p-Coumaric Acid Has Protective Effects against Mutant Copper–Zinc Superoxide Dismutase 1 via the Activation of Autophagy in N2a Cells


Abstract
:
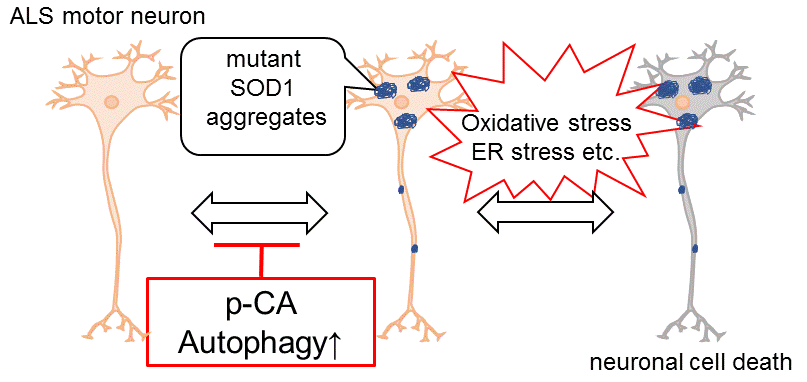{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. p-CA Reduced Cytoplasmic Aggregation of SOD1mut and Protected against SOD1mut-Associated Neurotoxicity
2.2. p-CA Attenuated SOD1G85R-Associated Oxidative and Endoplasmic Reticulum (ER) Stress
2.3. p-CA Exerted a Neuroprotective Effect against SOD1G85R Aggregates via the Activation of Autophagy
3. Discussion
4. Materials and Methods
4.1. Culture, Construct, and Transfection Cell Lines
4.2. Antibodies
4.3. Thiazolyl Blue Tetrazolium Bromide (MTT) Assay and Lactate Dehydrogenase (LDH) Release Assay
4.4. Measurement of the Aggregation Rate
4.5. Biochemical Analysis of Cell Culture Lysates
4.6. Reactive Oxygen Species (ROS) Production
4.7. ESR Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| SALS | sporadic amyotrophic lateral sclerosis |
| FALS | familial amyotrophic lateral sclerosis |
| SOD1 | superoxide dismutase 1 |
| UPS | ubiquitin proteasome system |
| EBGP | ethanol extract of Brazilian green propolis |
| p-CA | p-coumaric acid |
| ER | endoplasmic reticulum |
| N2a cells | Neuro2a cells |
| MTT assay | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide assay |
| DMPO | 5,5-Dimethyl-1-pyrroline N-Oxide |
| BiP | immunoglobulin heavy chain-binding protein |
| CHOP | transcription factor C/EBP homologous protein |
| CQ | chloroquine |
| AMPK | AMP-activated protein kinase |
References
- Julien, J.P. Amyotrophic lateral sclerosis. unfolding the toxicity of the misfolded. Cell 2001, 104, 581–591. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G.L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, R.; Beal, M.F.; Thomas, B. Autophagy in neurodegenerative disorders: Pathogenic roles and therapeutic implications. Trends Neurosci. 2010, 33, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Marino, M.; Tortarolo, M.; Veglianese, P.; De Biasi, S.; Fontana, E.; Zuccarello, L.V.; Maynard, C.J.; Dantuma, N.P.; Bendotti, C. Functional alterations of the ubiquitin-proteasome system in motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009, 18, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.; Heath, P.R.; Kirby, J.; Wharton, S.B.; Cookson, M.R.; Menzies, F.M.; Banks, R.E.; Shaw, P.J. Analysis of the cytosolic proteome in a cell culture model of familial amyotrophic lateral sclerosis reveals alterations to the proteasome, antioxidant defenses, and nitric oxide synthetic pathways. J. Biol. Chem. 2003, 278, 6371–6383. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, M.; Kurisu, J.; Tsukita, K.; Takahashi, R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J. Neurochem. 2002, 83, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Gal, J.; Kwinter, DM.; Liu, X.; Zhu, H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2010, 1802, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guégan, C.; Nagai, M.; Xu, Z.; Sosunov, A.A.; McKhann, G.M., 2nd; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. USA 2006, 103, 6025–6030. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef] [PubMed]
