Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy


C.U.R.E. University Centre for Liver Disease Research and Treatment, Department of Medical and Surgical Sciences, Institute of Internal Medicine, University of Foggia, 71122 Foggia, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(1), 49; https://doi.org/10.3390/ijms20010049
Submission received: 9 October 2018
/
Revised: 18 December 2018
/
Accepted: 19 December 2018
/
Published: 22 December 2018
(This article belongs to the Special Issue Hepatitis Virus Infection and Molecular Research 2018)
Abstract
:Chronic hepatitis C is associated with a high risk of developing hepatocellular carcinoma (HCC) because of a direct effect of the Hepatitis C Virus (HCV) proteins and an indirect oncogenic effect of chronic inflammation and impaired immune response. The treatment of chronic hepatitis C markedly reduces all-cause mortality; in fact, interferon-based treatment has shown a reduction of HCC incidence of more than 70%. The recent introduction of the highly effective direct-acting antivirals (DAAs) has completely changed the scenario of chronic hepatitis C (CHC) with rates of HCV cure over 90%. However, an unexpectedly high incidence of HCC recurrence was observed in patients after DAA treatment (27% versus 0.4–2% in patients who received interferon treatment). The mechanism that underlies the high rate of tumor relapse is currently unknown and is one of the main issues in hepatology. We reviewed the possible mechanisms involved in HCC recurrence after DAA treatment.
1. Introduction
Chronic viral hepatitis is a well-recognized risk factor for end-stage liver disease and liver cancer [1]. Among primary liver cancers, hepatocellular carcinoma (HCC) accounts for 70% to 85% of cases [2] and it is currently the fifth most common cancer [3] and the third leading cause of cancer mortality in the world [4].
Compared with other causes of cirrhosis, chronic hepatitis C is associated with a higher risk of developing HCC [5,7] because the Hepatitis C Virus (HCV) owns a direct oncogenic effect; moreover, chronic inflammation, impairment of immune response, cellular senescence, and proliferation indirectly depend on HCV disease [8,9,10].
Direct oncogenic effects of HCV are related to the cellular expression of viral proteins localized in the cytosol, lipid droplets, endoplasmic reticulum, mitochondria, and nuclei, affecting a variety of cellular functions [10].
Overexpression of HCV proteins, e.g., core, NS3, and NS5A, promotes cellular proliferation, transformation and tumor formation in mice, suggesting a direct effect in activating oncogenic pathways [10,11,12,13,14]. The core protein modulates p53 regulatory activity and directly influences the p53-related p73 protein [15,16] whereas NS3 and NS5A inhibit p53 [17,18]. NS5A promotes evasion from apoptosis by caspase-3 inhibition [19] and inhibits tumor necrosis factor-alpha (TNFα) mediated apoptosis [20], whereas NS5B inhibits the retinoblastoma-associated protein (RB1), which is involved in controlling cellular proliferation and apoptosis by regulating transcription factors [21].
Moreover, viral proteins indirectly regulate innate immune pathways: NS3 suppresses innate immunity by cleavage of the mitochondrial antiviral signaling protein (MAVS), a pivotal antiviral protein involved in interferon induction [22]; the binding of the hepatitis C virus envelope protein E2 to CD81 causes inhibition of natural killer (NK) cells contributing to immune evasion [23]; HCV core protein inhibits hepatocyte senescence, a physiological process providing a barrier to tumorigenesis [24].
HCV-induced HCC is, therefore, a model of chronic inflammation-driven tumor, where a complex interaction between viruses and hepatocytes occurs, promoting hepatocarcinogenesis [10].
The cure of HCV infection defined as the absence of circulating HCV RNA at least 12 weeks after treatment completion (sustained virologic response—SVR), is associated with a marked reduction of the all-cause mortality [25] and particularly, with a reduction of more than 70% of HCC [26]. Compared with interferon-based treatments, direct-acting antivirals (DAAs) achieve SVR rates in over 90% of patients, irrespective of fibrosis stage [27,28,29] and this may significantly improve the natural history of HCV infection. Therefore, DAAs are currently the most promising strategy for reducing the future burden of HCC [30,31]. However, several recent reports have raised concerns about DAA treatment because a higher incidence of HCC recurrence has been observed in patients during and after antiviral treatment [32,33,34,35,36,37,38,39]. These studies have also reported an unexpectedly high incidence of de novo HCC in addition to a particularly aggressive behavior of HCC relapse. A recurrence rate up to 27% (versus 0.4–2% in patients with SVR after interferon treatment) [36,40] has sparked debate on a possible role of DAAs on hepatocellular carcinoma progression and recurrence. Nonetheless, given the lack of robust evidence of a drug-related effect, many authors have investigated all factors potentially involved in promoting liver cancer during or after antiviral treatment.
2. Immune Cell Dysfunction during Chronic HCV Infection
The immune system plays a key role both in viral clearance during acute infection and in liver injury during chronic C hepatitis and, because of the lack of a cytopathic effect [53], both innate and adaptive immune responses are necessary to achieve a full recovery from viral infection [54]. However, the interaction between virus and immune system is contradictory, as HCV induces T cell exhaustion and, at the same time, it can stimulate autoantibody production [55] or cause autoimmune diseases [56] (Figure 2).
HCV infection is a leading cause of chronic hepatitis and liver cirrhosis, since in 70–80% of infected individuals, the virus persists after an acute phase without a spontaneous recovery [57]. Viral elimination during acute infection involves both a rapid induction of innate response and a delayed induction of adaptive immune responses.
Innate immune response is the first defense against viral infections and is maintained by interferons (IFNs) that are also able to regulate the cellular components of innate immunity, such as NK cells [58]. After viral infection and endosomal-mediated degradation of viral nucleic acids, macrophages, dendritic cells, NK, and antigen-specific T cells (CD4+ Th1 and CD8+ cytotoxic T lymphocytes) expose viral antigens to toll-like receptors (TLRs), triggering a nonspecific antiviral response, and induce the transcription of hundreds of genes, which are distinct for different IFNs and target cell types [57].
