Claudin-7 Modulates Cl− and Na+ Homeostasis and WNK4 Expression in Renal Collecting Duct Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
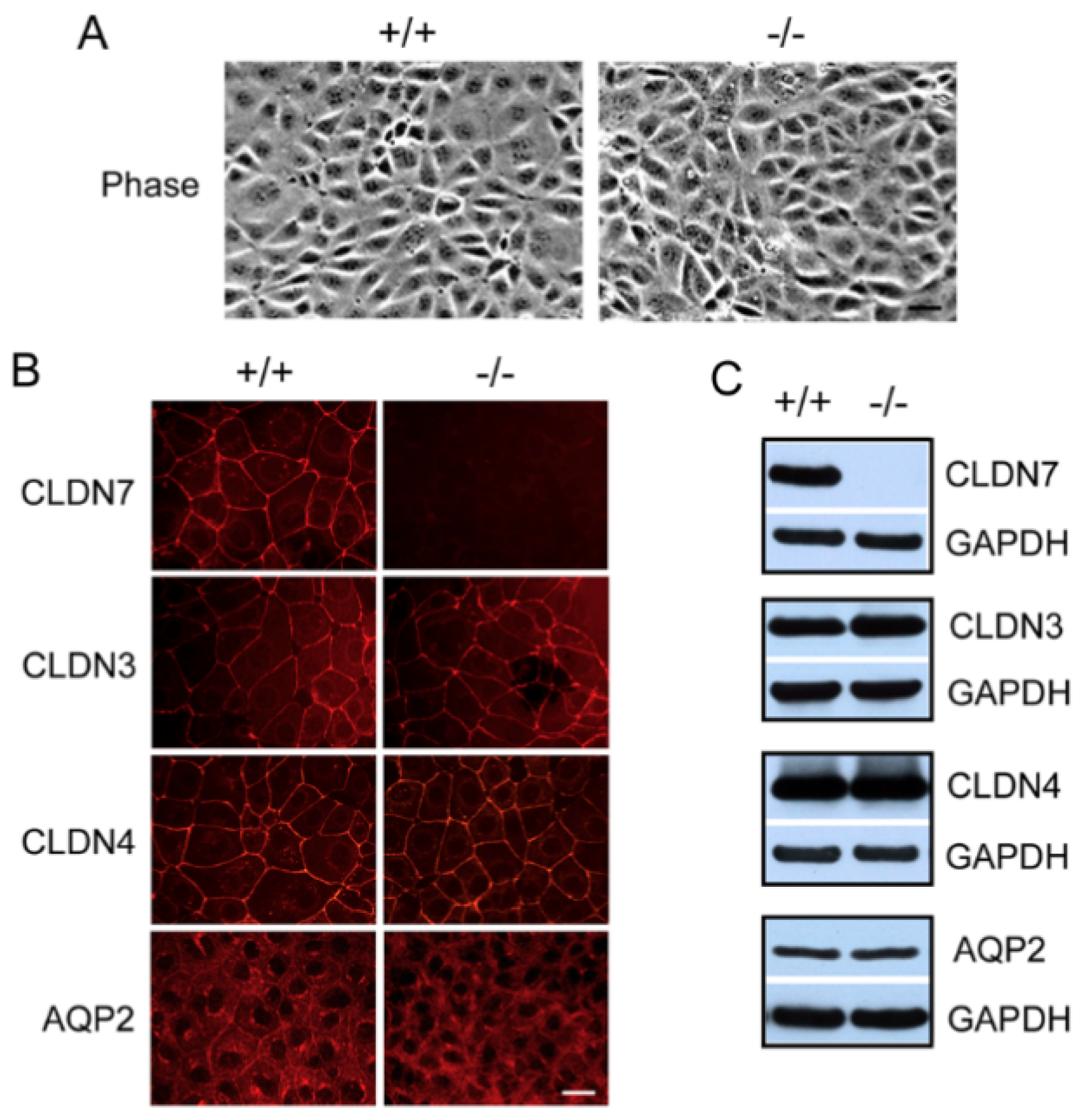
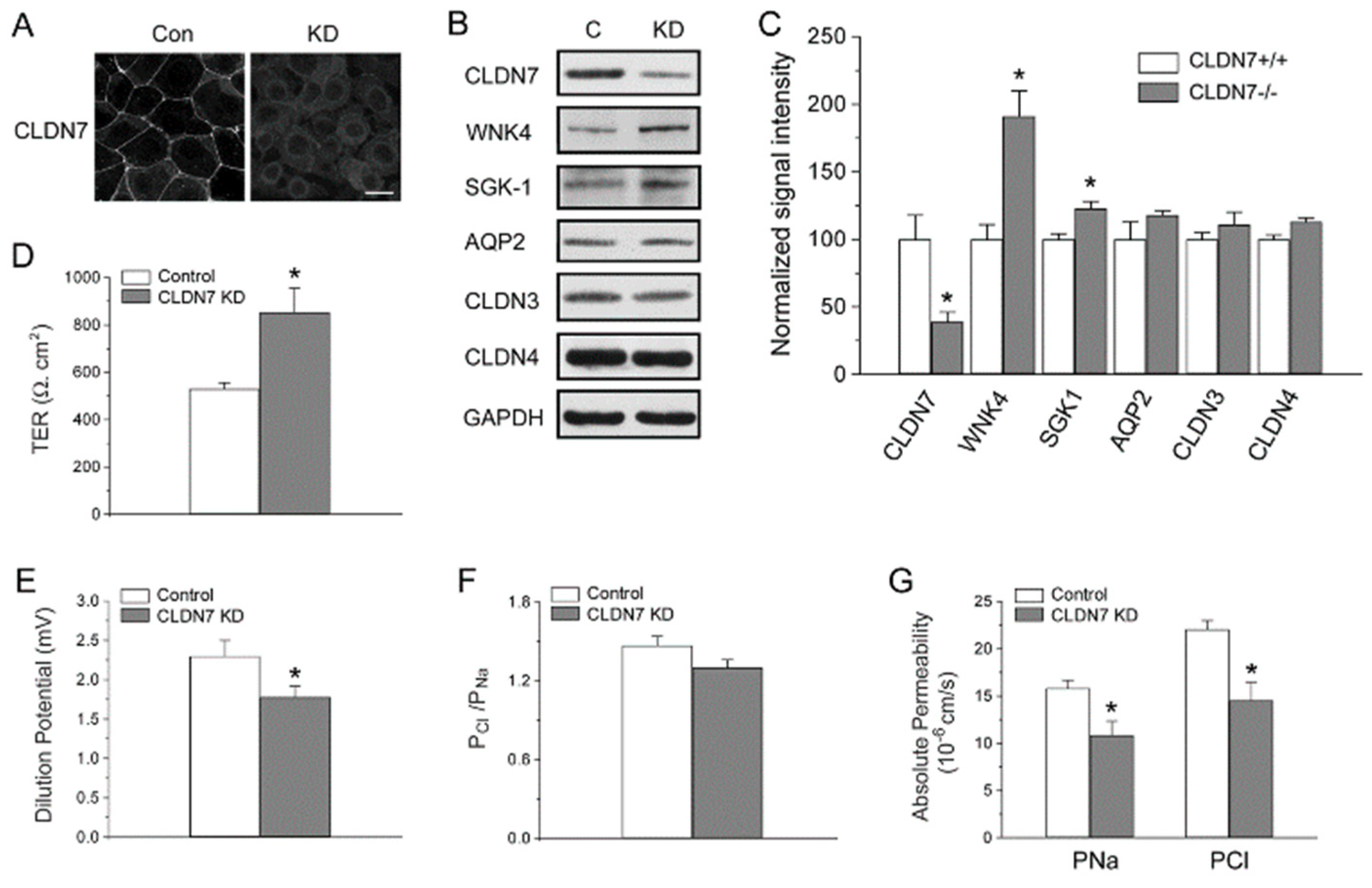
2.1. Generation of CD Cell Lines Isolated from CLDN7+/+ and CLDN7−/− Mouse Kidneys
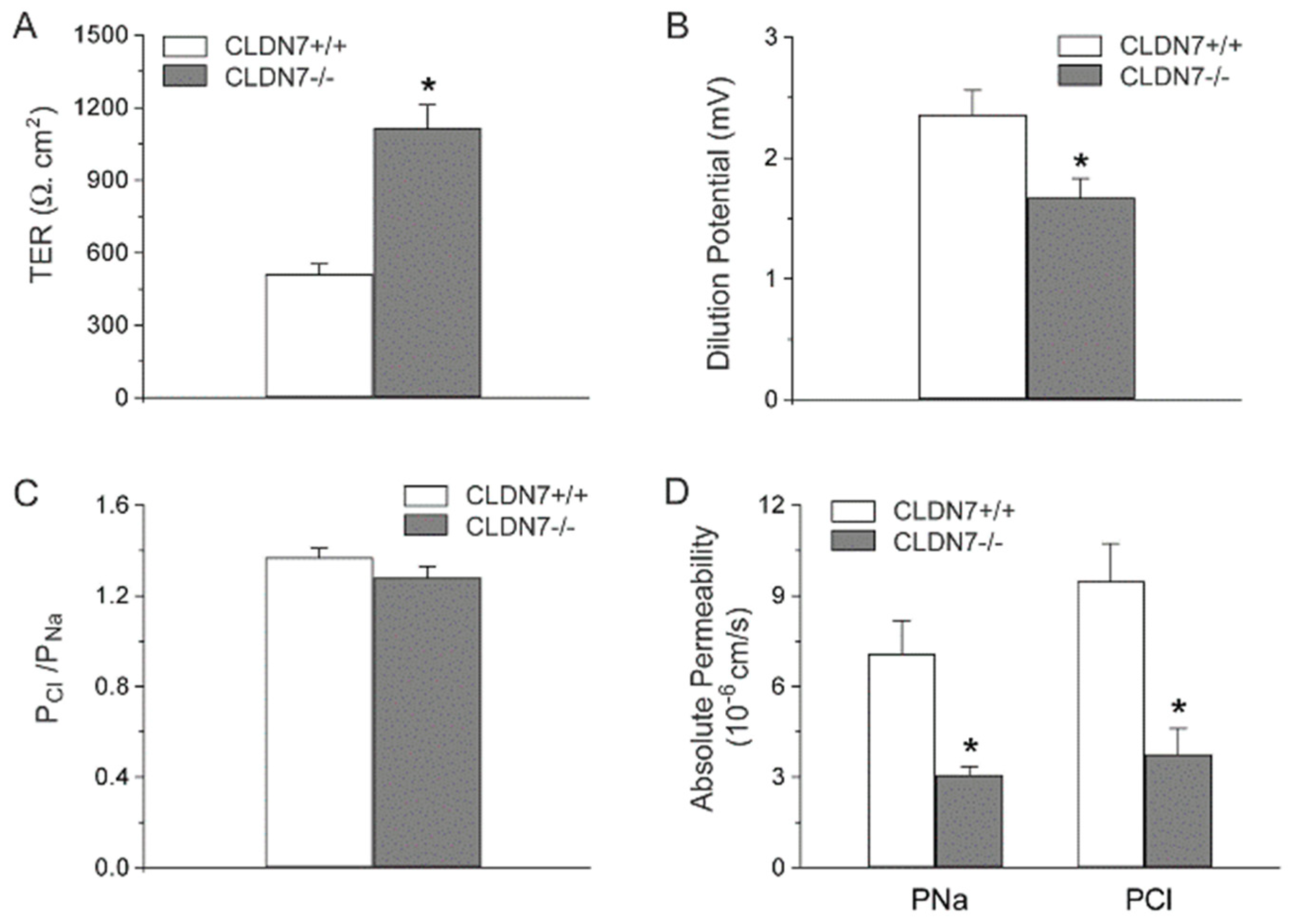
2.2. Decreased Paracellular Cl− and Na+ Permeability in CLDN7−/− CD Cells
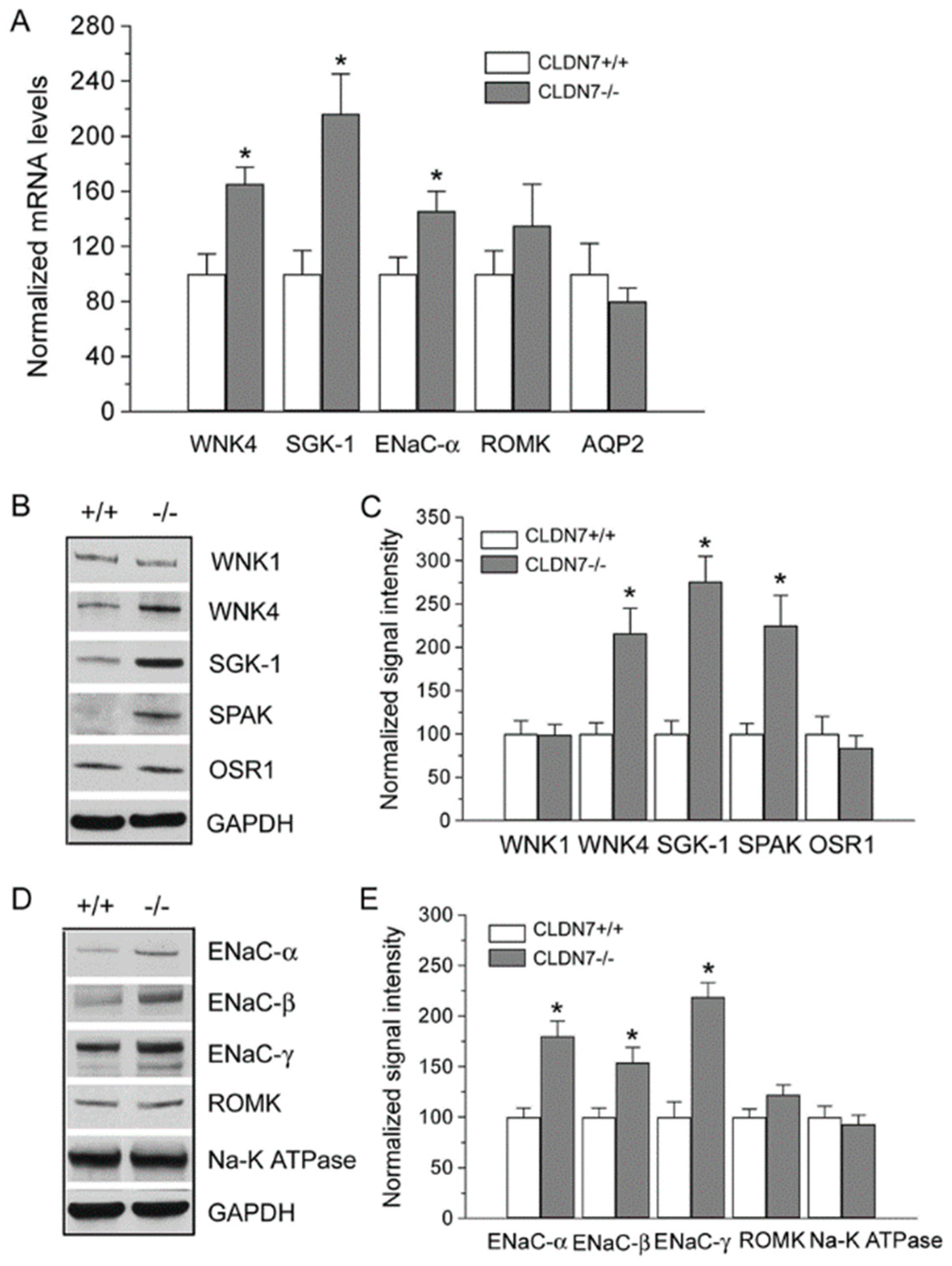
2.3. Increased Expression Levels of WNK4 and ENaC in CLDN7−/− CD Cells
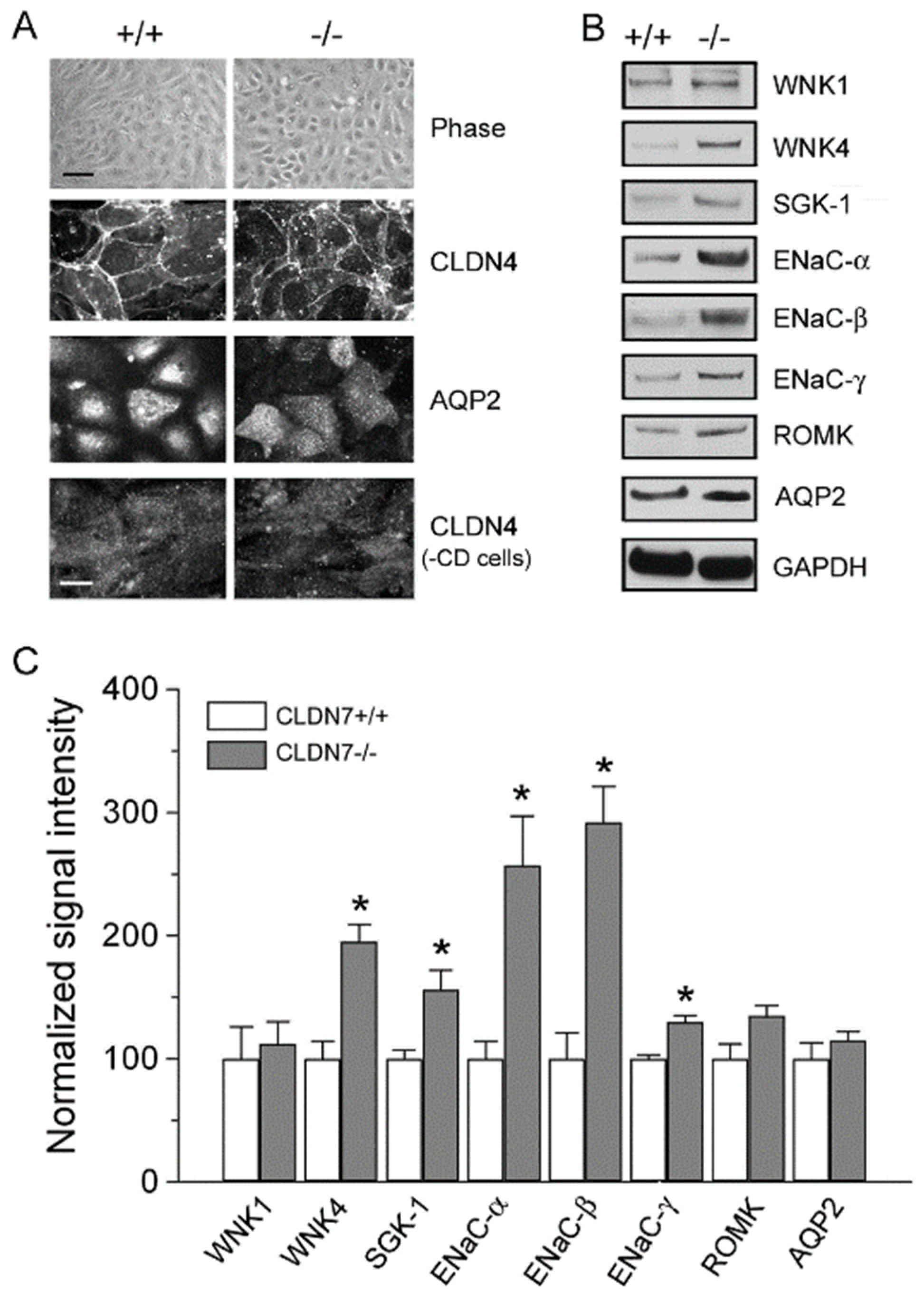
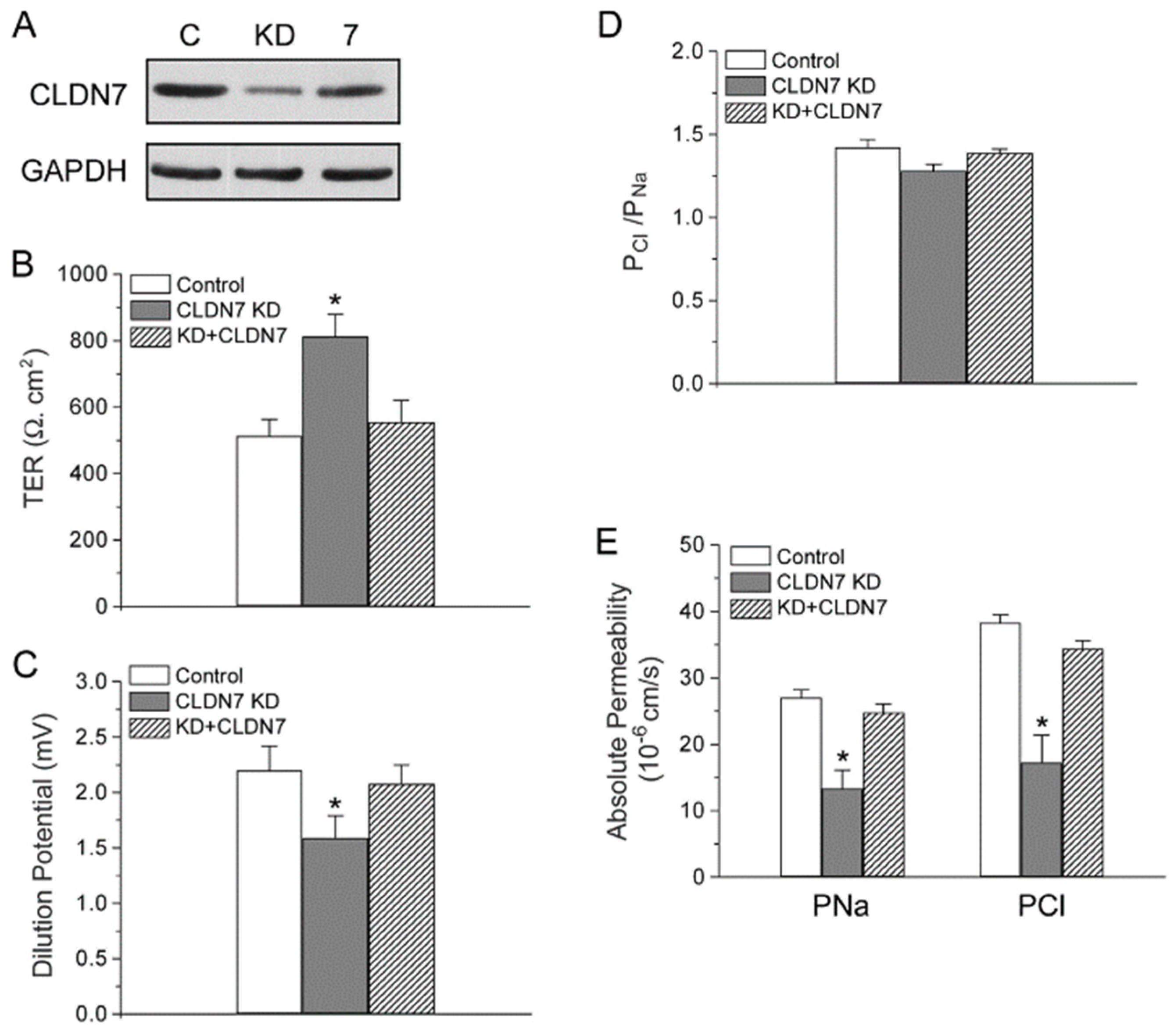
2.4. Rescued Function of Ion Permeability in Immortalized CLDN7+/+ CD Cells with CLDN7 Knockdown
3. Discussion
4. Materials and methods
4.1. Antibodies and Reagents
4.2. Isolation and Immortalization of CD Cells from CLDN7+/+ and CLDN7−/− Mouse Kidneys
4.3. Electrophysiological Measurements
4.4. RNA Extraction and Quantitative Real-Time PCR
4.5. Statistical Analysis
5. Conclusion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sansom, S.C.; Weinman, E.J.; O’Neil, R.G. Microelectrode assessment of chloride-conductive properties of cortical collecting duct. Am. J. Physiol. 1984, 247, F291–F302. [Google Scholar] [CrossRef] [PubMed]
- Schuster, V.L.; Stokes, J.B. Chloride transport by the cortical and outer medullary collecting duct. Am. J. Physiol. 1987, 253, F203–F212. [Google Scholar] [CrossRef] [PubMed]
