Co-Inhibition of the DNA Damage Response and CHK1 Enhances Apoptosis of Neuroblastoma Cells

 ,
,  and
and
Abstract
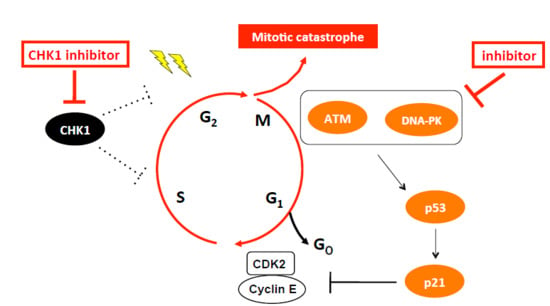
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Cell Viability Assays and Morphological Analysis
2.3. Microarray Analysis of Gene Expression
2.4. Quantitative Reverse-Transcription PCR
2.5. Immunoblotting
2.6. Immunofluorescence Microscopy
2.7. Flow Cytometry
2.8. RNA Interference
2.9. Statistical Analysis
3. Results
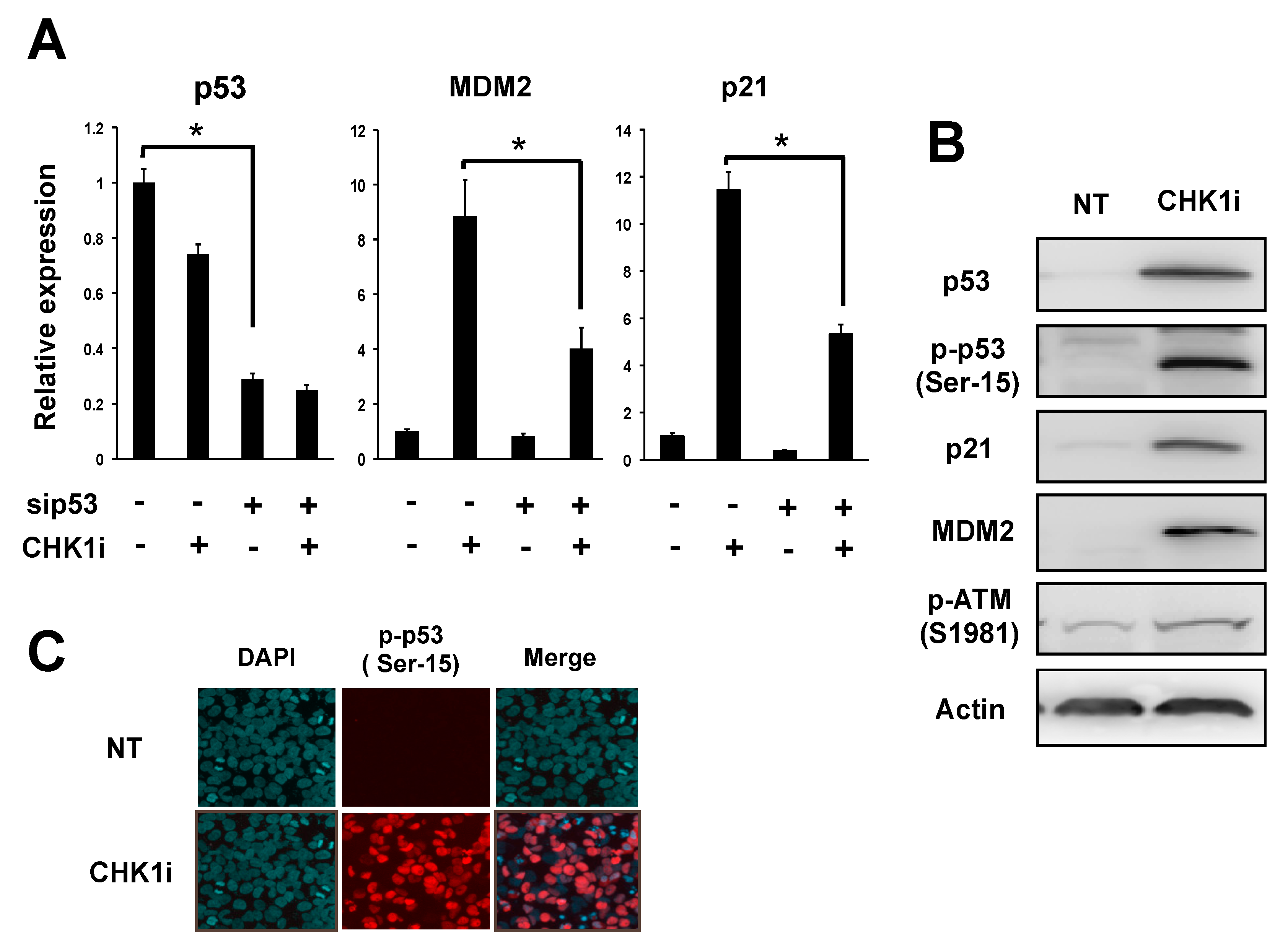
3.1. CHK1 Inhibition Activates Downstream Targets of p53 in NB Cells
3.2. CHK1 Inhibition Upregulates the ATM-p53 Axis in NB Cells
3.3. CHK1 Inhibition Induces DSB-Stimulated DDR Signaling
3.4. Co-Targeting of ATM and DNA-PK Overcomes CHK1 Inhibitor Insensitivity and Induces Apoptosis in NB Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | Ataxia telangiectasia mutated serine/threonine kinase |
| ATMi | ATM inhibitor |
| ATR | Ataxia telangiectasia mutated and Rad3-related serine/threonine kinase |
| CDK | Cyclin-dependent kinase |
| CHK1 | Checkpoint kinase 1 |
| CHK1i | CHK1 inhibitor |
| CHK2 | Checkpoint kinase 2 |
| DDR | DNA damage response |
| DNA-PKi | DNA-PK inhibitor |
| DSB | Double-strand break |
| NB | Neuroblastoma |
| NHEJ | Non-homologous end joining |
| PI | Propidium iodide |
| RS | Replication stress |
| SD | Standard deviation |
References
- Bartek, J.; Lukas, J. DNA repair: Damage alert. Nature 2003, 421, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Balestrini, A.; Garner, E.; Haber, J.E.; Costanzo, V. Mre11-Rad50-Nbs1-dependent processing of DNA breaks generates oligonucleotides that stimulate ATM activity. Embo J. 2008, 27, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Lobrich, M.; Jeggo, P.A. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat. Rev. Cancer 2007, 7, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Merry, C.; Fu, K.; Wang, J.; Yeh, I.J.; Zhang, Y. Targeting the checkpoint kinase Chk1 in cancer therapy. Cell Cycle 2010, 9, 279–283. [Google Scholar] [CrossRef]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Dai, Y.; Grant, S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 376–383. [Google Scholar] [CrossRef]
- Bucher, N.; Britten, C.D. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 2008, 98, 523–528. [Google Scholar] [CrossRef]