In contrast to the innate immune response induced within hours to days after infection, adaptive immune responses (humoral antibody and T cells responses) become detectable not before 6–8 weeks after viral infection [59,60,61,62,63,64]. Antibodies blocking virus binding, entry or uncoating were found in most patients with spontaneous viral clearance [65] and, similarly, a long lasting CD4+ and CD8+ T cell response targeting multiple viral epitopes persists after resolution of acute viral infection, suggesting a major role of adaptive immunity in hepatitis C infection [57,60,66,67,68,69,70,71].
In chronic hepatitis C, HCV typically escapes both innate and adaptive responses [57] thanks to the release of several factors promoting viral immune evasion and globally impairing viral clearance [72], a complex immune mechanism named “T cell exhaustion”, typically characterized by CD4+ T cell and CD8+ T cell dysfunction together with impaired cytokine production and lack of response to antigen stimulation [73,74].
In acute self-limiting HCV infection, circulating helper T-cells mostly produce IFNα, suggesting Th1 predominance, whereas during chronic infection, a limited Th1-response and a prevalent Th2 response has been reported [75,76,77].
A critical balance between initiation and downregulation of the immune response is required for immune homeostasis and chronic inflammation and autoimmunity may result from dysfunctions of immune resolution [78].
Treatment with anti-interleukin-10 monoclonal antibody enhances IFNα production in HCV patients [79] and a long-term treatment with recombinant IL-10 reduces inflammation in the same patients, suggesting a pivotal role of immunosuppressive cytokine IL-10 in antiviral response and Th1/Th2 balance [75].
Experimental data have shown that a significant decline in CD4+/CD25+ regulatory cells (also called Treg), typically involved in the maintenance of self-tolerance and suppression of self-reactive lymphocytes [80], is needed to induce suppression of CD8+ T cell proliferation and IFN-α production [81,82]. Tregs are involved in the long-term maintenance of memory T cells, ensuring protective immunity, and in controlling the intensity of T cell immune responses, even though this condition allows for inflammation to persist [80]. The presence of CD4+/CD25+ depends, almost partially, on the production of immunoregulatory cytokines [75]. In contrast to natural Treg cells, which develop in the thymus and play an important role in the maintenance of self-tolerance and immune homeostasis, induced Treg cells develop from nonregulatory T cells in the periphery [83]. Distinct subsets of induced regulatory T cells have been identified, and, among those, Treg cells secreting IL-10 play a pivotal role in viral persistence [83]. During chronic hepatitis C, effector T cells become exhausted and show reduced antiviral activity, while Tregs progressively infiltrate the liver [84]. A higher frequency of Treg in HCV-infected patients was reported by several authors [85,86]. Similarly, Cabrera et al. reported a positive correlation between Treg frequency and HCV RNA and an inverse relation with histologic inflammatory scores, confirming a significant role of regulatory T cells in viral persistence [76].
Therefore, during chronic hepatitis C, effector T cells become exhausted over time and show an impaired antiviral activity while CD4+ regulatory T cells (Tregs) accumulate in the liver [84]. Tregs are mainly regulated by interleukin (IL)-2 receptor α chain CD25 and Foxp3 [87]; once activated, Tregs induce expression of programmed cell death protein 1 (PD-1), programmed death-ligand 1 (PD-L1), contact-dependent regulatory molecules such as cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and ultimately cytokines such as IL-10 or transforming growth factor (TGF-β) [48,88,89]. Because of their extensive role in modifying multiple immune functions, such as T cell responses and natural killer (NK) cells function [48], Tregs are considered to be involved in immune surveillance of tumors.
Natural killer (NK) cells are the prototype of innate lymphoid cells with potent cytolytic function that provide viral and immune surveillance against cancer [90]; two subsets of NK cells are commonly recognized: CD56high/CD16low, which mainly produce chemokines and cytokines, and CD56low/CD16high, a phenotype with prevalent cytotoxic activity [91]. Generally, NK cells are an important part of the interferon-responsive innate population in the liver (30% of lymphocytes) in comparison to the blood (5–20%), and the percentage increases in viral hepatitis [92,93]. During chronic viral infection, they are permanently activated and polarized toward cytotoxicity with deficient IFNγ secretion because of chronic exposure to endogenous IFNα [94,95]. The result is a “functional dichotomy” presenting with enhanced cytolytic activity and failure to produce IFNγ and TNFα, with consequent inability to eradicate the virus [96]. A possible involvement of NK cells in immune surveillance of tumors might be considered [97].
3. DAAs Are Able to Modulate Immune Cell Response
Initial data on T cell function during and after direct-acting antiviral treatment were published in 2014 [98]. The authors analyzed virus-specific CD8+ T cells obtained from 51 previously untreated and chronically infected patients undergoing antiviral treatment with a combination of faldaprevir (a protease inhibitor) and deleobuvir (a non-nucleoside polymerase inhibitor) and observed a significant increase in the frequency of HCV-specific CD8+ T cells within 4 weeks of therapy in patients obtaining SVR, whereas no changes were observed in patients with treatment failure. This result indicated that the immune dysfunction induced by HCV infection was reversible and that even short therapy with novel DAAs was able to restore immune capacity, whereas the same effect has never been observed in patients treated with interferons [41,97,99,100].
Taken together, these first reports suggested that drugs targeted on HCV replication were able to modulate the immune system by changing T cell balance.
Several authors have addressed the role of NKs in chronic HCV patients during DAA therapy [93]. Before treatment, NK cells showed increased degranulation (studied by the expression of CD107a and tumor necrosis factor-related apoptosis-inducing ligand—TRAIL) and decreased production of cytokins such as IFNγ and TNFα; during treatment, the authors observed a decrease of NK cell-activating receptor expression together with decreased IFNγ production followed by normalization of degranulation. Restoration of immune function induced by DAAs persisted up to the end of treatment and maintained even after suggesting that antiviral treatment is also immunologically effective [96].