- Vehviläinen, P.; Koistinaho, J.; Gundars, G. Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front. Cell Neurosci. 2014, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress-and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhangani, D.; Endo, F.; Amanullah, A.; Upadhyay, A.; Watanabe, S.; Mishra, R.; Yamanaka, K.; Mishra, A. Mahogunin ring finger 1 confers cytoprotection against mutant SOD1 aggresomes and is defective in an ALS mouse model. Neurobiol. Dis. 2016, 86, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Drago, L.; Mombelli, B.; De Vecchi, E.; Fassina, M.C.; Tocalli, L.; Gismondo, M.R. In vitro antimicrobial activity of propolis dry extract. J. Chemother. 2000, 12, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Amoros, M.; Lurton, E.; Boustie, J.; Girre, L.; Sauvager, F.; Cormier, M. Comparison of the anti-herpes simplex virus activities of propolis and 3-methyl-but-2-enyl caffeate. J. Nat. Prod. 1994, 57, 644–647. [Google Scholar] [CrossRef]
- Paulino, N.; Teixeira, C.; Martins, R.; Scremin, A.; Dirsch, V.M.; Vollmar, A.M.; Abreu, S.R.; de Castro, S.L.; Marcucci, M.C. Evaluation of the analgesic and anti-inflammatory effects of a Brazilian green propolis. Plan. Med. 2006, 72, 899–906. [Google Scholar] [CrossRef]
- Xuan, H.; Li, Z.; Yan, H.; Sang, Q.; Wang, K.; He, Q.; Wang, Y.; Hu, F. Antitumor activity of Chinese propolis in human breast cancer MCF-7 and MDA-MB-231 Cells. Evid. Based Complement. Altern. Med. 2014, 280120. [Google Scholar] [CrossRef]
- Kudo, D.; Inden, M.; Sekine, S.; Tamaoki, N.; Iida, K.; Naito, E.; Watanabe, K.; Kamishina, H.; Shibata, T.; Hozumi, I. Conditioned medium of dental pulp cells stimulated by Chinese propolis show neuroprotection and neurite extension in vitro. Neurosci. Lett. 2015, 589, 92–97. [Google Scholar] [CrossRef]
- Naramoto, K.; Kato, M.; Ichihara, K. Effects of an ethanol extract of Brazilian green propolis on human cytochrome P450 enzyme activities in vitro. J. Agr. Food Chem. 2014, 62, 11296–11302. [Google Scholar] [CrossRef]
- Hata, T.; Tazawa, S.; Ohta, S.; Rhyu, M.R.; Misaka, T.; Ichihara, K.; Artepillin, C. A major ingredient of Brazilian propolis, induces a pungent taste by activating TRPA1 channels. PLoS ONE 2012, 7, e48072. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Inden, M.; Shirai, K.; Sekine, SI.; Masaki, Y.; Kurita, H.; Ichihara, K.; Inuzuka, T.; Hozumi, I. The effects of Brazilian green propolis that contains flavonols against mutant copper-zinc superoxide dismutase-mediated toxicity. Sci. Res. 2017, 7, 2882. [Google Scholar] [CrossRef] [PubMed]
- An, S.M.; Koh, J.S.; Boo, Y.C. P-coumaric acid not only inhibits human tyrosinase activity in vitro but also melanogenesis in cells exposed to UVB. Phytother. Res. 2010, 8, 1175–1180. [Google Scholar]
- Ferguson, L.R.; Lim, I.F.; Pearson, A.E.; Ralph, J.; Harris, P.J. Bacterial antimutagenesis by hydroxycinnamic acids from plant cell walls. Mutat. Res. 2003, 542, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Luceri, C.; Giannini, L.; Lodovici, M.; Antonucci, E.; Abbate, R.; Masini, E.; Dolara, P. P-Coumaric acid, a common dietary phenol, inhibits platelet activity in vitro and in vivo. Br. J. Nutr. 2007, 97, 458–463. [Google Scholar] [CrossRef]
- Navaneethan, D.; Rasool, M. P-Coumaric acid, a common dietary polyphenol, protects cadmium chloride-induced nephrotoxicity in rats. Ren. Fail. 2014, 36, 244–251. [Google Scholar] [CrossRef]
- Pragasam, S.J.; Venkatesan, V.; Rasool, M. Immunomodulatory and anti-inflammatory effect of p-coumaric acid, a common dietary polyphenol on experimental inflammation in rats. Inflammation 2013, 36, 169–176. [Google Scholar] [CrossRef]