- Hou, J. Paracellular transport in the collecting duct. Curr. Opin. Nephrol. Hypertens. 2016, 25, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Denker, B.M.; Sabath, E. The biology of epithelial cell tight junctions in the kidney. J. Am. Soc. Nephrol. 2011, 22, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Van Itallie, C.M.; Anderson, J.M. Claudins and epithelial paracellular transport. Annu. Rev. Physiol. 2006, 68, 403–429. [Google Scholar] [CrossRef] [PubMed]
- Balkovetz, D.F. Claudins at the gate: Determinants of renal epithelial tight junction paracellular permeability. Am. J. Physiol. Renal Physiol. 2006, 290, F572–F579. [Google Scholar] [CrossRef] [PubMed]
- Mineta, K.; Yamamoto, Y.; Yamazaki, Y.; Tanaka, H.; Tada, Y.; Saito, K.; Tamura, A.; Igarashi, M.; Endo, T.; Takeuchi, K.; et al. Predicted expansion of the claudin multigene family. FEBS Lett. 2011, 585, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi-Saishin, Y.; Gotoh, S.; Furuse, M.; Takasuga, A.; Tano, Y.; Tsukita, S. Differential expression patterns of claudins, tight junction membrane proteins, in mouse nephron segments. J. Am. Soc. Nephrol. 2002, 13, 875–886. [Google Scholar]
- Konrad, M.; Schaller, A.; Seelow, D.; Pandey, A.V.; Waldegger, S.; Lesslauer, A.; Vitzthum, H.; Suzuki, Y.; Luk, J.M.; Becker, C.; et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am. J. Hum. Genet. 2006, 79, 949–957. [Google Scholar] [CrossRef]
- Simon, D.B.; Lu, Y.; Choate, K.A.; Velazquez, H.; Al-Sabban, E.; Praga, M.; Casari, G.; Bettinelli, A.; Colussi, G.; Rodriguez-Soriano, J.; et al. Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 1999, 285, 103–106. [Google Scholar] [CrossRef]
