Cell Autonomous Dysfunction and Insulin Resistance in Pancreatic α Cells

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Physiological Roles of Pancreatic Islet Hormones on Glucose Metabolism
2. Cell Autonomous Dysfunction and Insulin Resistance in Pancreatic α Cells
2.1. Glucose-Regulated Glucagon Secretion in α Cells (Cell Autonomous)
2.2. Insulin Resistance in Pancreatic α Cells
3. Other Islet Cell Factors Regulating Glucagon Secretion
4. Diabetes Therapy Targeting Glucagon
5. Limitations of Pancreatic α Cell Research
6. Conclusions
- Type 2 diabetes is considered to be a “bi-hormonal disorder” rather than an “insulin-centric disorder,” suggesting that glucagon is as important as insulin. Recently, “glucagonocentric hypothesis”, in which glucagon contributes more to increase in blood sugar level than insulin, has attracted attention.
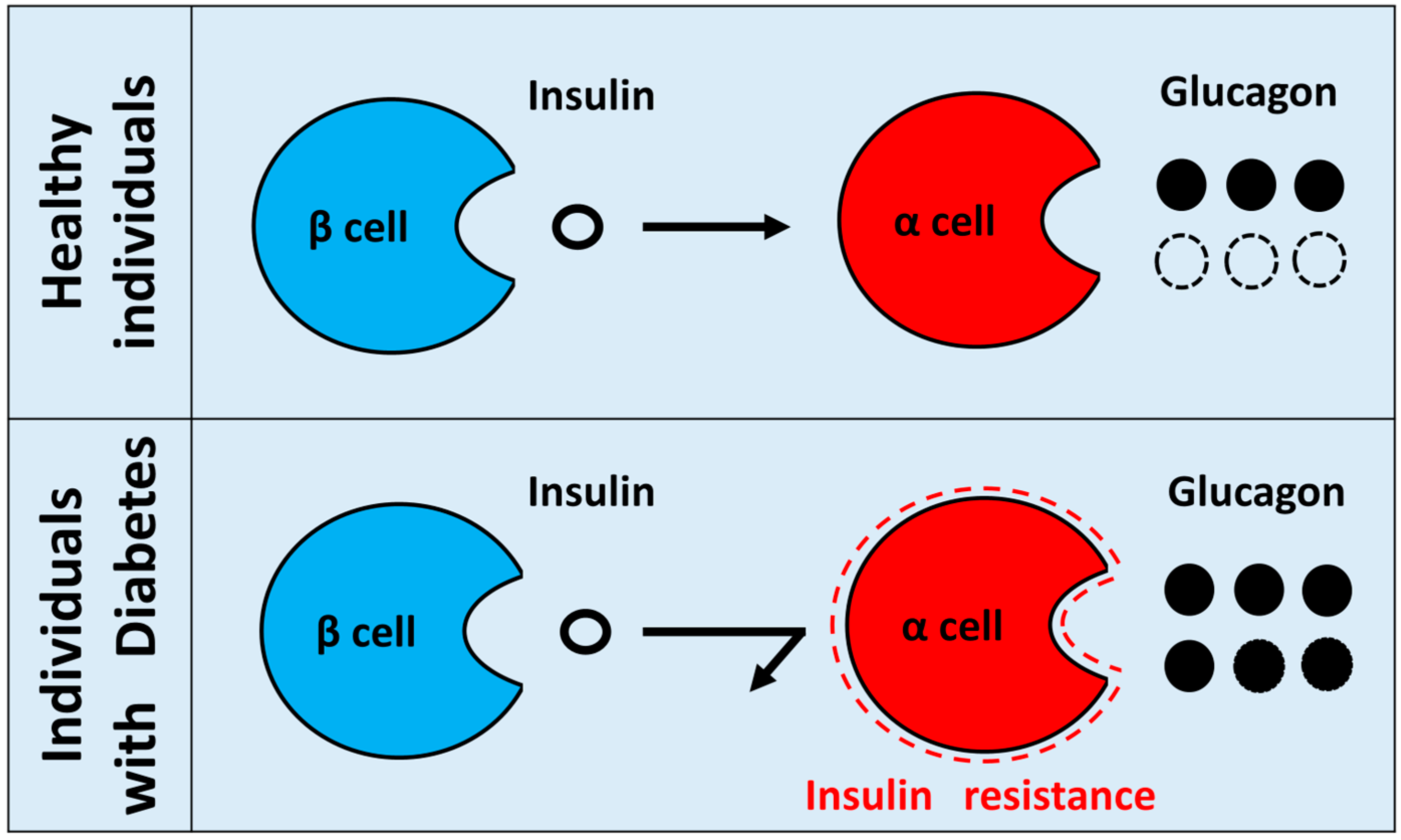
- Insulin resistance in pancreatic α cells means a state in which the insulin signal of α cells is attenuated. In normal conditions, insulin suppresses the secretion of glucagon from pancreatic α cells. However, when insulin resistance exists in diabetic pancreatic α cells, insulin can no longer suppress glucagon secretion from α cells, which results in hypersecretion of glucagon. Hypothetical paradoxical glucagon hypersecretion observed in diabetes.