- Dixon, H.; Norbury, C.J. Therapeutic exploitation of checkpoint defects in cancer cells lacking p53 function. Cell Cycle 2002, 1, 362–368. [Google Scholar] [CrossRef]
- Kawabe, T. G2 checkpoint abrogators as anticancer drugs. Mol. Cancer Ther. 2004, 3, 513–519. [Google Scholar]
- Hoglund, A.; Nilsson, L.M.; Muralidharan, S.V.; Hasvold, L.A.; Merta, P.; Rudelius, M.; Nikolova, V.; Keller, U.; Nilsson, J.A. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7067–7079. [Google Scholar] [CrossRef]
- Cole, K.A.; Huggins, J.; Laquaglia, M.; Hulderman, C.E.; Russell, M.R.; Bosse, K.; Diskin, S.J.; Attiyeh, E.F.; Sennett, R.; Norris, G.; et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3336–3341. [Google Scholar] [CrossRef]
- Xu, H.; Cheung, I.Y.; Wei, X.X.; Tran, H.; Gao, X.; Cheung, N.K. Checkpoint kinase inhibitor synergizes with DNA-damaging agents in G1 checkpoint-defective neuroblastoma. Int. J. Cancer. 2011, 129, 1953–1962. [Google Scholar] [CrossRef]
- Hong, D.S.; Moore, K.; Patel, M.; Grant, S.C.; Burris, H.A., 3rd; William, W.N., Jr.; Jones, S.; Meric-Bernstam, F.; Infante, J.; Golden, L.; et al. Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 3263–3272. [Google Scholar] [CrossRef]
- Lee, J.M.; Nair, J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Merino, M.J.; Swisher, E.M.; Harrell, M.I.; Trepel, J.B.; Lee, M.J.; et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: A first-in-class proof-of-concept phase 2 study. Lancet Oncol. 2018, 19, 207–215. [Google Scholar] [CrossRef]
- Lin, A.B.; McNeely, S.C.; Beckmann, R.P. Achieving Precision Death with Cell-Cycle Inhibitors that Target DNA Replication and Repair. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3232–3240. [Google Scholar] [CrossRef]
- Islam, A.; Kageyama, H.; Takada, N.; Kawamoto, T.; Takayasu, H.; Isogai, E.; Ohira, M.; Hashizume, K.; Kobayashi, H.; Kaneko, Y.; et al. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene 2000, 19, 617–623. [Google Scholar] [CrossRef]
- Suenaga, Y.; Kaneko, Y.; Matsumoto, D.; Hossain, M.S.; Ozaki, T.; Nakagawara, A. Positive auto-regulation of MYCN in human neuroblastoma. Biochem. Biophys. Res. Commun. 2009, 390, 21–26. [Google Scholar] [CrossRef]
- Niizuma, H.; Nakamura, Y.; Ozaki, T.; Nakanishi, H.; Ohira, M.; Isogai, E.; Kageyama, H.; Imaizumi, M.; Nakagawara, A. Bcl-2 is a key regulator for the retinoic acid-induced apoptotic cell death in neuroblastoma. Oncogene 2006, 25, 5046–5055. [Google Scholar] [CrossRef]