- Prasanna, N.; Krishnan, D.N.; Rasool, M. Sodium arsenite-induced cardiotoxicity in rats: Protective role of p-coumaric acid, a common dietary polyphenol. Toxicol. Mech. Methods. 2013, 23, 255–262. [Google Scholar] [CrossRef]
- Peng, J.; Zheng, T.T.; Liang, Y.; Duan, L.F.; Zhang, Y.D.; Wang, L.J.; He, G.M.; Xiao, H.T. P-Coumaric acid protects human lens epithelial cells against oxidative stress-induced apoptosis by MAPK signaling. Oxid. Med. Cell Longev. 2018, 2018, 8549052. [Google Scholar] [CrossRef]
- Sunitha, M.C.; Dhanyakrishnan, R.; PrakashKumar, B.; Nevin, K.G. P-Coumaric acid mediated protection of H9c2 cells from doxorubicin-induced cardiotoxicity: Involvement of augmented Nrf2 and autophagy. Biomed. Pharmacother. 2018, 102, 823–832. [Google Scholar] [CrossRef]
- Shailasree, S.; Venkataramana, M.; Niranjana, S.R.; Prakash, H.S. Cytotoxic effect of p-Coumaric acid on neuroblastoma, N2a cell via generation of reactive oxygen species leading to dysfunction of mitochondria inducing apoptosis and autophagy. Mol. Neurobiol. 2015, 51, 119–130. [Google Scholar] [CrossRef]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef]
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS. J. Clin. Invest. 2015, 125, 1767–1779. [Google Scholar] [CrossRef]
- Cao, X.; Antonyuk, S.V.; Seetharaman, S.V.; Whitson, L.J.; Taylor, A.B.; Holloway, S.P.; Strange, R.W.; Doucette, P.A.; Valentine, J.S.; Tiwari, A.; et al. Structures of the G85R variant of SOD1 in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2008, 283, 16169–16177. [Google Scholar] [CrossRef]
- Ueda, T.; Inden, M.; Asaka, Y.; Masaki, Y.; Kurita, H.; Tanaka, W.; Yamaguchi, E.; Itoh, A.; Hozumi, I. Effects of gem-dihydroperoxides against mutant copper-zinc superoxide dismutase-mediated neurotoxicity. Mol. Cell Neurosci. 2018, 92, 177–184. [Google Scholar] [CrossRef]
- Zimmerman, M.C.; Oberley, L.W.; Flanagan, S.W. Mutant SOD1-induced neuronal toxicity is mediated by increased mitochondrial superoxide levels. J. Neurochem. 2007, 102, 609–618. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Bosco, D.A.; Morfini, G.; Karabacak, N.M.; Song, Y.; Gros-Louis, F.; Pasinelli, P.; Goolsby, H.; Fontaine, B.A.; Lemay, N.; McKenna-Yasek, D.; et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010, 13, 1396–1403. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, K.; Jonsson, P.A.; Andersen, P.M.; Bergemalm, D.; Graffmo, K.S.; Hultdin, M.; Jacobsson, J.; Rosquist, R.; Marklund, S.L.; Brännström, T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 2010, 5, e11552. [Google Scholar] [CrossRef]
- Watanabe, S.; Ageta-Ishihara, N.; Nagatsu, S.; Takao, K.; Komine, O.; Endo, F.; Miyakawa, T.; Misawa, H.; Takahashi, R.; Kinoshita, M.; et al. SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol. Brain 2014, 7, 62. [Google Scholar] [CrossRef]
- Kang, S.W.; Kang, S.I.; Shin, H.S.; Yoon, S.A.; Kim, J.H.; Ko, H.C.; Kim, S.J. Sasa quelpaertensis Nakai extract and its constituent p-coumaric acid inhibit adipogenesis in 3T3-L1 cells through activation of the AMPK pathway. Food Chem. Toxicol. 2013, 59, 380–385. [Google Scholar] [CrossRef]
- Yoon, S.A.; Kang, S.I.; Shin, H.S.; Kang, S.W.; Kim, J.H.; Ko, H.C.; Kim, S.J. P-Coumaric acid modulates glucose and lipid metabolism via AMP-activated protein kinase in L6 skeletal muscle cells. Biochem. Biophys. Res. Commun. 2013, 432, 553–557. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [Green Version]