- The molecular mechanism of glucose-regulated glucagon secretion in pancreatic α-cells has not yet been elucidated, but the involvement of cAMP, ATP/ADP ratio, and KATP channel has been reported.
- The new drugs such as glucagon receptor antagonist and neutralizing antibody are currently developing.
- Glucagon studies have problems with measurement systems and materials. The problem of the measurement system is being solved, but the problem of the material has not been solved yet. Therefore, the establishment of new methods for isolating native α cells or construction of a new α cell line having similar properties with native α cells is required for future α cell research.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| Akt | Protein kinase B |
| AMP | Adenosine monophosphate |
| AMPK | Adenosine monophosphate-activated protein kinase |
| ATP | Adenosine triphosphate |
| cAMP | Cyclic adenosine monophosphate |
| CRTC | Cyclic adenosine monophosphate response element binding protein-regulated transcription coactivator |
| DPP-4 | Dipeptidyl peptidase-4 |
| ELISA | Enzyme-linked immuno sorbent assay |
| EphA | Epherin subtype A |
| ERK | Extracellular signal-regulated kinase |
| FOXO | Forkhead box protein O |
| GABA | Gamma-aminobutyric acid |
| GAD | Glutamic acid decarboxylase |
| GLUT | Glucose transporter |
| GLP-1 | Glucagon-like peptide-1 |
| GPase | Glycogen phosphorylase |
| G6-Pase | Glucose 6-phosphatase |
| IRs | Insulin receptors |
| IRS | Insulin receptor substrate |
| KO | Knockout |
| LC/MS | Liquid chromatography/mass spectrometry |
| MAPK | Mitogen-activated protein kinase |
| PAX | Paired box |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PI3K | Phosphatidylinositol-3 kinase |
| PKA | Protein kinase A |
| PPG | Preproglucagon |
| RIA | Radioimmunoassay |
| SGLT | Sodium glucose co-transporter |
| SSTR | Somatostatin receptor |
| STZ | Streptozotocin |
References
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. North Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A.A. Pancreatic extracts in the treatment of diabetes mellitus. Can. Med. Assoc. J. 1922, 12, 141–146. [Google Scholar] [PubMed]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signaling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Leto, D.; Saltiel, A.R. Regulation of glucose transport by insulin: Traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B. GLUT2, glucose sensing and glucose homeostasis. Diabetologia 2015, 58, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Kimball, C.P.; Murin, J.R. Aqueous extracts of pancreas III. Some precipitation reactions of insulin. J. Biol. Chem. 1923, 58, 337–346. [Google Scholar]
- Liu, Y.; Dentin, R.; Chen, D.; Hedrick, S.; Ravnskjaer, K.; Schenk, S.; Milne, J.; Meyers, D.J.; Cole, P.; Yates, J., III; et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature 2008, 456, 269–273. [Google Scholar] [CrossRef]
- Ravnskjaer, K.; Hogan, M.F.; Lackey, D.; Tora, L.; Dent, S.Y.; Olefsky, J.; Montminy, M. Glucagon regulates gluconeogenesis through KAT2B-and WDR5-mediated epigenetic effects. J. Clin. Investig. 2013, 123, 4318–4328. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yao, W.; Xia, J.; Wang, T.; Huang, F. Glucagon-induced acetylation of energy-sensing factors in control of hepatic metabolism. Int. J. Mol. Sci. 2019, 20, 1885. [Google Scholar] [CrossRef]