- Li, Z.; Takenobu, H.; Setyawati, A.N.; Akita, N.; Haruta, M.; Satoh, S.; Shinno, Y.; Chikaraishi, K.; Mukae, K.; Akter, J.; et al. EZH2 regulates neuroblastoma cell differentiation via NTRK1 promoter epigenetic modifications. Oncogene 2018, 37, 2714–2727. [Google Scholar] [CrossRef]
- Blasina, A.; Hallin, J.; Chen, E.; Arango, M.E.; Kraynov, E.; Register, J.; Grant, S.; Ninkovic, S.; Chen, P.; Nichols, T.; et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 2008, 7, 2394–2404. [Google Scholar] [CrossRef]
- Zhang, C.; Yan, Z.; Painter, C.L.; Zhang, Q.; Chen, E.; Arango, M.E.; Kuszpit, K.; Zasadny, K.; Hallin, M.; Hallin, J.; et al. PF-00477736 mediates checkpoint kinase 1 signaling pathway and potentiates docetaxel-induced efficacy in xenografts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 4630–4640. [Google Scholar] [CrossRef]
- Agarwal, S.; Milazzo, G.; Rajapakshe, K.; Bernardi, R.; Chen, Z.; Barberi, E.; Koster, J.; Perini, G.; Coarfa, C.; Shohet, J.M. MYCN acts as a direct co-regulator of p53 in MYCN amplified neuroblastoma. Oncotarget 2018, 9, 20323–20338. [Google Scholar]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef]
- Andorfer, P.; Rotheneder, H. EAPP: Gatekeeper at the crossroad of apoptosis and p21-mediated cell-cycle arrest. Oncogene 2011, 30, 2679–2690. [Google Scholar] [CrossRef]
- Derenzini, E.; Agostinelli, C.; Imbrogno, E.; Iacobucci, I.; Casadei, B.; Brighenti, E.; Righi, S.; Fuligni, F.; Ghelli Luserna Di Rora, A.; Ferrari, A.; et al. Constitutive activation of the DNA damage response pathway as a novel therapeutic target in diffuse large B-cell lymphoma. Oncotarget 2015, 6, 6553–6569. [Google Scholar] [CrossRef]
- Kozlov, S.V.; Graham, M.E.; Jakob, B.; Tobias, F.; Kijas, A.W.; Tanuji, M.; Chen, P.; Robinson, P.J.; Taucher-Scholz, G.; Suzuki, K.; et al. Autophosphorylation and ATM activation: Additional sites add to the complexity. J. Biol. Chem. 2011, 286, 9107–9119. [Google Scholar] [CrossRef]
- Bryant, C.; Rawlinson, R.; Massey, A.J. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer 2014, 14, 570. [Google Scholar] [CrossRef]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef]
- Lowery, C.D.; Dowless, M.; Renschler, M.; Blosser, W.; VanWye, A.B.; Stephens, J.R.; Iversen, P.W.; Lin, A.B.; Beckmann, R.P.; Krytska, K.; et al. Broad spectrum activity of the checkpoint kinase 1 inhibitor prexasertib as a single agent or chemopotentiator across a range of preclinical pediatric tumor models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2278–2289. [Google Scholar] [CrossRef]