These first observations were very impressive, since they did not occur during interferon therapy, where an increase in cytotoxic NK cell activity observed soon after starting therapy lasted only a few hours before leaving a refractory state [101].
In addition, Burchill et al. analyzed the composition of the memory CD4+ and CD8+ lymphocyte compartment on peripheral blood from nineteen chronic hepatitis C (CHC) patients treated with DAAs. After the rapid eradication of the virus, the frequency of NK cells was unchanged, whereas a significant reduction of the expression of PD1 on the HCV-specific T cells was found, suggesting a partial restoration in the functional capacity of HCV-specific T cells [42].
NK cell frequencies similar to healthy controls were also shown after 12 weeks of interferon-free treatment, adding new reports on the effect of viral load decline on the NK cell compartment. The frequencies of CD56high NK cells (NKbright) were typically higher in CHC, whereas CD56low NK cell (NKdim) frequencies were lower compared to healthy controls. After treatment with DAAs, NK cell subgroups returned to healthy control levels [43]. Cytokines involved in NK cell activation (IL-12p40 and IL-18), typically higher during chronic hepatitis C, normalized after DAA treatment [43]; the same observation was reported after interferon treatment [102]. Very interestingly, Chu et al. have reported that the pretreatment frequency of CD56high/CD16dim NK cells was significantly higher and that of CD16high/CD56dim NK cells was significantly lower in patients developing HCC after DAA treatment, showing a significant imbalance in NK cell subgroups. Moreover, the expression of NKG2D was significantly decreased during treatment, even more so in patients who developed HCC [45].
Several studies have reported a decrease of frequencies and activation of monocytes during DAA treatment [46,47], and other reports have suggested the involvement of neutrophils in the vascular endothelial growth factor (VEGF) and proteases secretion that may play a role in cancer cells spreading [44].
All these findings provide a possible rationale to support immune modification induced by DAA treatment, but further studies are needed to clarify the underlying molecular mechanism.
4. Cytokine Network Imbalance during Chronic C Hepatitis: Effect of DAAs
Cytokines are low-molecular-weight proteins mainly secreted by macrophages and lymphocytes and involved in cell-to-cell communication [103,104]; they also regulate proliferation, differentiation, migration, and death of immune cells [103].
A typical cytokine pattern of activated T-cell response has been described in chronic HCV patients, with an elevated level of serum IL-2, IL-4, TNFα other than IFN-α and IL-10 [75].
IL-10 is produced by several immune cells, including T and B cells, DC, and macrophages [105], and plays a key role as immunoregulatory molecule [72]; it suppresses antigen-presenting cells and T cell by inhibition of pro-inflammatory cytokine and chemokine production and, finally, by inhibition of costimulation and MHC class II expression [106,107,108]; moreover, viral persistence is associated with a high level of circulating DC-produced IL-10 and activation of PD-1/PD-ligand1 pathway [109,110,111,112,113,114]. In later stages, both NK and CD4+ T cells become the main IL-10 producers and play a pivotal role in the regulation of immune response [112,115].
A specific subset of T-cells also produces IL-10, named Tr1 [116], distinct from thymic-derived and naturally occurring CD25+Foxp3+ Tregs [117]. Tr1 have been found in patients with chronic HCV infection [118,119,120] and a subset of HCV-specific IL-10+ CD8+ Tr1 has been also described in the liver of such patients [121,122,123].
Since cytokines play a central role in the cross-talk among immune cells during chronic inflammation, some authors have suggested that modification in the circulating level of several cytokines, particularly TNFα, IL-6, and IL-10, may contribute to cancer promotion and progression [124,125,126]. TNFα is one of the cytokines more studied in cancer and has been correlated to chronic lymphocytic leukemia, Barret’s adenocarcinoma, prostate cancer, breast cancer, and cervical cancer [127,128,129,130].
A recent study has shown that, independently from HCV–RNA drop, TNFα remained stable or even higher in those developing HCC as compared to CHC who did not develop HCC [50], but conclusive data are lacking.
IL-6 is another cytokine which plays a significant role in the acute phase response and shows a pro-tumorigenic effect [103]; indeed, specific circulating levels are crucial for hepatocyte homeostasis because of powerful mitogen activity [131]. However, persistent activation of the IL-6 pathway in the liver is associated with the development of liver tumors [131] through a mechanism of hypermethylation of tumor suppressor genes. Very interestingly, serum IL-6 level has been reported to be increased in patients treated with DAAs and developing HCC recurrence [50].
Few data are available on the IL-18 circulating level during DAA treatment. It is considered a pro-inflammatory cytokine, synthesized and secreted by monocytes/macrophages and Kupffer cells. It activates nuclear factor (NF)-κB, which in turn activates cell proliferation, cycle progression, overexpression of angiogenic genes, and apoptosis inhibition [132,133,134]. IL-18 is upregulated in HCV-infected patients and its receptor is commonly expressed in HCC cells.
5. Potential Effect of DAAs in the Modulation of Angiogenesis Signaling
Angiogenesis is a dynamic process in which new vascular structures develop from preexisting vessels [135]. Key regulators of this process are hypoxia and inflammation, which stimulate the release of the hypoxia inducing factor (HIF), angiogenic cytokines, and growth factors from different cell types, including endothelial cells (ECs), monocytes, platelets, and smooth muscle, as well as tumor cells, and promote endothelial cell proliferation and stabilization of neovessels [135,136,137,138].
The most relevant angiogenic factor is VEGF (vascular endothelial growth factor), which has a key role in angiogenesis during inflammation and new vessel formation in tumors [139,140]. It is a potent growth factor produced mainly by tumor cells, macrophages, and platelets, and it is a highly specific mitogen for endothelial cells [141,142]. VEGF is activated by oncogenes and several cytokines [142] and functions as a cytokine promoting endothelial cell proliferation, vessel permeability, disruption of tight junction, and finally, the proliferative activity and the neoangiogenic potential of the tumor [143,144,145]. The effects of VEGF are mainly mediated in endothelial cells via VEGFR2. The activation of VEGFR2 receptor induces dilation of vessels, which also became leaky in response to VEGF signaling.