- Hancock, A.S.; Du, A.; Liu, J.; Miller, M.; May, C.L. Glucagon deficiency reduces hepatic glucose production and improves glucose tolerance in adult mice. Mol. Endocrinol. 2010, 24, 1605–1614. [Google Scholar] [CrossRef]
- Lee, Y.; Wang, M.Y.; Du, X.Q.; Charron, M.J.; Unger, R.H. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 2011, 60, 391–397. [Google Scholar] [CrossRef]
- Lee, Y.; Berglund, E.D.; Wang, M.Y.; Fu, X.; Yu, X.; Charron, M.J.; Burgess, S.C.; Unger, R.H. Metabolic manifestations of insulin deficiency do not occur without glucagon action. Proc. Natl. Acad. Sci. USA 2012, 109, 14972–14976. [Google Scholar] [CrossRef]
- Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef]
- Maruyama, H.; Tominaga, M.; Bolli, G.; Orci, L.; Unger, R.H. The alpha cell response to glucose change during perfusion of anti-insulin serum in pancreas isolated from normal rats. Diabetologia 1985, 28, 836–840. [Google Scholar] [CrossRef]
- Kawamori, D.; Kurpad, A.J.; Hu, J.; Liew, C.W.; Hih, J.L.; Ford, E.L.; Herrera, P.L.; Polonsky, K.S.; McGuinness, O.P.; Kulkarni, R.N. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab. 2009, 9, 351–361. [Google Scholar] [CrossRef]
- Heimberg, H.; De Vos, A.; Pipeleers, D.; Thorens, B.; Schuit, F. Differences in glucose transporter gene expression between rat pancreatic alpha- and beta- cells are correlated to differences in glucose transport but not in glucose utilization. J. Biol. Chem. 1995, 270, 8971–8975. [Google Scholar] [CrossRef]
- Bonner, C.; Kerr-Conte, J.; Gmyr, V.; Queniat, G.; Moerman, E.; Thevenet, J.; Beaucamps, C.; Delalleau, N.; Popescu, I.; Malaisse, W.J.; et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat. Med. 2015, 21, 512–517. [Google Scholar] [CrossRef]
- Benner, C.; van der Meulen, T.; Caceres, E.; Tigyi, K.; Donaldson, C.J.; Huising, M.O. The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC. Genom. 2014, 15, 620. [Google Scholar] [CrossRef]
- Blodgett, D.M.; Nowosielska, A.; Afik, S.; Pechhold, S.; Cura, A.J.; Kennedy, N.J.; Kim, S.; Kucukural, A.; Davis, R.J.; Kent, S.C.; et al. Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes 2015, 64, 3172–3181. [Google Scholar] [CrossRef]
- Suga, T.; Kikuchi, O.; Kobayashi, M.; Matsui, S.; Yokota-Hashimoto, H.; Wada, E.; Kohno, D.; Sasaki, T.; Takeuchi, K.; Kakizaki, S.; et al. SGLT1 in pancreatic α cells regulates glucagon secretion in mice, possibly explaining the distinct effects of SGLT2 inhibitors on plasma glucagon levels. Mol. Metab. 2019, 19, 1–12. [Google Scholar] [CrossRef]
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; Braun, M.; Brereton, M.; Collins, S.; Galvanovskis, J.; et al. Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013, 18, 871–882. [Google Scholar] [CrossRef]
- Zhang, Q.; Chibalina, M.V.; Bengtsson, M.; Groschner, L.N.; Ramracheya, R.; Rorsman, N.J.; Leiss, V.; Nassar, M.A.; Welling, A.; Gribble, F.M.; et al. Na+ current properties in islet alpha- and beta- cells reflect cell-specific Scn3a and Scn9a expression. J. Physiol. 2014, 592, 4677–4696. [Google Scholar] [CrossRef]
- Barg, S.; Galvanovskis, J.; Göpel, S.O.; Rorsman, P.; Eliasson, L. Tight Coupling Between Electrical Activity and Exocytosis in Mouse Glucagon-Secreting α-Cells. Diabetes 2000, 49, 1500–1510. [Google Scholar] [CrossRef]