- Oo, Z.Y.; Stevenson, A.J.; Proctor, M.; Daignault, S.M.; Walpole, S.; Lanagan, C.; Chen, J.; Skalamera, D.; Spoerri, L.; Ainger, S.A.; et al. Endogenous Replication Stress Marks Melanomas Sensitive to CHEK1 Inhibitors In Vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2901–2912. [Google Scholar] [CrossRef]
- Sen, T.; Tong, P.; Stewart, C.A.; Cristea, S.; Valliani, A.; Shames, D.S.; Redwood, A.B.; Fan, Y.H.; Li, L.; Glisson, B.S.; et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res. 2017, 77, 3870–3884. [Google Scholar] [CrossRef]
- Carrassa, L.; Chila, R.; Lupi, M.; Ricci, F.; Celenza, C.; Mazzoletti, M.; Broggini, M.; Damia, G. Combined inhibition of Chk1 and Wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012, 11, 2507–2517. [Google Scholar] [CrossRef]
- Russell, M.R.; Levin, K.; Rader, J.; Belcastro, L.; Li, Y.; Martinez, D.; Pawel, B.; Shumway, S.D.; Maris, J.M.; Cole, K.A. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013, 73, 776–784. [Google Scholar] [CrossRef]
- Sanjiv, K.; Hagenkort, A.; Calderon-Montano, J.M.; Koolmeister, T.; Reaper, P.M.; Mortusewicz, O.; Jacques, S.A.; Kuiper, R.V.; Schultz, N.; Scobie, M.; et al. Cancer-Specific Synthetic Lethality between ATR and CHK1 Kinase Activities. Cell Rep. 2016, 14, 298–309. [Google Scholar] [CrossRef]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Hills, S.A.; Diffley, J.F. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Sorensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef]
- Gadhikar, M.A.; Zhang, J.; Shen, L.; Rao, X.; Wang, J.; Zhao, M.; Kalu, N.N.; Johnson, F.M.; Byers, L.A.; Heymach, J.; et al. CDKN2A/p16 Deletion in Head and Neck Cancer Cells Is Associated with CDK2 Activation, Replication Stress, and Vulnerability to CHK1 Inhibition. Cancer Res. 2018, 78, 781–797. [Google Scholar] [CrossRef]
- Brooks, K.; Oakes, V.; Edwards, B.; Ranall, M.; Leo, P.; Pavey, S.; Pinder, A.; Beamish, H.; Mukhopadhyay, P.; Lambie, D.; et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene 2013, 32, 788–796. [Google Scholar] [CrossRef]
- McNeely, S.; Conti, C.; Sheikh, T.; Patel, H.; Zabludoff, S.; Pommier, Y.; Schwartz, G.; Tse, A. Chk1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle 2010, 9, 995–1004. [Google Scholar] [CrossRef]