Both VEGF and Angiopoietin-2 (Ang2), a growth factor specific for the vascular endothelium and expressed during vascular remodeling in tumors [146,147], are involved in the dissolution of the vascular basement membrane and assembly into vascular networks [139] and serum concentration have been investigated to assess possible implications in liver diseases.
Hepatic neoangiogenesis has been described in liver diseases, such as viral hepatitis, cirrhosis, and HCC [148,149], and hepatitis C virus infection was found to induce production of TGF beta [137] and stabilization of HIF, resulting in the release of angiogenic cytokines [150] and proliferation of human endothelial cells [135,151].
Among the factors contributing to liver damage during chronic hepatitis C, angiogenesis seems to play a pivotal role [138,149,152] and the degree of microvessel density is increased in HCV-positive patients [153].
The serum concentrations of VEGF and Ang2 are also increased in patients with HCC compared to patients without liver cancer [154].
We observed a rapid increase of VEGF level in patients treated with all DAA regimens. After 4 weeks of treatment, in comparison to baseline, serum VEGF remained higher until the end of treatment and returned to baseline concentration after stopping the therapy [49]. Our findings were recently confirmed by Faillaci et al., who analyzed serum liver angiopoietin-2 and VEGF levels in 242 DAA-treated patients and found a DAA-mediated increase of VEGF and angiopoietin-2 supported the increased risk of HCC recurrence/occurrence during antiviral treatment [52].
Debes et al. confirmed a potential role of VEGF in risk of hepatocellular carcinoma, but they showed an alteration of baseline serum VEGF concentration in patients who developed HCC de novo, supporting the possible pre-existing increased VEGF level without a direct role of DAAs in liver cancer [50]. Further studies are required to definitively clarify the role of DAAs on tumor recurrence.
6. Conclusions
DAA treatment has completely changed the natural history of chronic hepatitis C, with the rate of cure over 90%. However, some authors have reported a worrying increase in HCC recurrence. Among the pathophysiological hypotheses, the most appealing one suggests that DAAs induce dramatic HCV clearance that in turn may induce immune cell alteration, imbalance of cytokine network, and angiogenesis that could explain, almost in part, the HCC recurrence observed after DAAs. Data are not conclusive, and the final word remains to be said.
Acknowledgments
We are sincerely grateful to Francesca Tursi for editing the English language in the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| HCC | hepatocellular carcinoma |
| CHC | chronic hepatitis C |
| NS3 | nonstructural protein 3 |
| NS5A | nonstructural protein 5A |
| TNFα | tumor necrosis factor-alpha |
| NS5B | nonstructural protein 5B |
| RB1 | retinoblastoma-associated protein |
| MAVS | mitochondrial antiviral signaling protei |
| NK | natural killer |
| SVR | sustained virologic response |
| DAAs | direct-acting antivirals |
| IL12p40 | p40 subunit of IL12 |
| Treg | regulatory T cell |
| VEGF | vascular endothelial growth factor |
| IFN | interferon |
| TLR | toll-like receptor |
| IFNα | interferon alpha |
| TGFβ | transforming growth factor beta |
| PD-L1 | programmed death-ligand1 |
| CTLA-4 | cytotoxic Tl-lymphocyte associated protein 4 |
| IFNγ | interferon gamma |
| TRAIL | tumor necrosis factor-related apoptosis-inducing ligand |
| PD-1 | programmed death 1 |
| Ang-2 | angiopoietin |
| VEGFR2 | vascular endothelial growth factor receptor 2 |
| IL-2 | interlukin 2 |
| IL-4 | interlukin 4 |
| IL-6 | interleukin 6 |
| IL-10 | interleukin 10 |
| IL-18 | interleukin 18 |
References
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B. Epidemiology of hepatocellular carcinoma. Clin. Liver Dis. 2001, 5, 87–107. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, I.I.; Tillmann, T.; Banerjee, A. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar] [CrossRef]
- Bialecki, E.S.; Di Bisceglie, A.M. Clinical presentation and natural course of hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2005, 17, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Blum, H.E. Pathogenesis of hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2005, 17, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Koike, K. Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S108–S111. [Google Scholar] [CrossRef] [PubMed]
- Farinati, F.; Cardin, R.; Bortolami, M.; Burra, P.; Russo, F.P.; Rugge, M.; Guido, M.; Sergio, A.; Naccarato, R. Hepatitis C virus: From oxygen free radicals to hepatocellular carcinoma. J. Viral Hepat. 2007, 14, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Fuchs, B.C.; Bardeesy, N.; Baumert, T.F.; Chung, R.T. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J. Hepatol. 2014, 61, S79–S90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemel, R.; Gerechet, S.; Greif, H.; Bachmatove, L.; Birk, Y.; Golan-Goldhirsh, A.; Kunin, M.; Berdichevsky, Y.; Benhar, I.; Tur-Kaspa, R. Cell transformation induced by hepatitis C virus NS3 serine protease. J. Viral Hepat. 2001, 8, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Arima, N.; Kao, C.Y.; Licht, T.; Padmanabhan, R.; Sasaguri, Y.; Padmanabhan, R. Modulation of cell growth by the hepatitis C virus nonstructural protein NS5A. J. Biol. Chem. 2001, 276, 12675–12684. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, T.; Zhou, Y.; Kawai, S.; Eguchi, H.; Wands, J.R.; Li, J. Hepatitis C virus core protein stimulates hepatocyte growth: Correlation with upregulation of wnt-1 expression. Hepatology 2005, 41, 1096–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.F.; Chen, S.Y.; Chen, J.Y.; Wu Lee, Y.H. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene 2004, 23, 2472–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alisi, A.; Giambartolomei, S.; Cupelli, F.; Merlo, P.; Fontemaggi, G.; Spaziani, A.; Balsano, C. Physical and functional interaction between HCV core protein and the different p73 isoforms. Oncogene 2003, 22, 2573–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Nagano-Fujii, M.; Tanaka, M.; Nomura-Takigawa, Y.; Ikeda, M.; Kato, N.; Sada, K.; Hotta, H. NS3 protein of Hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J. Gen. Virol. 2006, 87, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Street, A.; Macdonald, A.; McCormick, C.; Harris, M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 2005, 79, 5006–5016. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Majumder, M.; Steele, R.; Meyer, K.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein protects against TNF-α mediated apoptotic cell death. Virus Res. 2000, 67, 173–178. [Google Scholar] [CrossRef]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.T.; Klimpel, G.R. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J. Exp. Med. 2002, 195, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Park, S.H.; Jang, K.L. Hepatitis C virus Core protein overcomes stress-induced premature senescence by down-regulating p16 expression via DNA methylation. Cancer Lett. 2012, 321, 154–161. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, A.J.; Veldt, B.J.; Feld, J.J.; Wedemeyer, H.; Dufour, J.F.; Lammert, F.; Duarte-Rojo, A.; Heathcote, E.J.; Manns, M.P.; Kuske, L.; et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. JAMA 2012, 308, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.L.; Baack, B.; Smith, B.D.; Yartel, A.; Pitasi, M.; Falck-Ytter, Y. Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma: A meta-analysis of observational studies. Ann. Intern. Med. 2013, 158, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Bourliere, M.; Bronowicki, J.P.; de Ledinghen, V.; Hezode, C.; Zoulim, F.; Mathurin, P.; Tran, A.; Larrey, D.G.; Ratziu, V.; Alric, L.; et al. Ledipasvir-sofosbuvir with or without ribavirin to treat patients with HCV genotype 1 infection and cirrhosis non-responsive to previous protease-inhibitor therapy: A randomised, double-blind, phase 2 trial (SIRIUS). Lancet Infect. Dis. 2015, 15, 397–404. [Google Scholar] [CrossRef]
- Leroy, V.; Angus, P.; Bronowicki, J.P.; Dore, G.J.; Hezode, C.; Pianko, S.; Pol, S.; Stuart, K.; Tse, E.; McPhee, F.; et al. Daclatasvir, sofosbuvir, and ribavirin for hepatitis C virus genotype 3 and advanced liver disease: A randomized phase III study (ALLY-3+). Hepatology 2016, 63, 1430–1441. [Google Scholar] [CrossRef] [Green Version]
- Poordad, F.; Hezode, C.; Trinh, R.; Kowdley, K.V.; Zeuzem, S.; Agarwal, K.; Shiffman, M.L.; Wedemeyer, H.; Berg, T.; Yoshida, E.M.; et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N. Engl. J. Med. 2014, 370, 1973–1982. [Google Scholar] [CrossRef]
- Belli, L.S.; Berenguer, M.; Cortesi, P.A.; Strazzabosco, M.; Rockenschaub, S.R.; Martini, S.; Morelli, C.; Donato, F.; Volpes, R.; Pageaux, G.P.; et al. Delisting of liver transplant candidates with chronic hepatitis C after viral eradication: A European study. J. Hepatol. 2016, 65, 524–531. [Google Scholar] [CrossRef]
- Sievert, W.; Razavi, H.; Estes, C.; Thompson, A.J.; Zekry, A.; Roberts, S.K.; Dore, G.J. Enhanced antiviral treatment efficacy and uptake in preventing the rising burden of hepatitis C-related liver disease and costs in Australia. J. Gastroenterol. Hepatol. 2014, 29 (Suppl. S1), 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Kozbial, K.; Moser, S.; Schwarzer, R.; Laferl, H.; Al-Zoairy, R.; Stauber, R.; Stattermayer, A.F.; Beinhardt, S.; Graziadei, I.; Freissmuth, C.; et al. Unexpected high incidence of hepatocellular carcinoma in cirrhotic patients with sustained virologic response following interferon-free direct-acting antiviral treatment. J. Hepatol. 2016, 65, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, H.; Vale, A.M.; Rodrigues, S.; Goncalves, R.; Albuquerque, A.; Pereira, P.; Lopes, S.; Silva, M.; Andrade, P.; Morais, R.; et al. High incidence of hepatocellular carcinoma following successful interferon-free antiviral therapy for hepatitis C associated cirrhosis. J. Hepatol. 2016, 65, 1070–1071. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.C.M.; Walker, A.J.; Hudson, B.E.; Verma, S.; McLauchlan, J.; Mutimer, D.J.; Brown, A.; Gelson, W.T.H.; MacDonald, D.C.; Agarwal, K.; et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis C and decompensated cirrhosis. J. Hepatol. 2016, 65, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reig, M.; Marino, Z.; Perello, C.; Inarrairaegui, M.; Ribeiro, A.; Lens, S.; Diaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Aqel, B.A.; Pungpapong, S.; Gores, G.J.; Roberts, L.R.; Leise, M.D. Direct acting antiviral therapy and tumor recurrence after liver transplantation for hepatitis C-associated hepatocellular carcinoma. J. Hepatol. 2016, 65, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Zavaglia, C.; Okolicsanyi, S.; Cesarini, L.; Mazzarelli, C.; Pontecorvi, V.; Ciaccio, A.; Strazzabosco, M.; Belli, L.S. Is the risk of neoplastic recurrence increased after prescribing direct-acting antivirals for HCV patients whose HCC was previously cured? J. Hepatol. 2017, 66, 236–237. [Google Scholar] [CrossRef]
