Pathogenic Variants in STXBP1 and in Genes for GABAa Receptor Subunities Cause Atypical Rett/Rett-like Phenotypes

 , , , , and
, , , , and
Abstract
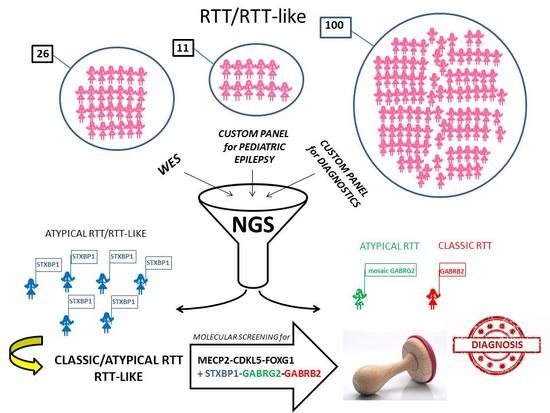
:
1. Introduction
2. Results
2.1. STXBP1 Variants
2.2. Clinical Features of Girls with STXBP1 Variants
2.3. GABAa Receptors Genes Variants
2.4. Clinical Features of Girls with Variants in GABAa Receptors Genes
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Methods
4.2.1. WES
Sequencing Data Analysis
Selection of Potentially Causative Variants
4.2.2. NGS Custom Panel for Pediatric Epilepsy
4.2.3. NGS Custom Panel for Diagnostic Analysis
4.2.4. Variants Validation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RTT | Rett Syndrome |
| MECP2 | Methyl CpG-binding protein 2 |
| CREB1 | cAMP Responsive Element Binding Protein 1 |
| mTOR | Mammalian Target of Rapamycin |
| GABA | Gamma-aminobutyric acid |
| CDKL5 | Cyclin-dependent kinase-like 5 |
| FOXG1 | Forkhead box G1 gene |
| NGS | Next generation sequencing |
| WES | Whole Exome Sequencing |
| DEE | Developmental and Epileptic Encephalopaty |
| ID | Intellectual Disability |
| EE | Epileptic Encephalopaty |
| EOEE | Early Onset Epileptic Encephalopaty |
| EIEE | Early Infantil Epileptic Encephalopaty |
| STXBP1 | Syntaxin-binding protein 1 |
| GABRB2 | Gamma-aminobutyric acid type A receptor beta2 |
| GABRG2 | Gamma-aminobutyric acid type A receptor gamma2 |
| EME | Early Myoclonic Encephalopathy |
| SNARE | Soluble NSF Attachment Protein REceptor |
| Munc18-1 | Mammalian uncoordinated-18-1 |
| CNS | Central Nervous System |
| FS | Febrile Seizures |
| CAE | Childhood Absence Epilepsy |
| GEFS + | Generalized Epilepsy with Febrile Seizures plus |
| DS | Dravet syndrome |
| TM | Transmembrane Domain |
| ER | Endoplasmic Reticulum |
| IECEE2 | Infantile or Early Childhood Epilepsy Encephalopaty type 2 |
| NTS | Nucleus of the solitary tract |
References
- Laurvick, C.L.; de Klerk, N.; Bower, C.; Christodoulou, J.; Ravine, D.; Ellaway, C.; Williamson, S.; Leonard, H. Rett syndrome in Australia: A review of the epidemiology. J. Pediatrics 2006, 148, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Glaze, D.G.; Percy, A.K.; Motil, K.J.; Lane, J.B.; Isaacs, J.S.; Schultz, R.J.; Barrish, J.O.; Neul, J.L.; O’Brien, W.E.; Smith, E.O. A study of the treatment of Rett syndrome with folate and betaine. J. Child. Neurol. 2009, 24, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, F.; Coort, S.L.; Cirillo, E.; Smeets, E.; Evelo, C.T.; Curfs, L.M. Rett syndrome - biological pathways leading from MECP2 to disorder phenotypes. Orphanet J. Rare Dis. 2016, 11, 158. [Google Scholar] [CrossRef] [PubMed]
- Weissman, J.; Naidu, S.; Bjornsson, H.T. Abnormalities of the DNA methylation mark and its machinery: An emerging cause of neurologic dysfunction. Semin. Neurol. 2014, 34, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MECP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.L.; Wang, Z.; Liao, Q.; Zhu, Y.; Zhou, W.H.; Xu, W.; Qiu, Z. MECP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev. Cell 2014, 28, 547–560. [Google Scholar] [CrossRef]