- Yu, Q.; Shuai, H.; Ahooghalandari, P.; Gylfe, E.; Tengholm, A. Glucose controls glucagon secretion by directly modulating cAMP in alpha cells. Diabetologia 2019, 62, 1212–1224. [Google Scholar] [CrossRef]
- Knudsen, J.G.; Hamilton, A.; Ramracheya, R.; Tarasov, A.I.; Brereton, M.; Haythorne, E.; Chibalina, M.V.; Spegel, P.; Mulder, H.; Zhang, Q.; et al. Dysregulation of glucagon secretion by hyperglycemia-induced sodium-dependent reduction of ATP production. Cell Metab. 2019, 2019. 29, 430–442. [Google Scholar] [CrossRef]
- Gerich, J.E.; Langlois, M.; Noacco, C.; Karam, J.H.; Forsham, P.H. Lack of glucagon response to hypoglycemia in diabetes: Evidence for an intrinsic pancreatic alpha cell defect. Science 1973, 182, 171–173. [Google Scholar] [CrossRef]
- Unger, R.H.; Orci, L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1975, 1, 14–16. [Google Scholar] [CrossRef]
- Cryer, P.E. Mechanisms of hypoglycemia-associated autonomic failure in diabetes. N. Engl. J. Med. 2013, 369, 362–372. [Google Scholar] [CrossRef]
- Reaven, G. The metabolic syndrome or the insulin resistance syndrome? Different names, different concepts, and different goals. Endocrinol. Metab. Clin. North Am. 2004, 33, 283–303. [Google Scholar] [CrossRef]
- Braaten, J.T.; Faloona, G.R.; Unger, R.H. The Effect of Insulin on the Alpha-Cell Response to Hyperglycemia in Long-Standing Alloxan Diabetes. J. Clin. Investig. 1974, 53, 1017–1021. [Google Scholar] [CrossRef]
- Gromada, J.; Ma, X.; Høy, M.; Bokvist, K.; Salehi, A.; Berggren, P.O.; Rorsman, P. ATP-Sensitive K+ Channel–Dependent Regulation of Glucagon Release and Electrical Activity by Glucose in Wild-Type and SUR1-/- Mouse α-Cells. Diabetes 2004, 53, S181–S189. [Google Scholar] [CrossRef]
- Briant, L.J.B.; Reinbothe, T.M.; Spiliotis, I.; Miranda, C.; Rodriguez, B.; Rorsman, P. Delta-cells and beta-cells are electrically coupled and regulate alpha-cell activity via somatostatin. J. Physiol. 2018, 596, 197–215. [Google Scholar] [CrossRef]
- Elliott, A.D.; Ustione, A.; Piston, D.W. Somatostatin and insulin mediate glucose-inhibited glucagon secretion in the pancreatic alpha-cell by lowering cAMP. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E130–E143. [Google Scholar] [CrossRef]
- Franklin, I.; Gromada, J.; Gjinovci, A.; Theander, S.; Wollheim, C.B. β-Cell secretory products activate α-Cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 2005, 54, 1808–1815. [Google Scholar] [CrossRef]
- Leung, Y.M.; Ahmed, I.; Sheu, L.; Gao, X.; Hara, M.; Tsushima, R.G.; Diamant, N.E.; Gaisano, H.Y. Insulin regulates islet alpha-cell function by reducing KATP channel sensitivity to adenosine 5′-triphosphate inhibition. Endocrinology 2006, 147, 2155–2162. [Google Scholar] [CrossRef]
- Xu, E.; Kumar, M.; Zhang, Y.; Ju, W.; Obata, T.; Zhang, N.; Liu, S.; Wendt, A.; Deng, S.; Ebina, Y.; et al. Intra-islet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab. 2006, 3, 47–58. [Google Scholar] [CrossRef]
- Katsura, T.; Kawamori, D.; Aida, E.; Matsuoka, T.A.; Shimomura, I. Glucotoxicity induces abnormal glucagon secretion through impaired insulin signaling in InR1G cells. PLoS ONE 2017, 12, e0176271. [Google Scholar] [CrossRef]
- Lee, Y.; Berglund, E.D.; Yu, X.; Wang, M.Y.; Evans, M.R.; Scherer, P.E.; Holland, W.L.; Charron, M.J.; Roth, M.G.; Unger, R.H. Hyperglycemia in rodent models of type 2 diabetes requires insulin-resistant alpha cells. Proc. Natl. Acad. Sci. USA 2014, 111, 13217–13222. [Google Scholar] [CrossRef]