- Hou, J.; Shan, Q.; Wang, T.; Gomes, A.S.; Yan, Q.; Paul, D.L.; Bleich, M.; Goodenough, D.A. Transgenic RNAi depletion of claudin-16 and the renal handling of magnesium. J. Biol. Chem. 2007, 282, 17114–17122. [Google Scholar] [CrossRef] [PubMed]
- Muto, S.; Hata, M.; Taniguchi, J.; Tsuruoka, S.; Moriwaki, K.; Saitou, M.; Furuse, K.; Sasaki, H.; Fujimura, A.; Imai, M.; et al. Claudin-2-deficient mice are defective in the leaky and cation-selective paracellular permeability properties of renal proximal tubules. Proc. Natl. Acad. Sci. USA 2010, 107, 8011–8016. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Gunzel, D.; Conrad, M.P.; Rosenthal, R.; Fromm, A.; Amasheh, S.; Schulzke, J.D.; Fromm, M. Claudin-17 forms tight junction channels with distinct anion selectivity. Cell Mol. Life Sci. 2012, 69, 2765–2778. [Google Scholar] [CrossRef] [PubMed]
- Breiderhoff, T.; Himmerkus, N.; Stuiver, M.; Mutig, K.; Will, C.; Meij, I.C.; Bachmann, S.; Bleich, M.; Willnow, T.E.; Muller, D. Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc. Natl. Acad. Sci. USA 2012, 109, 14241–14246. [Google Scholar] [CrossRef]
- Breiderhoff, T.; Himmerkus, N.; Drewell, H.; Plain, A.; Gunzel, D.; Mutig, K.; Willnow, T.E.; Muller, D.; Bleich, M. Deletion of claudin-10 rescues claudin-16-deficient mice from hypomagnesemia and hypercalciuria. Kidney Int. 2018, 93, 580–588. [Google Scholar] [CrossRef]