- Bedogni, F.; Rossi, R.L.; Galli, F.; Cobolli Gigli, C.; Gandaglia, A.; Kilstrup-Nielsen, C.; Landsberger, N. Rett syndrome and the urge of novel approaches to study MeCP2 functions and mechanisms of action. Neurosci. Biobehav. Rev. 2014, 46, 187–201. [Google Scholar] [CrossRef]
- Landucci, E.; Brindisi, M.; Bianciardi, L.; Catania, L.M.; Daga, S.; Croci, S.; Frullanti, E.; Fallerini, C.; Butini, S.; Brogi, S.; et al. iPSC -derived neurons profiling reveals GABAergic circuit disruption and acetylated alpha-tubulin defect which improves after iHDAC6 treatment in Rett syndrome. Exp. Cell Res. 2018, 368, 225–235. [Google Scholar] [CrossRef]
- Operto, F.F.; Mazza, R.; Pastorino, G.M.G.; Verrotti, A.; Coppola, G. Epilepsy and genetic in Rett syndrome: A review. Brain Behav. 2019, 9, e01250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percy, A.K. Rett syndrome: Recent research progress. J. Child. Neurol. 2008, 23, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, F.; Sangani, N.B.; Curfs, L.M.G. Current developments in the genetics of Rett and Rett-like syndrome. Curr. Opin. Psychiatry 2018, 31, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Mari, F.; Azimonti, S.; Bertani, I.; Bolognese, F.; Colombo, E.; Caselli, R.; Scala, E.; Longo, I.; Grosso, S.; Pescucci, C.; et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 2005, 14, 1935–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scala, E.; Ariani, F.; Mari, F.; Caselli, R.; Pescucci, C.; Longo, I.; Meloni, I.; Giachino, D.; Bruttini, M.; Hayek, G.; et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 2005, 42, 103–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef]
- Zhu, X.; Petrovski, S.; Xie, P.; Ruzzo, E.K.; Lu, Y.F.; McSweeney, K.M.; Ben-Zeev, B.; Nissenkorn, A.; Anikster, Y.; Oz-Levi, D.; et al. Whole-exome sequencing in undiagnosed genetic diseases: Interpreting 119 trios. Genet. Med. 2015, 17, 774–781. [Google Scholar] [CrossRef]
- Lucariello, M.; Vidal, E.; Vidal, S.; Saez, M.; Roa, L.; Huertas, D.; Pineda, M.; Dalfo, E.; Dopazo, J.; Jurado, P.; et al. Whole exome sequencing of Rett syndrome-like patients reveals the mutational diversity of the clinical phenotype. Hum. Genet. 2016, 135, 1343–1354. [Google Scholar] [CrossRef] [Green Version]
- Percy, A.K.; Lane, J.; Annese, F.; Warren, H.; Skinner, S.A.; Neul, J.L. When Rett syndrome is due to genes other than MECP2. Transl. Sci. Rare Dis. 2018, 3, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Lopes, F.; Barbosa, M.; Ameur, A.; Soares, G.; de Sa, J.; Dias, A.I.; Oliveira, G.; Cabral, P.; Temudo, T.; Calado, E.; et al. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 2016, 53, 190–199. [Google Scholar] [CrossRef]
- Kulikovskaja, L.; Sarajlija, A.; Savic-Pavicevic, D.; Dobricic, V.; Klein, C.; Westenberger, A. WDR45 mutations may cause a MECP2 mutation-negative Rett syndrome phenotype. Neurol. Genet. 2018, 4, e227. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, M.W.; Ravn, K.; Paus, B.; von Tetzchner, S.; Skjeldal, O.H. De novo mutations in SCN1A are associated with classic Rett syndrome: A case report. Bmc Med. Genet. 2018, 19, 184. [Google Scholar] [CrossRef] [PubMed]