- Kong, C.S.; Jeong, C.H.; Choi, J.S.; Kim, K.J.; Jeong, J.W. Antiangiogenic effects of p-coumaric acid in human endothelial cells. Phytother. Res. 2013, 27, 317–323. [Google Scholar] [CrossRef]
- Lim, M.A.; Selak, M.A.; Xiang, Z.; Krainc, D.; Neve, R.L.; Kraemer, B.C.; Watts, J.L.; Kalb, R.G. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J. Neurosci. 2012, 32, 1123–1141. [Google Scholar] [CrossRef]
- Watanabe, S.; Hayakawa, T.; Wakasugi, K.; Yamanaka, K. Cystatin C protects neuronal cells against mutant copper-zinc superoxide dismutase-mediated toxicity. Cell Death Dis. 2014, 5, e1497. [Google Scholar] [CrossRef]
- Watanabe, S.; Komine, O.; Endo, F.; Wakasugi, K.; Yamanaka, K. Intracerebroventricular administration of Cystatin C ameliorates disease in SOD1-linked amyotrophic lateral sclerosis mice. J. Neurochem. 2018, 145, 80–89. [Google Scholar] [CrossRef]
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 2018, 14, 534–551. [Google Scholar] [CrossRef]
- Kim, S.H.; Jung, S.Y.; Lee, K.W.; Lee, S.H.; Cai, M.; Choi, S.M.; Yang, E.J. Bee venom effects on ubiquitin proteasome system in hSOD1(G85R)-expressing NSC34 motor neuron cells. BMC Complement. Altern. Med. 2013, 13, 179. [Google Scholar] [CrossRef]
- Kabuta, T.; Suzuki, Y.; Wada, K. Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J. Biol. Chem. 2006, 281, 30524–30533. [Google Scholar] [CrossRef]
- Mehta, P.; Kaye, W.; Raymond, J.; Punjani, R.; Larson, T.; Cohen, J.; Muravov, O.; Horton, K. Prevalence of amyotrophic lateral sclerosis—United States, 2015. Morb. Mortal. Wkly. Rep. 2018, 67, 1285–1289. [Google Scholar] [CrossRef]
- Dohare, P.; Hyzinski-García, M.C.; Vipani, A.; Bowens, N.H.; Nalwalk, J.W.; Feustel, P.J.; Keller, R.W., Jr.; Jourd’heuil, D.; Mongin, A.A. The neuroprotective properties of the superoxide dismutase mimetic tempol correlate with its ability to reduce pathological glutamate release in a rodent model of stroke. Free Radic. Biol. Med. 2014, 77, 168–182. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Pasinelli, P.; Trotti, D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1842, 1295–1301. [Google Scholar] [CrossRef] [Green Version]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol improves motoneuron function and extends survival in SOD1 (G93A) ALS mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar]
- Daniel, B.; Green, O.; Viskind, O.; Gruzman, A. Riluzole increases the rate of glucose transport in L6 myotubes and NSC-34 motor neuron-like cells via AMPK pathway activation. Amyotroph. Lateral Scler. Frontotemporal Degener. 2013, 17, 434–443. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueda, T.; Ito, T.; Kurita, H.; Inden, M.; Hozumi, I. p-Coumaric Acid Has Protective Effects against Mutant Copper–Zinc Superoxide Dismutase 1 via the Activation of Autophagy in N2a Cells. Int. J. Mol. Sci. 2019, 20, 2942. https://doi.org/10.3390/ijms20122942
Ueda T, Ito T, Kurita H, Inden M, Hozumi I. p-Coumaric Acid Has Protective Effects against Mutant Copper–Zinc Superoxide Dismutase 1 via the Activation of Autophagy in N2a Cells. International Journal of Molecular Sciences. 2019; 20(12):2942. https://doi.org/10.3390/ijms20122942
Chicago/Turabian StyleUeda, Tomoyuki, Taisei Ito, Hisaka Kurita, Masatoshi Inden, and Isao Hozumi. 2019. "p-Coumaric Acid Has Protective Effects against Mutant Copper–Zinc Superoxide Dismutase 1 via the Activation of Autophagy in N2a Cells" International Journal of Molecular Sciences 20, no. 12: 2942. https://doi.org/10.3390/ijms20122942