- Furgeson, S.B.; Linas, S. Mechanisms of type I and type II pseudohypoaldosteronism. J. Am. Soc. Nephrol. 2010, 21, 1842–1845. [Google Scholar] [CrossRef]
- Healy, J.K. Pseudohypoaldosteronism type II: History, arguments, answers, and still some questions. Hypertension 2014, 63, 648–654. [Google Scholar] [CrossRef]
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; et al. Human hypertension caused by mutations in WNK kinases. Science 2001, 293, 1107–1112. [Google Scholar] [CrossRef]
- Arroyo, J.P.; Gamba, G. Advances in WNK signaling of salt and potassium metabolism: Clinical implications. Am. J. Nephrol. 2012, 35, 379–386. [Google Scholar] [CrossRef]
- Welling, P.A.; Chang, Y.P.; Delpire, E.; Wade, J.B. Multigene kinase network, kidney transport, and salt in essential hypertension. Kidney Int. 2010, 77, 1063–1069. [Google Scholar] [CrossRef][Green Version]
- Kahle, K.T.; Ring, A.M.; Lifton, R.P. Molecular physiology of the WNK kinases. Annu. Rev. Physiol. 2008, 70, 329–355. [Google Scholar] [CrossRef]
- Wang, W.H.; Giebisch, G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch. 2009, 458, 157–168. [Google Scholar] [CrossRef]
- Kahle, K.T.; Wilson, F.H.; Leng, Q.; Lalioti, M.D.; O’Connell, A.D.; Dong, K.; Rapson, A.K.; MacGregor, G.G.; Giebisch, G.; Hebert, S.C.; et al. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat. Genet. 2003, 35, 372–376. [Google Scholar] [CrossRef]
- Chen, J.C.; Lo, Y.F.; Lin, Y.W.; Lin, S.H.; Huang, C.L.; Cheng, C.J. WNK4 kinase is a physiological intracellular chloride sensor. Proc. Natl. Acad. Sci. USA 2019, 116, 4502–4507. [Google Scholar] [CrossRef]