- Allou, L.; Julia, S.; Amsallem, D.; El Chehadeh, S.; Lambert, L.; Thevenon, J.; Duffourd, Y.; Saunier, A.; Bouquet, P.; Pere, S.; et al. Rett-like phenotypes: Expanding the genetic heterogeneity to the KCNA2 gene and first familial case of CDKL5-related disease. Clin. Genet. 2017, 91, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Stamberger, H.; Nikanorova, M.; Willemsen, M.H.; Accorsi, P.; Angriman, M.; Baier, H.; Benkel-Herrenbrueck, I.; Benoit, V.; Budetta, M.; Caliebe, A.; et al. STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology 2016, 86, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Myers, C.T.; Cossette, P.; Lemay, P.; Spiegelman, D.; Laporte, A.D.; Nassif, C.; Diallo, O.; Monlong, J.; Cadieux-Dion, M.; et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am. J. Hum. Genet. 2017, 101, 664–685. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE commission for classification and terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, F.F.; Piton, A.; Gauthier, J.; Lortie, A.; Dubeau, F.; Dobrzeniecka, S.; Spiegelman, D.; Noreau, A.; Pellerin, S.; Cote, M.; et al. De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann. Neurol. 2009, 65, 748–753. [Google Scholar] [CrossRef]
- Baldridge, D.; Heeley, J.; Vineyard, M.; Manwaring, L.; Toler, T.L.; Fassi, E.; Fiala, E.; Brown, S.; Goss, C.W.; Willing, M.; et al. The exome clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet. Med. 2017, 19, 1040–1048. [Google Scholar] [CrossRef]
- Sajan, S.A.; Jhangiani, S.N.; Muzny, D.M.; Gibbs, R.A.; Lupski, J.R.; Glaze, D.G.; Kaufmann, W.E.; Skinner, S.A.; Annese, F.; Friez, M.J.; et al. Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet. Med. 2017, 19, 13–19. [Google Scholar] [CrossRef] [Green Version]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Dawidowski, D.; Cafiso, D.S. Munc18-1 and the Syntaxin-1 n terminus regulate open-closed states in a t-SNARE complex. Structure 2016, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, J.; Maroteaux, G.; Schut, D.; Loos, M.; Dubey, M.; Pitsch, J.; Remmelink, E.; Koopmans, B.; Crowley, J.; Cornelisse, L.N.; et al. Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. Brain 2018, 141, 1350–1374. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Q. Defects at the crossroads of GABAergic signaling in generalized genetic epilepsies. Epilepsy Res. 2017, 137, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Toonen, R.F.; Wierda, K.; Sons, M.S.; de Wit, H.; Cornelisse, L.N.; Brussaard, A.; Plomp, J.J.; Verhage, M. Munc18-1 expression levels control synapse recovery by regulating readily releasable pool size. Proc. Natl. Acad. Sci. USA 2006, 103, 18332–18337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitsu, H.; Kato, M.; Shimono, M.; Senju, A.; Tanabe, S.; Kimura, T.; Nishiyama, K.; Yoneda, Y.; Kondo, Y.; Tsurusaki, Y.; et al. Association of genomic deletions in the STXBP1 gene with Ohtahara syndrome. Clin. Genet. 2012, 81, 399–402. [Google Scholar] [CrossRef]
- Mastrangelo, M.; Peron, A.; Spaccini, L.; Novara, F.; Scelsa, B.; Introvini, P.; Raviglione, F.; Faiola, S.; Zuffardi, O. Neonatal suppression-burst without epileptic seizures: Expanding the electroclinical phenotype of STXBP1-related, early-onset encephalopathy. Epileptic Disord. 2013, 15, 55–61. [Google Scholar] [PubMed]