- Minami, T.; Tateishi, R.; Nakagomi, R.; Fujiwara, N.; Sato, M.; Enooku, K.; Nakagawa, H.; Asaoka, Y.; Kondo, Y.; Shiina, S.; et al. The impact of direct-acting antivirals on early tumor recurrence after radiofrequency ablation in hepatitis C-related hepatocellular carcinoma. J. Hepatol. 2016, 65, 1272–1273. [Google Scholar] [CrossRef]
- Nault, J.C.; Colombo, M. Hepatocellular carcinoma and direct acting antiviral treatments: Controversy after the revolution. J. Hepatol. 2016, 65, 663–665. [Google Scholar] [CrossRef]
- Meissner, E.G.; Wu, D.; Osinusi, A.; Bon, D.; Virtaneva, K.; Sturdevant, D.; Porcella, S.; Wang, H.; Herrmann, E.; McHutchison, J.; et al. Endogenous intrahepatic IFNs and association with IFN-free HCV treatment outcome. J. Clin. Investig. 2014, 124, 3352–3363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchill, M.A.; Golden-Mason, L.; Wind-Rotolo, M.; Rosen, H.R. Memory re-differentiation and reduced lymphocyte activation in chronic HCV-infected patients receiving direct-acting antivirals. J. Viral Hepat. 2015, 22, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Spaan, M.; van Oord, G.; Kreefft, K.; Hou, J.; Hansen, B.E.; Janssen, H.L.; de Knegt, R.J.; Boonstra, A. Immunological Analysis During Interferon-Free Therapy for Chronic Hepatitis C Virus Infection Reveals Modulation of the Natural Killer Cell Compartment. J. Infect. Dis. 2016, 213, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Casadei Gardini, A.; Conti, F.; Foschi, F.G.; Brillanti, S.; Andreone, P.; Mazzella, G.; Ravaioli, F. Imbalance of Neutrophils and Lymphocyte Counts Can Be Predictive of Hepatocellular Carcinoma Occurrence in Hepatitis C-related Cirrhosis Treated With Direct-acting Antivirals. Gastroenterology 2018, 154, 2281–2282. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.S.; Nakamoto, N.; Taniki, N.; Ojiro, K.; Amiya, T.; Makita, Y.; Murata, H.; Yamaguchi, A.; Shiba, S.; Miyake, R.; et al. On-treatment decrease of NKG2D correlates to early emergence of clinically evident hepatocellular carcinoma after interferon-free therapy for chronic hepatitis C. PLoS ONE 2017, 12, e0179096. [Google Scholar] [CrossRef] [PubMed]
- Ning, G.; Li, Y.T.; Chen, Y.M.; Zhang, Y.; Zeng, Y.F.; Lin, C.S. Dynamic Changes of the Frequency of Classic and Inflammatory Monocytes Subsets and Natural Killer Cells in Chronic Hepatitis C Patients Treated by Direct-Acting Antiviral Agents. Can. J. Gastroenterol. Hepatol. 2017, 2017, 3612403. [Google Scholar] [CrossRef]
- Meissner, E.G.; Kohli, A.; Higgins, J.; Lee, Y.J.; Prokunina, O.; Wu, D.; Orr, C.; Masur, H.; Kottilil, S. Rapid changes in peripheral lymphocyte concentrations during interferon-free treatment of chronic hepatitis C virus infection. Hepatol. Commun. 2017, 1, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Langhans, B.; Nischalke, H.D.; Kramer, B.; Hausen, A.; Dold, L.; van Heteren, P.; Huneburg, R.; Nattermann, J.; Strassburg, C.P.; Spengler, U. Increased peripheral CD4(+) regulatory T cells persist after successful direct-acting antiviral treatment of chronic hepatitis C. J. Hepatol. 2017, 66, 888–896. [Google Scholar] [CrossRef]
- Villani, R.; Facciorusso, A.; Bellanti, F.; Tamborra, R.; Piscazzi, A.; Landriscina, M.; Vendemiale, G.; Serviddio, G. DAAs Rapidly Reduce Inflammation but Increase Serum VEGF Level: A Rationale for Tumor Risk during Anti-HCV Treatment. PLoS ONE 2016, 11, e0167934. [Google Scholar] [CrossRef]
- Debes, J.D.; van Tilborg, M.; Groothuismink, Z.M.A.; Hansen, B.E.; Schulze Zur Wiesch, J.; von Felden, J.; de Knegt, R.J.; Boonstra, A. Levels of Cytokines in Serum Associate With Development of Hepatocellular Carcinoma in Patients With HCV Infection Treated With Direct-Acting Antivirals. Gastroenterology 2018, 154, 515–517. [Google Scholar] [CrossRef]
- Carlin, A.F.; Aristizabal, P.; Song, Q.; Wang, H.; Paulson, M.S.; Stamm, L.M.; Schooley, R.T.; Wyles, D.L. Temporal dynamics of inflammatory cytokines/chemokines during sofosbuvir and ribavirin therapy for genotype 2 and 3 hepatitis C infection. Hepatology 2015, 62, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Faillaci, F.; Marzi, L.; Critelli, R.; Milosa, F.; Schepis, F.; Turola, E.; Andreani, S.; Vandelli, G.; Bernabucci, V.; Lei, B.; et al. Liver Angiopoietin-2 is a key predictor of de novo or recurrent hepatocellular cancer after HCV direct-acting antivirals. Hepatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Koziel, M.J. Cellular immune responses against hepatitis C virus. Clin. Infect. Dis. 2005, 41 (Suppl. S1), S25–S31. [Google Scholar] [CrossRef] [PubMed]
- Terilli, R.R.; Cox, A.L. Immunity and hepatitis C: A review. Curr. HIV/AIDS Rep. 2013, 10, 51–58. [Google Scholar] [CrossRef]
- Rigopoulou, E.I.; Zachou, K.; Gatselis, N.; Koukoulis, G.K.; Dalekos, G.N. Autoimmune hepatitis in patients with chronic HBV and HCV infections: Patterns of clinical characteristics, disease progression and outcome. Ann. Hepatol. 2013, 13, 127–135. [Google Scholar] [PubMed]
- Strassburg, C.P.; Vogel, A.; Manns, M.P. Autoimmunity and hepatitis C. Autoimmun. Rev. 2003, 2, 322–331. [Google Scholar] [CrossRef]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, S14–S25. [Google Scholar] [CrossRef]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef]