- Piro, S.; Maniscalchi, E.T.; Monello, A.; Andini, G.; Mascali, L.G.; Rabuazzo, A.M.; Purrello, F. Palmitate affects insulin receptor phosphorylation and intracellular insulin signal in a pancreatic a-cell line. Endocrinology 2010, 151, 4197–4206. [Google Scholar] [CrossRef]
- Diao, J.; Asghar, Z.; Chan, C.B.; Wheeler, M.B. Glucose-regulated glucagon secretion requires insulin receptor expression in pancreatic alpha-cells. J. Biol. Chem. 2005, 280, 33487–33496. [Google Scholar] [CrossRef]
- Gromada, J.; Franklin, I.; Wollheim, C.B. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr. Rev. 2007, 28, 84–116. [Google Scholar] [CrossRef]
- Stagner, J.I.; Samols, E. Retrograde perfusion as a model for testing the relative effects of glucose versus insulin on the A cell. J. Clin. Investig. 1986, 77, 1034–1037. [Google Scholar] [CrossRef]
- Bosco, D.; Armanet, M.; Morel, P.; Niclauss, N.; Sgroi, A.; Muller, Y.D.; Giovannoni, L.; Parnaud, G.; Berney, T. Unique arrangement of alpha- and beta-cells in human islets of Langerhans. Diabetes 2010, 59, 1202–1210. [Google Scholar] [CrossRef]
- Okada, Y.; Taniguchi, H.; Schimada, C. High concentration of GABA and high glutamate decarboxylase activity in rat pancreatic islets and human insulinoma. Science 1976, 194, 620–622. [Google Scholar] [CrossRef]
- Briel, G.; Gylfe, E.; Hellman, B.; Neuhoff, V. Microdetermination of free amino acids in pancreatic islets isolated from obese-hyperglycemic mice. Acta Physiol. Scand. 1972, 84, 247–253. [Google Scholar] [CrossRef]
- Gylfe, E.; Hellman, B. Role of glucose as a regulator and precursor of amino acids in the pancreatic beta-cells. Endocrinology 1974, 94, 1150–1156. [Google Scholar] [CrossRef]
- Reetz, A.; Solimena, M.; Matteoli, M.; Folli, F.; Takei, K.; De Camilli, P. GABA and pancreatic beta-cells: Colocalization of glutamic acid decarboxylase (GAD) and GABA with synaptic-like microvesicles suggests their role in GABA storage and secretion. Embo J. 1991, 10, 1275–1284. [Google Scholar] [CrossRef]
- Rorsman, P.; Berggren, P.O.; Bokvist, K.; Ericson, H.; Mohler, H.; Ostenson, C.G.; Smith, P.A. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 1989, 341, 233–236. [Google Scholar] [CrossRef]
- Bailey, S.J.; Ravier, M.A.; Rutter, G.A. Glucose-dependent regulation of gamma-aminobutyric acid (GABA A) receptor expression in mouse pancreatic islet alpha-cells. Diabetes 2007, 56, 320–327. [Google Scholar] [CrossRef]
- Ishihara, H.; Maechler, P.; Gjinovci, A.; Herrera, P.L.; Wollheim, C.B. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat. Cell Biol. 2003, 5, 330–335. [Google Scholar] [CrossRef]
- Ravier, M.A.; Rutter, G.A. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha-cells. Diabetes 2005, 54, 1789–1797. [Google Scholar] [CrossRef]
- Nicolson, T.J.; Bellomo, E.A.; Wijesekara, N.; Loder, M.K.; Baldwin, J.M.; Gyulkhandanyan, A.V.; Koshkin, V.; Tarasov, A.I.; Carzaniga, R.; Kronenberger, K.; et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 2009, 58, 2070–2083. [Google Scholar] [CrossRef]
- Hutchens, T.; Piston, D.W. EphA 4 receptor forward signaling inhibits glucagon secretion from αcells. Diabetes 2015, 64, 3839–3851. [Google Scholar] [CrossRef]