- Aravindhan, A.; Shah, K.; Pak, J.; Veerapandiyan, A. Early-onset epileptic encephalopathy with myoclonic seizures related to 9q33.3-q34.11 deletion involving STXBP1 and SPTAN1 genes. Epileptic Disord. 2018, 20, 214–218. [Google Scholar] [Green Version]
- Deprez, L.; Weckhuysen, S.; Holmgren, P.; Suls, A.; Van Dyck, T.; Goossens, D.; Del-Favero, J.; Jansen, A.; Verhaert, K.; Lagae, L.; et al. Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology 2010, 75, 1159–1165. [Google Scholar] [CrossRef]
- Otsuka, M.; Oguni, H.; Liang, J.S.; Ikeda, H.; Imai, K.; Hirasawa, K.; Imai, K.; Tachikawa, E.; Shimojima, K.; Osawa, M.; et al. STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome--Result of Japanese cohort study. Epilepsia 2010, 51, 2449–2452. [Google Scholar] [CrossRef]
- Carvill, G.L.; Weckhuysen, S.; McMahon, J.M.; Hartmann, C.; Moller, R.S.; Hjalgrim, H.; Cook, J.; Geraghty, E.; O’Roak, B.J.; Petrou, S.; et al. GABRA1 and STXBP1: Novel genetic causes of Dravet syndrome. Neurology 2014, 82, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdan, F.F.; Gauthier, J.; Dobrzeniecka, S.; Lortie, A.; Mottron, L.; Vanasse, M.; D’Anjou, G.; Lacaille, J.C.; Rouleau, G.A.; Michaud, J.L. Intellectual disability without epilepsy associated with STXBP1 disruption. Eur. J. Hum. Genet. 2011, 19, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.M.; Yatsenko, S.A.; Hixson, P.; Reimschisel, T.; Thomas, M.; Wilson, W.; Dayal, U.; Wheless, J.W.; Crunk, A.; Curry, C.; et al. Novel 9q34.11 gene deletions encompassing combinations of four mendelian disease genes: STXBP1, SPTAN1, ENG, and TOR1a. Genet. Med. 2012, 14, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Wieczorek, D.; Graf, E.; Wieland, T.; Endele, S.; Schwarzmayr, T.; Albrecht, B.; Bartholdi, D.; Beygo, J.; Di Donato, N.; et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: An exome sequencing study. Lancet 2012, 380, 1674–1682. [Google Scholar] [CrossRef]
- Khaikin, Y.; Mercimek-Mahmutoglu, S. STXBP1 encephalopathy with epilepsy. In Genereviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews® [Internet]: Washington, DC, USA, 2016. [Google Scholar]
- Yuge, K.; Iwama, K.; Yonee, C.; Matsufuji, M.; Sano, N.; Saikusa, T.; Yae, Y.; Yamashita, Y.; Mizuguchi, T.; Matsumoto, N.; et al. A novel STXBP1 mutation causes typical Rett syndrome in a Japanese girl. Brain Dev. 2018, 40, 493–497. [Google Scholar] [CrossRef]
- Romaniello, R.; Saettini, F.; Panzeri, E.; Arrigoni, F.; Bassi, M.T.; Borgatti, R. A de-novo STXBP1 gene mutation in a patient showing the Rett syndrome phenotype. Neuroreport 2015, 26, 254–257. [Google Scholar] [CrossRef]
- Olson, H.E.; Tambunan, D.; LaCoursiere, C.; Goldenberg, M.; Pinsky, R.; Martin, E.; Ho, E.; Khwaja, O.; Kaufmann, W.E.; Poduri, A. Mutations in epilepsy and intellectual disability genes in patients with features of Rett syndrome. Am. J. Med. Genet. 2015, 167, 2017–2025. [Google Scholar] [CrossRef] [Green Version]