- Thimme, R.; Bukh, J.; Spangenberg, H.C.; Wieland, S.; Pemberton, J.; Steiger, C.; Govindarajan, S.; Purcell, R.H.; Chisari, F.V. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15661–15668. [Google Scholar] [CrossRef] [Green Version]
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Binder, M.; Bartenschlager, R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol. Rev. 2012, 36, 663–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.C.; Park, S.H.; Demino, M.; Nascimbeni, M.; Mihalik, K.; Major, M.; Veerapu, N.S.; Heller, T.; Feinstone, S.M.; Rice, C.M.; et al. Delayed induction, not impaired recruitment, of specific CD8(+) T cells causes the late onset of acute hepatitis C. Gastroenterology 2011, 141, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Klenerman, P.; Thimme, R. T cell responses in hepatitis C: The good, the bad and the unconventional. Gut 2012, 61, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Pestka, J.M.; Zeisel, M.B.; Blaser, E.; Schurmann, P.; Bartosch, B.; Cosset, F.L.; Patel, A.H.; Meisel, H.; Baumert, J.; Viazov, S.; et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. USA 2007, 104, 6025–6030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimme, R.; Oldach, D.; Chang, K.M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Erickson, A.L.; Adams, E.J.; Kansopon, J.; Weiner, A.J.; Chien, D.Y.; Houghton, M.; Parham, P.; Walker, C.M. Analysis of a successful immune response against hepatitis C virus. Immunity 1999, 10, 439–449. [Google Scholar] [CrossRef]
- Takaki, A.; Wiese, M.; Maertens, G.; Depla, E.; Seifert, U.; Liebetrau, A.; Miller, J.L.; Manns, M.P.; Rehermann, B. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat. Med. 2000, 6, 578–582. [Google Scholar] [CrossRef]
- Diepolder, H.M.; Gerlach, J.T.; Zachoval, R.; Hoffmann, R.M.; Jung, M.C.; Wierenga, E.A.; Scholz, S.; Santantonio, T.; Houghton, M.; Southwood, S.; et al. Immunodominant CD4+ T-cell epitope within nonstructural protein 3 in acute hepatitis C virus infection. J. Virol. 1997, 71, 6011–6019. [Google Scholar]
- Lechner, F.; Wong, D.K.; Dunbar, P.R.; Chapman, R.; Chung, R.T.; Dohrenwend, P.; Robbins, G.; Phillips, R.; Klenerman, P.; Walker, B.D. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 2000, 191, 1499–1512. [Google Scholar] [CrossRef]
- Missale, G.; Bertoni, R.; Lamonaca, V.; Valli, A.; Massari, M.; Mori, C.; Rumi, M.G.; Houghton, M.; Fiaccadori, F.; Ferrari, C. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J. Clin. Investig. 1996, 98, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.M.; Avia, M.; Martin, V.; Sevilla, N. IL-10: A Multifunctional Cytokine in Viral Infections. J. Immunol. Res. 2017, 2017, 6104054. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.G.; Teyton, L.; Oldstone, M.B.; McGavern, D.B. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J. Virol. 2005, 79, 10514–10527. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Tu, Z.; Soldevila-Pico, C.; Abdelmalek, M.; Zhu, H.; Xu, Y.L.; Cabrera, R.; Liu, C.; Davis, G.L. Long-term interleukin 10 therapy in chronic hepatitis C patients has a proviral and anti-inflammatory effect. Hepatology 2003, 38, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, R.; Tu, Z.; Xu, Y.; Firpi, R.J.; Rosen, H.R.; Liu, C.; Nelson, D.R. An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology 2004, 40, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.L.; Liaw, Y.F.; Chen, M.H.; Huang, C.Y.; Kuo, G.C. Detection of type 2-like T-helper cells in hepatitis C virus infection: Implications for hepatitis C virus chronicity. Hepatology 1997, 25, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Tuma, R.A.; Pamer, E.G. Homeostasis of naive, effector and memory CD8 T cells. Curr. Opin. Immunol. 2002, 14, 348–353. [Google Scholar] [CrossRef]
- Piazzolla, G.; Tortorella, C.; Schiraldi, O.; Antonaci, S. Relationship between interferon-gamma, interleukin-10, and interleukin-12 production in chronic hepatitis C and in vitro effects of interferon-alpha. J. Clin. Immunol. 2000, 20, 54–61. [Google Scholar] [CrossRef]
- Sakaguchi, S. Regulatory T cells: Mediating compromises between host and parasite. Nat. Immunol. 2003, 4, 10–11. [Google Scholar] [CrossRef]
- Sakaguchi, S. Regulatory T cells: Key controllers of immunologic self-tolerance. Cell 2000, 101, 455–458. [Google Scholar] [CrossRef]
- Piccirillo, C.A.; Shevach, E.M. Cutting edge: Control of CD8+ T cell activation by CD4+CD25+ immunoregulatory cells. J. Immunol. 2001, 167, 1137–1140. [Google Scholar] [CrossRef]
- Billerbeck, E.; Bottler, T.; Thimme, R. Regulatory T cells in viral hepatitis. World J. Gastroenterol. 2007, 13, 4858–4864. [Google Scholar] [CrossRef]
- Barjon, C.; Dahlqvist, G.; Calmus, Y.; Conti, F. Role of regulatory T-cells during hepatitis C infection: From the acute phase to post-transplantation recurrence. Dig. Liver Dis. 2015, 47, 913–917. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ikeda, F.; Stadanlick, J.; Nunes, F.A.; Alter, H.J.; Chang, K.M. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology 2003, 38, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Rushbrook, S.M.; Ward, S.M.; Unitt, E.; Vowler, S.L.; Lucas, M.; Klenerman, P.; Alexander, G.J. Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J. Virol. 2005, 79, 7852–7859. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; Thornton, A.M. tTregs, pTregs, and iTregs: Similarities and differences. Immunol. Rev. 2014, 259, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wan, L.; Zhang, C.; Zheng, X.; Li, J.; Chen, Z.K. Tim-3-Galectin-9 pathway involves the suppression induced by CD4+CD25+ regulatory T cells. Immunobiology 2009, 214, 342–349. [Google Scholar] [CrossRef]