- Gerich, J.E.; Langlois, M.; Noacco, C.; Lorenzi, M.; Karam, J.H.; Korsham, P.H. Comparison of the suppressive effects of elevated plasma glucose and free fatty acid levels on glucagon secretion in normal and insulin-dependent diabetic subjects. Evidence for selective alpha-cell insensitivity to glucose in diabetes mellitus. J. Clin. Investig. 1976, 58, 320–325. [Google Scholar] [CrossRef]
- Sakurai, H.; Dobbs, R.; Unger, R.H. Somatostatin-induced changes in insulin and glucagon secretion in normal and diabetic dogs. J. Clin. Investig. 1974, 54, 1395–1402. [Google Scholar] [CrossRef]
- Cejvan, K.; Coy, D.H.; Efendic, S. Intra-islet somatostatin regulates glucagon release via type 2 somatostatin receptors in rats. Diabetes 2003, 52, 1176–1181. [Google Scholar] [CrossRef]
- Starke, A.; Imamura, T.; Unger, R.H. Relationship of glucagon suppression by insulin and somatostatin to the ambient glucose concentration. J. Clin. Investig. 1987, 79, 20–24. [Google Scholar] [CrossRef]
- Strowski, M.Z.; Parmar, R.M.; Blake, A.D.; Schaeffer, J.M. Somatostatin inhibits insulin and glucagon secretion via two receptor subtypes: An in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology 2000, 141, 111–117. [Google Scholar] [CrossRef]
- Chen, L.; Philippe, J.; Unger, R.H. Glucagon responses of isolated alpha cells to glucose, insulin, somatostatin, and leptin. Endocr. Pr. 2011, 17, 819–825. [Google Scholar] [CrossRef]
- Hauge-Evans, A.C.; King, A.J.; Carmignac, D.; Richardson, C.C.; Robinson, I.C.; Low, M.J.; Christie, M.R.; Persaud, S.J.; Jones, P.M. Somatostatin secreted by islet delta-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 2009, 58, 403–411. [Google Scholar] [CrossRef]
- Yue, J.T.; Burdett, E.; Coy, D.H.; Giacca, A.; Efendic, S.; Vranic, M. Somatostatin receptor type 2 antagonism improves glucagon and corticosterone counterregulatory responses to hypoglycemia in streptozotocin-induced diabetic rats. Diabetes 2012, 61, 197–207. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Y.; Gromada, J.; Sewing, S.; Berggren, P.O.; Buschard, K.; Salehi, A.; Vikman, J.; Rorsman, P.; Eliasson, L. Glucagon stimulates exocytosis in mouse and rat pancreatic alpha-cells by binding to glucagon receptors. Mol. Endocrinol. 2005, 19, 198–212. [Google Scholar] [CrossRef]
- Tian, G.; Sandler, S.; Gylfe, E.; Tengholm, A. Glucose- and hormone-induced cAMP oscillations in alpha- and beta-cells within intact pancreatic islets. Diabetes 2011, 60, 1535–1543. [Google Scholar] [CrossRef]
- Liu, Z.; Kim, W.; Chen, Z.; Shin, Y.K.; Carlson, O.D.; Fiori, J.L.; Xin, L.; Napora, J.K.; Short, R.; Odetunde, J.O.; et al. Insulin and glucagon regulate pancreatic alpha-cell proliferation. Plos ONE 2011, 6, e16096. [Google Scholar]
- Leibiger, B.; Moede, T.; Muhandiramlage, T.P.; Kaiser, D.; Vaca Sanchez, P.; Leibiger, I.B.; Berggren, P.O. Glucagon regulates its own synthesis by autocrine signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 20925–20930. [Google Scholar] [CrossRef]
- Hare, K.J.; Vilsboll, T.; Asmar, M.; Deacon, C.F.; Knop, F.K.; Holst, J.J. The glucagonostatic and insulinotropic effects of glucagon-like peptide 1 contribute equally to its glucose-lowering action. Diabetes 2010, 59, 1765–1770. [Google Scholar] [CrossRef]
- De Marinis, Y.Z.; Salehi, A.; Ward, C.E.; Zhang, Q.; Abdulkader, F.; Bengtsson, M.; Braha, O.; Braun, M.; Ramracheya, R.; Amisten, S.; et al. GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab. 2010, 11, 543–553. [Google Scholar] [CrossRef]