- Allen, N.M.; Conroy, J.; Shahwan, A.; Lynch, B.; Correa, R.G.; Pena, S.D.; McCreary, D.; Magalhaes, T.R.; Ennis, S.; Lynch, S.A.; et al. Unexplained early onset epileptic encephalopathy: Exome screening and phenotype expansion. Epilepsia 2016, 57, e12–e17. [Google Scholar] [CrossRef]
- Fitzgerald, T.W.; Gerety, S.S.; Jones, W.D.; van Kogelenberg, M.; King, D.A.; McRae, J.; Morley, K.I.; Parthiban, V.; Al-Turki, S.; Ambridge, K.; et al. Deciphering Developmental Disorders, S. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar]
- Barcia, G.; Chemaly, N.; Gobin, S.; Milh, M.; Van Bogaert, P.; Barnerias, C.; Kaminska, A.; Dulac, O.; Desguerre, I.; Cormier, V.; et al. Early epileptic encephalopathies associated with STXBP1 mutations: Could we better delineate the phenotype? Eur. J. Med. Genet. 2014, 57, 15–20. [Google Scholar] [CrossRef]
- Keogh, M.J.; Daud, D.; Pyle, A.; Duff, J.; Griffin, H.; He, L.; Alston, C.L.; Steele, H.; Taggart, S.; Basu, A.P.; et al. A novel de novo STXBP1 mutation is associated with mitochondrial complex I deficiency and late-onset juvenile-onset parkinsonism. Neurogenetics 2015, 16, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.E.; Kelly, M.; LaCoursiere, C.M.; Pinsky, R.; Tambunan, D.; Shain, C.; Ramgopal, S.; Takeoka, M.; Libenson, M.H.; Julich, K.; et al. Genetics and genotype-phenotype correlations in early onset epileptic encephalopathy with burst suppression. Ann. Neurol. 2017, 81, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Ohashi, T.; Akasaka, N.; Tohyama, J. Congenital variant of Rett syndrome due to an intragenic large deletion in MECP2. Brain Dev. 2012, 34, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Rajaei, S.; Erlandson, A.; Kyllerman, M.; Albage, M.; Lundstrom, I.; Karrstedt, E.L.; Hagberg, B. Early infantile onset “congenital” Rett syndrome variants: Swedish experience through four decades and mutation analysis. J. Child. Neurol. 2011, 26, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Einspieler, C.; Marschik, P.B. Regression in Rett syndrome: Developmental pathways to its onset. Neurosci. Biobehav. Rev. 2019, 98, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Kaminska, A.; Boddaert, N.; Rio, M.; Afenjar, A.; Gerard, M.; Giuliano, F.; Motte, J.; Heron, D.; Morel, M.A.; et al. The three stages of epilepsy in patients with CDKL5 mutations. Epilepsia 2008, 49, 1027–1037. [Google Scholar] [CrossRef]
- Pintaudi, M.; Calevo, M.G.; Vignoli, A.; Parodi, E.; Aiello, F.; Baglietto, M.G.; Hayek, Y.; Buoni, S.; Renieri, A.; Russo, S.; et al. Epilepsy in Rett syndrome: Clinical and genetic features. Epilepsy Behav. 2010, 19, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Rezazadeh, A.; Uddin, M.; Snead, O.C., 3rd; Lira, V.; Silberberg, A.; Weiss, S.; Donner, E.J.; Zak, M.; Bradbury, L.; Scherer, S.W.; et al. STXBP1 encephalopathy is associated with awake bruxism. Epilepsy Behav. 2019, 92, 121–124. [Google Scholar] [CrossRef]
- Sarto-Jackson, I.; Sieghart, W. Assembly of GABA(a) receptors (review). Mol. Membr. Biol. 2008, 25, 302–310. [Google Scholar] [CrossRef]
- Kang, J.Q.; MacDonald, R.L. Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to Dravet syndrome. Jama Neurol. 2016, 73, 1009–1016. [Google Scholar] [CrossRef]