- Kahle, K.T.; Macgregor, G.G.; Wilson, F.H.; Van Hoek, A.N.; Brown, D.; Ardito, T.; Kashgarian, M.; Giebisch, G.; Hebert, S.C.; Boulpaep, E.L.; et al. Paracellular Cl- permeability is regulated by WNK4 kinase: Insight into normal physiology and hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 14877–14882. [Google Scholar] [CrossRef]
- Yamauchi, K.; Rai, T.; Kobayashi, K.; Sohara, E.; Suzuki, T.; Itoh, T.; Suda, S.; Hayama, A.; Sasaki, S.; Uchida, S. Disease-causing mutant WNK4 increases paracellular chloride permeability and phosphorylates claudins. Proc. Natl. Acad. Sci. USA 2004, 101, 4690–4694. [Google Scholar] [CrossRef]
- Tatum, R.; Zhang, Y.; Lu, Q.; Kim, K.; Jeansonne, B.G.; Chen, Y.H. WNK4 phosphorylates ser(206) of claudin-7 and promotes paracellular Cl(-) permeability. FEBS Lett. 2007, 581, 3887–3891. [Google Scholar] [CrossRef]
- Hou, J.; Renigunta, A.; Yang, J.; Waldegger, S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc. Natl. Acad. Sci. USA 2010, 107, 18010–18015. [Google Scholar] [CrossRef]
- Alexandre, M.D.; Jeansonne, B.G.; Renegar, R.H.; Tatum, R.; Chen, Y.H. The first extracellular domain of claudin-7 affects paracellular Cl- permeability. Biochem. Biophys. Res. Commun. 2007, 357, 87–91. [Google Scholar] [CrossRef]
- Alexandre, M.D.; Lu, Q.; Chen, Y.H. Overexpression of claudin-7 decreases the paracellular Cl- conductance and increases the paracellular Na+ conductance in LLC-PK1 cells. J. Cell. Sci. 2005, 118, 2683–2693. [Google Scholar] [CrossRef]