- De Heer, J.; Rasmussen, C.; Coy, D.H.; Holst, J.J. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 2008, 51, 2263–2270. [Google Scholar] [CrossRef]
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 818–837. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2014, 494, 256–260. [Google Scholar] [CrossRef]
- Kazda, C.M.; Ding, Y.; Kelly, R.P.; Garhyan, P.; Shi, C.; Lim, C.N.; Fu, H.; Watson, D.E.; Lewin, A.J.; Landschulz, W.H.; et al. Response to comment on kazda et al. evaluation of efficacy and safety of the glucagon receptor antagonist ly2409021 in patients with type 2 diabetes: 12- and 24-week phase 2 studies. Diabetes Care 2016, 39, 1241–1249. [Google Scholar] [CrossRef]
- Kazda, C.M.; Frias, J.; Foga, I.; Cui, X.; Guzman, C.B.; Garhyan, P.; Heilmann, C.; Yang, J.A.; Hardy, T.A. Treatment with the glucagon receptor antagonist LY2409021 increases ambulatory blood pressure in patients with type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1071–1077. [Google Scholar] [CrossRef]
- Guzman, C.B.; Zhang, X.M.; Liu, R.; Regev, A.; Shankar, S.; Garhyan, P.; Pillai, S.G.; Kazda, C.; Chalasani, N.; Hardy, T.A. Treatment with LY2409021, a glucagon receptor antagonist, increases liver fat in patients with type 2 diabetes. Diabetes Obes. Metab. 2017, 19, 1521–1528. [Google Scholar] [CrossRef]
- Sharma, A.X.; Quittner-Strom, E.B.; Lee, Y.; Johnson, J.A.; Martin, S.A.; Yu, X.; Li, J.; Lu, J.; Cai, Z.; Chen, S.; et al. Glucagon receptor antagonism improves glucose metabolism and cardiac function by promoting amp-mediated protein kinase in diabetic mice. Cell Rep. 2018, 22, 1760–1773. [Google Scholar] [CrossRef]
- Takaki, R.; Ono, J.; Nakamura, M.; Yokogawa, Y.; Kumae, S.; Hiraoka, T.; Yamaguchi, K.; Hamaguchi, K.; Uchida, S. Isolation of glucagon-secreting cell lines by cloning insulinoma cells. In Vitro Cell Dev. Biol. 1986, 22, 120–126. [Google Scholar] [CrossRef]
- Shennan, K.I.; Holst, J.J.; Docherty, K. Proglucagon expression, posttranslational processing and secretion in SV40-transformed islet cells. Mol. Cell Endocrinol. 1989, 67, 93–99. [Google Scholar] [CrossRef]
- Hamaguchi, K.; Leiter, E.H. Comparison of cytokine effects on mouse pancreatic alpha-cell and beta-cell lines. Viability, secretory function, and MHC antigen expression. Diabetes 1990, 39, 415–425. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honzawa, N.; Fujimoto, K.; Kitamura, T. Cell Autonomous Dysfunction and Insulin Resistance in Pancreatic α Cells. Int. J. Mol. Sci. 2019, 20, 3699. https://doi.org/10.3390/ijms20153699
Honzawa N, Fujimoto K, Kitamura T. Cell Autonomous Dysfunction and Insulin Resistance in Pancreatic α Cells. International Journal of Molecular Sciences. 2019; 20(15):3699. https://doi.org/10.3390/ijms20153699
Chicago/Turabian StyleHonzawa, Norikiyo, Kei Fujimoto, and Tadahiro Kitamura. 2019. "Cell Autonomous Dysfunction and Insulin Resistance in Pancreatic α Cells" International Journal of Molecular Sciences 20, no. 15: 3699. https://doi.org/10.3390/ijms20153699
APA StyleHonzawa, N., Fujimoto, K., & Kitamura, T. (2019). Cell Autonomous Dysfunction and Insulin Resistance in Pancreatic α Cells. International Journal of Molecular Sciences, 20(15), 3699. https://doi.org/10.3390/ijms20153699