- Alldred, M.J.; Mulder-Rosi, J.; Lingenfelter, S.E.; Chen, G.; Luscher, B. Distinct gamma2 subunit domains mediate clustering and synaptic function of postsynaptic GABAa receptors and gephyrin. J. Neurosci. 2005, 25, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.C.; Kong, W.; Hu, N.; Zhang, Y.; Shen, W.; Jackson, L.; Liu, X.; Jiang, Y.; Macdonald, R.L. Altered channel conductance states and gating of GABAa receptors by a pore mutation linked to Dravet syndrome. eNeuro 2017, 4. [Google Scholar] [CrossRef]
- Shen, D.; Hernandez, C.C.; Shen, W.; Hu, N.; Poduri, A.; Shiedley, B.; Rotenberg, A.; Datta, A.N.; Leiz, S.; Patzer, S.; et al. De novo GABRG2 mutations associated with epileptic encephalopathies. Brain 2017, 140, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; McWalter, K.; Schmidt, L.; Decker, A.; Picker, J.D.; Lincoln, S.; Sweetser, D.A.; Briere, L.C.; Harini, C.; Members of the Undiagnosed Diseases, N.; et al. Expanding the phenotypic spectrum of GABRG2 variants: A recurrent GABRG2 missense variant associated with a severe phenotype. J. Neurogenet. 2017, 31, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Stosser, M.B.; Lindy, A.S.; Butler, E.; Retterer, K.; Piccirillo-Stosser, C.M.; Richard, G.; McKnight, D.A. High frequency of mosaic pathogenic variants in genes causing epilepsy-related neurodevelopmental disorders. Genet. Med. 2018, 20, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.T.; Hollingsworth, G.; Muir, A.M.; Schneider, A.L.; Thuesmunn, Z.; Knupp, A.; King, C.; Lacroix, A.; Mehaffey, M.G.; Berkovic, S.F.; et al. Parental mosaicism in "de novo" epileptic encephalopathies. New Engl. J. Med. 2018, 378, 1646–1648. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.M.; King, C.; Schneider, A.L.; Buttar, A.S.; Scheffer, I.E.; Sadleir, L.G.; Mefford, H.C. Double somatic mosaicism in a child with Dravet syndrome. Neurol. Genet. 2019, 5, e333. [Google Scholar] [CrossRef] [Green Version]
- Jacob, T.C.; Moss, S.J.; Jurd, R. Gaba(a) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 2008, 9, 331–343. [Google Scholar] [CrossRef]
- Srivastava, S.; Cohen, J.; Pevsner, J.; Aradhya, S.; McKnight, D.; Butler, E.; Johnston, M.; Fatemi, A. A novel variant in GABRB2 associated with intellectual disability and epilepsy. Am. J. Med. Genet. 2014, 164, 2914–2921. [Google Scholar] [CrossRef]
- Ishii, A.; Kang, J.Q.; Schornak, C.C.; Hernandez, C.C.; Shen, W.; Watkins, J.C.; Macdonald, R.L.; Hirose, S. A de novo missense mutation of GABRB2 causes early myoclonic encephalopathy. J. Med. Genet. 2017, 54, 202–211. [Google Scholar] [CrossRef]
- Chao, H.T.; Chen, H.; Samaco, R.C.; Xue, M.; Chahrour, M.; Yoo, J.; Neul, J.L.; Gong, S.; Lu, H.C.; Heintz, N.; et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010, 468, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Di Lucente, J.; Lin, Y.C.; Lien, C.C.; Rogawski, M.A.; Maezawa, I.; Jin, L.W. Defective GABAergic neurotransmission in the nucleus tractus solitarius in MECP2-null mice, a model of Rett syndrome. Neurobiol. Dis. 2018, 109, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila Melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. Dbnsfp v2.0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
|---|---|---|---|---|---|---|---|---|---|