- Tatum, R.; Zhang, Y.; Salleng, K.; Lu, Z.; Lin, J.J.; Lu, Q.; Jeansonne, B.G.; Ding, L.; Chen, Y.H. Renal salt wasting and chronic dehydration in claudin-7-deficient mice. Am. J. Physiol. Renal Physiol. 2010, 298, F24–34. [Google Scholar] [CrossRef]
- Li, W.Y.; Huey, C.L.; Yu, A.S. Expression of claudin-7 and -8 along the mouse nephron. Am. J. Physiol. Renal Physiol. 2004, 286, F1063–F1071. [Google Scholar] [CrossRef]
- Muto, S.; Yasoshima, K.; Yoshitomi, K.; Imai, M.; Asano, Y. Electrophysiological identification of alpha- and beta-intercalated cells and their distribution along the rabbit distal nephron segments. J. Clin. Invest. 1990, 86, 1829–1839. [Google Scholar] [CrossRef]
- Lu, Z.; Ding, L.; Hong, H.; Hoggard, J.; Lu, Q.; Chen, Y.H. Claudin-7 inhibits human lung cancer cell migration and invasion through ERK/MAPK signaling pathway. Exp. Cell Res. 2011, 317, 1935–1946. [Google Scholar] [CrossRef]
- Light, D.B.; Schwiebert, E.M.; Fejes-Toth, G.; Naray-Fejes-Toth, A.; Karlson, K.H.; McCann, F.V.; Stanton, B.A. Chloride channels in the apical membrane of cortical collecting duct cells. Am. J. Physiol. 1990, 258, F273–F280. [Google Scholar] [CrossRef]
- Lopez-Cayuqueo, K.I.; Chavez-Canales, M.; Pillot, A.; Houillier, P.; Jayat, M.; Baraka-Vidot, J.; Trepiccione, F.; Baudrie, V.; Busst, C.; Soukaseum, C.; et al. A mouse model of pseudohypoaldosteronism type II reveals a novel mechanism of renal tubular acidosis. Kidney Int. 2018, 94, 514–523. [Google Scholar] [CrossRef]
