647. [Google Scholar] [CrossRef]
- Kruth, H. Histochemical detection of esterified cholesterol within human atherosclerotic lesions using the fluorescent probe filipin. Atherosclerosis 1984, 51, 281–292. [Google Scholar] [CrossRef]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniels, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Basseri, S.; Austin, R.C. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 2012, 841362. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.N.; Soccio, R.E.; Duncan, E.M.; Sehayek, E.; Breslow, J.L. Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J. Lipid Res. 2003, 44, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, H.; Goldstein, J.L.; Brown, M.S.; Liang, G. LXR-SREBP-1c-phospholipid transfer protein axis controls very low density lipoprotein (VLDL) particle size. J. Biol. Chem. 2010, 285, 6801–6810. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Suto, F.; Suga, T.; Yamada, J. Sterol regulatory element-binding protein-2 modulates human brain acyl-CoA hydrolase gene transcription. Mol. Cell Biochem. 2005, 275, 199–206. [Google Scholar] [CrossRef]
- Liesa, M.; Palacin, M.; Zorzano, A. mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef]
- Legros, F.; Lombes, A.; Frachon, P.; Rojo, M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell. 2002, 13, 4343–4354. [Google Scholar] [CrossRef]
- Dagda, R.K.; Cherra, S.J., 3rd; Kulich, S.M.; Tandon, A.; Park, D.; Chu, C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009, 284, 13843–13855. [Google Scholar] [CrossRef]
- Zorzano, A.; Liesa, M.; Sebastian, D.; Segales, J.; Palacin, M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin. Cell Dev. Biol. 2010, 21, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, D.; Topczewski, F.; Pagliassotti, M.J. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E275–E281. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.; Lee, A.S. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007, 581, 3641–3651. [Google Scholar] [CrossRef] [PubMed]
- Deberardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; White, E. Eat this, not that! How selective autophagy helps cancer cells survive. Mol. Cell. Oncol. 2014, 2, e975638. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Vander Heiden, M.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lee, H.J.; Wu, X.; Huo, L.; Kim, S.J.; Xu, L.; Wang, Y.; He, J.; Bollu, L.R.; Gao, G.; et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015, 75, 554–565. [Google Scholar] [CrossRef]
- Dietschy, J. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar] [CrossRef]
- Villa, G.; Hulce, J.; Zanca, C.; Bi, J.; Ikegami, S.; Cahill, G.; Gu, Y.; Lum, K.; Cravatt, B.; Mischel, P. An LXR-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell 2016, 30, 683–693. [Google Scholar] [CrossRef]
- Nieva, C.; Marro, M.; Santana-Codina, N.; Rao, S.; Petrov, D.; Sierra, A. The lipid phenotype of breast cancer cells characterized by Raman microspectroscopy: Towards a stratification of malignancy. PLoS ONE 2012, 7, e46456. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Cantley, L.C. Decoding key nodes in the metabolism of cancer cells: Sugar & spice and all things nice. F1000 Biol. Rep. 2012, 4, 2. [Google Scholar] [PubMed]
- Kuhajda, F.P.; Jenner, K.; Wood, F.D.; Hennigar, R.A.; Jacobs, L.B.; Dick, J.D.; Pasternack, G.R. Fatty acid synthesis: A potential selective target for antineoplastic therapy. Proc. Natl. Acad. Sci. USA 1994, 91, 6379–6383. [Google Scholar] [CrossRef] [PubMed]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Futur. Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R.; Colomer, R. Inhibition of tumor-associated fatty acid synthase hyperactivity induces synergistic chemosensitization of HER −2/neu-overexpressing human breast cancer cells to docetaxel (taxotere). Breast Cancer Res. Treat. 2004, 84, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Warmoes, M.; Jaspers, J.E.; Xu, G.; Sampadi, B.K.; Pham, T.V.; Knol, J.C.; Piersma, S.R.; Boven, E.; Jonkers, J.; Rottenberg, S.; et al. Proteomics of genetically engineered mouse mammary tumors identifies fatty acid metabolism members as predictive markers for cisplatin resistance. Mol. Cell. Proteomics 2013, 12, 1319–1334. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.E.; Wheeler, F.B.; Davis, A.L.; Thomas, M.J.; Ntambi, J.M.; Seals, D.F.; Kridel, S.J. Metabolic regulation of invadopodia and invasion by acetyl-CoA carboxylase 1 and de novo lipogenesis. PLoS ONE 2012, 7, e29761. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.Y.; Kim, H.M.; Koo, J.S. Expression of lipid metabolism-related proteins in metastatic breast cancer. PLoS ONE 2015, 22, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cheung, H.; Tu, J.; Miu, K.; Chang, W. New insights into the unfolded protein response in stem cells. Oncotarget 2016, 7, 54010–54027. [Google Scholar] [CrossRef] [PubMed]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Van den Eynde, K.; et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018, 9, 2500. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Chou, S.; Yen, M.; Kuo, P. Regulatory mechanism of fatty acid-CoA metabolic enzymes under endoplasmic reticulum stress in lung cancer. Oncol. Rep. 2018, 40, 2674–2682. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Lee, H.; Hwang, H.; Parl, H.; Moon, Y.; Kim, B.; Lee, H.; Kim, K.; Kin, Y.; Lee, B.; et al. Acyl-CoA thioesterase 7 is involved in cell cycle progression via regulation of PKCζ–p53–p21 signaling pathway. Cell Death Dis. 2017, 8, e2793. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.I.; Hewel, J.; Krueger, J.S.; Tiraby, C.; Weber, M.R.; Kralli, A.; Becker, K.; Yates, J.R., 3rd; Felding-Habermann, B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007, 67, 1472–1486. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Lee, C.; Tu, C.; Wu, C.; Huang, M.; Wei, P.; Chang, Y. Glucose-regulated protein 94 mediates progression and metastasis of esophageal squamous cell carcinoma via mitochondrial function and the NF-kB/COX-2/VEGF axis. Oncotarget 2018, 9, 9425–9441. [Google Scholar] [CrossRef]
- Mendez, O.; Fernandez, Y.; Peinado, M.A.; Moreno, V.; Sierra, A. Anti-apoptotic proteins induce non-random genetic alterations that result in selecting breast cancer metastatic cells. Clin. Exp. Metastasis 2005, 22, 297–307. [Google Scholar] [CrossRef]
- Martinez-Aranda, A.; Hernandez, V.; Picon, C.; Modolell, I.; Sierra, A. Development of a preclinical therapeutic model of human brain metastasis with chemoradiotherapy. Int. J. Mol. Sci. 2013, 14, 8306–8327. [Google Scholar] [CrossRef]
- Collins, C.L.; Bode, B.P.; Souba, W.W.; Abcouwer, S.F. Multiwell 14CO2-capture assay for evaluation of substrate oxidation rates of cells in culture. Biotechniques 1998, 24, 803–808. [Google Scholar] [CrossRef]
- Bocklitz, T.; Walter, A.; K, H.; Rösch, P.; Popp, J. How to pre-process raman spectra for reliable and stable models? Anal. Chim. Acta. 2011, 704, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Hilvo, M.; Denkert, C.; Lehtinen, L.; Muller, B.; Brockmoller, S.; Seppanen-Laakso, T.; Budczies, J.; Bucher, E.; Yetukuri, L.; Castillo, S.; et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011, 71, 3236–3245. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Location | Function |
|---|---|---|---|
| ACOT7 | Acyl coenzyme A thioester hydrolase | Cytoplasmic | Acyl-CoA hydrolysis |
| PCYT2 | Ethanolamine-phosphate cytidylyltransferase | Cytoplasmic | Phospholipid biosynthesis |
| SLC25A1 | Tricarboxylate transport protein | Mitochondrial | Citrate/malate exchange, FA synthesis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana-Codina, N.; Marcé-Grau, A.; Muixí, L.; Nieva, C.; Marro, M.; Sebastián, D.; Muñoz, J.P.; Zorzano, A.; Sierra, A. GRP94 Is Involved in the Lipid Phenotype of Brain Metastatic Cells. Int. J. Mol. Sci. 2019, 20, 3883. https://doi.org/10.3390/ijms20163883
Santana-Codina N, Marcé-Grau A, Muixí L, Nieva C, Marro M, Sebastián D, Muñoz JP, Zorzano A, Sierra A. GRP94 Is Involved in the Lipid Phenotype of Brain Metastatic Cells. International Journal of Molecular Sciences. 2019; 20(16):3883. https://doi.org/10.3390/ijms20163883
Chicago/Turabian StyleSantana-Codina, Naiara, Anna Marcé-Grau, Laia Muixí, Claudia Nieva, Mónica Marro, David Sebastián, Juan Pablo Muñoz, Antonio Zorzano, and Angels Sierra. 2019. "GRP94 Is Involved in the Lipid Phenotype of Brain Metastatic Cells" International Journal of Molecular Sciences 20, no. 16: 3883. https://doi.org/10.3390/ijms20163883
APA StyleSantana-Codina, N., Marcé-Grau, A., Muixí, L., Nieva, C., Marro, M., Sebastián, D., Muñoz, J. P., Zorzano, A., & Sierra, A. (2019). GRP94 Is Involved in the Lipid Phenotype of Brain Metastatic Cells. International Journal of Molecular Sciences, 20(16), 3883. https://doi.org/10.3390/ijms20163883