| Sex, Current Age (years) | F (18 y) | F (11 y) | F (19 y) | F (29 y) | F (7y) | F (9 y) | F (38 y) | F (42 y) | |
| Molecular Approach | NGS-pediatric epilepsy | WES-RTT | WES-RTT | NGS -pediatric epilepsy | NGS-diagnostic | NGS-diagnostic | NGS -pediatric epilepsy | WES-RTT | |
| Mutation/Inheritance Pattern | STXBP1 NC_000009.11: g.130423471 C>T, NM_003165.3: c.416C>T: p.(Pro139Leu), de novo | STXBP1 NC_000009.11: g.130435529 C>T, NM_003165.3: c.1099C>T: p.(Arg367Ter), de novo | STXBP1 NC_000009.11: g.130416077 T>C, (NM_003165.3): c.169+2T>C, r.([169_170 ins [gc;169+3_169+1168]; 169_170ins [gc; 169+3_169+1334]]),p.(Ile57Serfs7*) de novo | STXBP1 NC_000009.11: g.130428548 T>C, NM_003165.3: c.767T>C, p.(Leu256Pro), de novo | STXBP1 NC_000009.11: g.130444840 G>A, (NM_003165.3) c.1702+1G>A, r. [1585_1702del117] p.(Glu530_Gly 568del) de novo | STXBP1 NC_000009.11: g.130438188 C>T, NM_003165.3: c.1216 C>T, p.(Arg406Cys), de novo | GABRG2 NC_000005.9: g.161576128_161576129 delinsGG, NM_000816.3: c.937_938 delinsGG, p.(Leu313Gly), de novo mosaic | GABRB2 NC_000005.9: g.160758063 C>T, NM_021911.2: c.904G>A p.(Val302Met), de novo | |
| Regression (age indicated) Followed by Recovery or Stabilization | No cdv | No cdv | No cdv | No cdv | No | Yes (6 months) | Yes (12 months) | Yes (9 months) | |
| Main Criteria | Partial or Complete Loss of Acquired Purposeful Hand Skills | No: not lost, but NevAcq (grabs food and takes it to her mouth) | No: not lost, but NevAcq (grasping and manipulation disturbed by involuntary movements) | No: not lost, but NevAcq (grasping disturbed by tremors and stereotypies) | No | No: very limited hand skills | No: not lost, but Nev completely Acq | Yes: very limited hand skills | Yes: (since 2 years leaves behind and drops things) |
| Partial or Complete loss of Acquired Spoken Language | No: not lost, but NevAcq | No: not lost, vocalisms and only ten words | No: not lost, but NevAcq | No: not lost, only a few words | No: not lost, but Nev Acq, only vocalisms | No: not lost, but NevAcq(vocalism) | No: not lost, but NevAcq | Yes (only “Mum” and “Dad”, then lost) | |
| Gait Abnormalities: Impaired or Absence of Ability | Yes (ataxic-dyspraxic, unstable and only for short distances: since 4 years) | Yes (ataxic with axillary support: since 4 years) | Yes absent (only standing with axillary support) | Yes ataxic (walking with enlarged base and out of rotation of feet: since 3 years) | Yes (walking with enlarged base/ not apraxic: since 3 years) | Yes (Nev Acq) | Yes (ataxic: since 6 years) | Yes (apraxic, slow but autonomous, since 16 months, climbs the stairs) | |
| Stereotypic Hand Movements (type) | Yes frequent (brings her hands to mouth and bites fingers) | Yes (not typical for RTT, beats her head: since 3 years) | Yes (hand washing) | Yes (hand rocking) | Yes (hand washing, clapping, tapping right hand on table/books, tapping the forehead with the right upper limb, upper limb flickering) | Yes (upper limbs tremors, upper limb flickering, and dyskinesias) | Yes (tapping her right hand on her teeth: since 18 months), | Yes (upper limbs flickering) | |