- Susa, K.; Sohara, E.; Rai, T.; Zeniya, M.; Mori, Y.; Mori, T.; Chiga, M.; Nomura, N.; Nishida, H.; Takahashi, D.; et al. Impaired degradation of WNK1 and WNK4 kinases causes PHAII in mutant KLHL3 knock-in mice. Hum. Mol. Genet. 2014, 23, 5052–5060. [Google Scholar] [CrossRef]
- Alvarez de la Rosa, D.; Zhang, P.; Naray-Fejes-Toth, A.; Fejes-Toth, G.; Canessa, C.M. The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J. Biol. Chem. 1999, 274, 37834–37839. [Google Scholar] [CrossRef]
- Faletti, C.J.; Perrotti, N.; Taylor, S.I.; Blazer-Yost, B.L. sgk: An essential convergence point for peptide and steroid hormone regulation of ENaC-mediated Na+ transport. Am. J. Physiol. Cell Physiol. 2002, 282, C494–500. [Google Scholar] [CrossRef]
- Loffing, J.; Zecevic, M.; Feraille, E.; Kaissling, B.; Asher, C.; Rossier, B.C.; Firestone, G.L.; Pearce, D.; Verrey, F. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: Possible role of SGK. Am. J. Physiol. Renal. Physiol. 2001, 280, F675–682. [Google Scholar] [CrossRef]
- Jeansonne, B.; Lu, Q.; Goodenough, D.A.; Chen, Y.H. Claudin-8 interacts with multi-PDZ domain protein 1 (MUPP1) and reduces paracellular conductance in epithelial cells. Cell Mol. Biol. (Noisy-le-grand) 2003, 49, 13–21. [Google Scholar]
- Bens, M.; Duong Van Huyen, J.P.; Cluzeaud, F.; Teulon, J.; Vandewalle, A. CFTR disruption impairs cAMP-dependent Cl(-) secretion in primary cultures of mouse cortical collecting ducts. Am. J. Physiol. Renal Physiol. 2001, 281, F434–442. [Google Scholar] [CrossRef]