- Scagliotti, G.; Kang, J.H.; Smith, D.; Rosenberg, R.; Park, K.; Kim, S.W.; Su, W.C.; Boyd, T.E.; Richards, D.A.; Novello, S.; et al. Phase II evaluation of LY2603618, a first-generation CHK1 inhibitor, in combination with pemetrexed in patients with advanced or metastatic non-small cell lung cancer. Investig. New Drugs 2016, 34, 625–635. [Google Scholar] [CrossRef]
- Wehler, T.; Thomas, M.; Schumann, C.; Bosch-Barrera, J.; Vinolas Segarra, N.; Dickgreber, N.J.; Dalhoff, K.; Sebastian, M.; Corral Jaime, J.; Alonso, M.; et al. A randomized, phase 2 evaluation of the CHK1 inhibitor, LY2603618, administered in combination with pemetrexed and cisplatin in patients with advanced nonsquamous non-small cell lung cancer. Lung Cancer 2017, 108, 212–216. [Google Scholar] [CrossRef]
- Webster, J.A.; Tibes, R.; Morris, L.; Blackford, A.L.; Litzow, M.; Patnaik, M.; Rosner, G.L.; Gojo, I.; Kinders, R.; Wang, L.; et al. Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk. Res. 2017, 61, 108–116. [Google Scholar] [CrossRef]
- Chen, C.C.; Kennedy, R.D.; Sidi, S.; Look, A.T.; D’Andrea, A. CHK1 inhibition as a strategy for targeting Fanconi Anemia (FA) DNA repair pathway deficient tumors. Mol. Cancer 2009, 8, 24. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ando, K.; Nakamura, Y.; Nagase, H.; Nakagawara, A.; Koshinaga, T.; Wada, S.; Makishima, M. Co-Inhibition of the DNA Damage Response and CHK1 Enhances Apoptosis of Neuroblastoma Cells. Int. J. Mol. Sci. 2019, 20, 3700. https://doi.org/10.3390/ijms20153700
Ando K, Nakamura Y, Nagase H, Nakagawara A, Koshinaga T, Wada S, Makishima M. Co-Inhibition of the DNA Damage Response and CHK1 Enhances Apoptosis of Neuroblastoma Cells. International Journal of Molecular Sciences. 2019; 20(15):3700. https://doi.org/10.3390/ijms20153700
Chicago/Turabian StyleAndo, Kiyohiro, Yohko Nakamura, Hiroki Nagase, Akira Nakagawara, Tsugumichi Koshinaga, Satoshi Wada, and Makoto Makishima. 2019. "Co-Inhibition of the DNA Damage Response and CHK1 Enhances Apoptosis of Neuroblastoma Cells" International Journal of Molecular Sciences 20, no. 15: 3700. https://doi.org/10.3390/ijms20153700
APA StyleAndo, K., Nakamura, Y., Nagase, H., Nakagawara, A., Koshinaga, T., Wada, S., & Makishima, M. (2019). Co-Inhibition of the DNA Damage Response and CHK1 Enhances Apoptosis of Neuroblastoma Cells. International Journal of Molecular Sciences, 20(15), 3700. https://doi.org/10.3390/ijms20153700