| Exclusion Criteria | Brain Injury: Peri or Postnatal Trauma, Neurometabolic Disease or Severe Infection | No | No | No | No | No | No | No | No |
| Grossly Abnormal Psychomotor Development in First 6 Months of Life: Exam at the Birth | hypotonia | normal | normal | hypotonia, hyperexcitability, inconsolable crying | normal | normal | normal | normal | |
| Supportive Criteria | Breathing Disturbances | No | Yes | No | No | No | No | Yes (mild cyanosis and apneas) | Yes (hyperventilation) |
| Bruxism when Awake | No | No | Yes | No | Yes | Yes | Yes | Yes (significant) | |
| Impaired Sleep Pattern | Yes (sleeplessness and nocturnal agitation) | Yes (seizures) | Yes (nocturnal bruxism) | No | Yes (several and prolonged nocturnal awakenings) | No | Yes | No | |
| Abnormal Muscle Tone | Yes (proximal hypotonia) | Yes | Yes | No | No | Yes (axial hypotonia, hypertonus of the limbs) | Yes mild hypertonus (hypotonia in the first years of life) | No | |
| Peripheral Vasomotor Disturbances | No | No | Yes | No | No | No | cold and bluish hands and feet without trophic changes | No | |
| Scoliosis/Kyphosis | Yes (lumbar hyperlordosis) | Yes (mild) | Yes | No | No | No | Yes (mild kyphosis) | No (only scoliotic attitude) | |
| Growth Retardation | No | No | No | hypostaturism and obesity | No | Yes | Yes mild | No | |
| Small Cold Hands/Feet | Yes | No | Yes | No (but short and stubby fingers) | No | Yes (small, not cold) | Yes | Yes (cold feet) | |
| Inappropriate Laughing /Screaming Spells | Yes | No | Yes (screams) | No | Yes | nd | Yes frequent | Yes rare | |
| Diminished Response to Pain | Yes | No | No | nd | Yes | nd | nd | No | |
| Intense Eye Communication | No | Yes | No | No | Yes | No | Yes | Yes | |
| Microcephaly: if Yes Indicate if Acquired | Yes acquired | No | No | No | No | Yes acquired | No | Yes acquired | |
| Clinical Diagnosis at Referral | RTT atypical | RTT atypical Hanefeld | RTT atypical congenital | RTT-like-EOEE (West>Lennox-Gastaut) | RTT atypical | RTT-like (myoclonic epileptic encephalopathy) | RTT atypical | RTT classic | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cogliati, F.; Giorgini, V.; Masciadri, M.; Bonati, M.T.; Marchi, M.; Cracco, I.; Gentilini, D.; Peron, A.; Savini, M.N.; Spaccini, L.; et al. Pathogenic Variants in STXBP1 and in Genes for GABAa Receptor Subunities Cause Atypical Rett/Rett-like Phenotypes. Int. J. Mol. Sci. 2019, 20, 3621. https://doi.org/10.3390/ijms20153621
Cogliati F, Giorgini V, Masciadri M, Bonati MT, Marchi M, Cracco I, Gentilini D, Peron A, Savini MN, Spaccini L, et al. Pathogenic Variants in STXBP1 and in Genes for GABAa Receptor Subunities Cause Atypical Rett/Rett-like Phenotypes. International Journal of Molecular Sciences. 2019; 20(15):3621. https://doi.org/10.3390/ijms20153621
Chicago/Turabian StyleCogliati, Francesca, Valentina Giorgini, Maura Masciadri, Maria Teresa Bonati, Margherita Marchi, Irene Cracco, Davide Gentilini, Angela Peron, Miriam Nella Savini, Luigina Spaccini, and et al. 2019. "Pathogenic Variants in STXBP1 and in Genes for GABAa Receptor Subunities Cause Atypical Rett/Rett-like Phenotypes" International Journal of Molecular Sciences 20, no. 15: 3621. https://doi.org/10.3390/ijms20153621