- Yu, A.S. Electrophysiological characterization of claudin ion permeability using stably transfected epithelial cell lines. Methods Mol. Biol. 2011, 762, 27–41. [Google Scholar]
- Ding, L.; Lu, Z.; Foreman, O.; Tatum, R.; Lu, Q.; Renegar, R.; Cao, J.; Chen, Y.H. Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology 2012, 142, 305–315. [Google Scholar] [CrossRef]
- Dussault, A.A.; Pouliot, M. Rapid and simple comparison of messenger RNA levels using real-time PCR. Biol. Proced. Online 2006, 8, 1–10. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, J.; Tatum, R.; Hoggard, J.; Chen, Y.-H. Claudin-7 Modulates Cl− and Na+ Homeostasis and WNK4 Expression in Renal Collecting Duct Cells. Int. J. Mol. Sci. 2019, 20, 3798. https://doi.org/10.3390/ijms20153798
Fan J, Tatum R, Hoggard J, Chen Y-H. Claudin-7 Modulates Cl− and Na+ Homeostasis and WNK4 Expression in Renal Collecting Duct Cells. International Journal of Molecular Sciences. 2019; 20(15):3798. https://doi.org/10.3390/ijms20153798
Chicago/Turabian StyleFan, Junming, Rodney Tatum, John Hoggard, and Yan-Hua Chen. 2019. "Claudin-7 Modulates Cl− and Na+ Homeostasis and WNK4 Expression in Renal Collecting Duct Cells" International Journal of Molecular Sciences 20, no. 15: 3798. https://doi.org/10.3390/ijms20153798
APA StyleFan, J., Tatum, R., Hoggard, J., & Chen, Y.-H. (2019). Claudin-7 Modulates Cl− and Na+ Homeostasis and WNK4 Expression in Renal Collecting Duct Cells. International Journal of Molecular Sciences, 20(15), 3798. https://doi.org/10.3390/ijms20153798