- Dilek, N.; Poirier, N.; Hulin, P.; Coulon, F.; Mary, C.; Ville, S.; Vie, H.; Clemenceau, B.; Blancho, G.; Vanhove, B. Targeting CD28, CTLA-4 and PD-L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T-cells. PLoS ONE 2013, 8, e83139. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Chiu, J.; Ernst, D.M.; Keating, A. Acquired Natural Killer Cell Dysfunction in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Front. Immunol. 2018, 9, 267. [Google Scholar] [CrossRef]
- Doherty, D.G.; O’Farrelly, C. Innate and adaptive lymphoid cells in the human liver. Immunol. Rev. 2000, 174, 5–20. [Google Scholar] [CrossRef]
- Serti, E.; Chepa-Lotrea, X.; Kim, Y.J.; Keane, M.; Fryzek, N.; Liang, T.J.; Ghany, M.; Rehermann, B. Successful Interferon-Free Therapy of Chronic Hepatitis C Virus Infection Normalizes Natural Killer Cell Function. Gastroenterology 2015, 149, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Oliviero, B.; Varchetta, S.; Paudice, E.; Michelone, G.; Zaramella, M.; Mavilio, D.; De Filippi, F.; Bruno, S.; Mondelli, M.U. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 2009, 137, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, G.; Titerence, R.H.; Koh, C.; Edlich, B.; Feld, J.J.; Rotman, Y.; Ghany, M.G.; Hoofnagle, J.H.; Liang, T.J.; Heller, T.; et al. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology 2010, 138, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Mondelli, M.U. Direct-Acting Antivirals Cure Innate Immunity in Chronic Hepatitis C. Gastroenterology 2015, 149, 25–28. [Google Scholar] [CrossRef]
- Missale, G.; Pilli, M.; Zerbini, A.; Penna, A.; Ravanetti, L.; Barili, V.; Orlandini, A.; Molinari, A.; Fasano, M.; Santantonio, T.; et al. Lack of full CD8 functional restoration after antiviral treatment for acute and chronic hepatitis C virus infection. Gut 2012, 61, 1076–1084. [Google Scholar] [CrossRef]
- Martin, B.; Hennecke, N.; Lohmann, V.; Kayser, A.; Neumann-Haefelin, C.; Kukolj, G.; Bocher, W.O.; Thimme, R. Restoration of HCV-specific CD8+ T cell function by interferon-free therapy. J. Hepatol. 2014, 61, 538–543. [Google Scholar] [CrossRef]
- Abdel-Hakeem, M.S.; Bedard, N.; Badr, G.; Ostrowski, M.; Sekaly, R.P.; Bruneau, J.; Willems, B.; Heathcote, E.J.; Shoukry, N.H. Comparison of immune restoration in early versus late alpha interferon therapy against hepatitis C virus. J. Virol. 2010, 84, 10429–10435. [Google Scholar] [CrossRef]
- Seigel, B.; Bengsch, B.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Thimme, R. Factors that determine the antiviral efficacy of HCV-specific CD8(+) T cells ex vivo. Gastroenterology 2013, 144, 426–436. [Google Scholar] [CrossRef]
- Ahlenstiel, G. The natural killer cell response to HCV infection. Immune Netw. 2013, 13, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Novick, D.; Rubinstein, M.; Siegmund, B.; Enrich, B.; Koch, R.O.; Vogel, W.; Kim, S.H.; Dinarello, C.A.; Tilg, H. Interferon-alpha induces interleukin-18 binding protein in chronic hepatitis C patients. Clin. Exp. Immunol. 2002, 129, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Wherry, E.J. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. 2007, 15, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; O’Garra, A.; de Waal Malefyt, R.; Vieira, P.; Mosmann, T.R. Interleukin-10. Annu. Rev. Immunol. 1993, 11, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Sarkar, D.; Walter, M.R.; Shi, Y.; Fisher, P.B. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 2004, 22, 929–979. [Google Scholar] [CrossRef]
- Ejrnaes, M.; Filippi, C.M.; Martinic, M.M.; Ling, E.M.; Togher, L.M.; Crotty, S.; von Herrath, M.G. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 2006, 203, 2461–2472. [Google Scholar] [CrossRef] [Green Version]
- Snell, L.M.; Osokine, I.; Yamada, D.H.; De la Fuente, J.R.; Elsaesser, H.J.; Brooks, D.G. Overcoming CD4 Th1 Cell Fate Restrictions to Sustain Antiviral CD8 T Cells and Control Persistent Virus Infection. Cell Rep. 2016, 16, 3286–3296. [Google Scholar] [CrossRef] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Flynn, J.K.; Dore, G.J.; Hellard, M.; Yeung, B.; Rawlinson, W.D.; White, P.A.; Kaldor, J.M.; Lloyd, A.R.; Ffrench, R.A.; Group, A.S. Early IL-10 predominant responses are associated with progression to chronic hepatitis C virus infection in injecting drug users. J. Viral Hepat. 2011, 18, 549–561. [Google Scholar] [CrossRef] [PubMed]
- De Maria, A.; Fogli, M.; Mazza, S.; Basso, M.; Picciotto, A.; Costa, P.; Congia, S.; Mingari, M.C.; Moretta, L. Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur. J. Immunol. 2007, 37, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockman, M.A.; Kwon, D.S.; Tighe, D.P.; Pavlik, D.F.; Rosato, P.C.; Sela, J.; Porichis, F.; Le Gall, S.; Waring, M.T.; Moss, K.; et al. IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood 2009, 114, 346–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Parish, I.A.; Marshall, H.D.; Staron, M.M.; Lang, P.A.; Brustle, A.; Chen, J.H.; Cui, W.; Tsui, Y.C.; Perry, C.; Laidlaw, B.J.; et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J. Clin. Investig. 2014, 124, 3455–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. The origin of FOXP3-expressing CD4+ regulatory T cells: Thymus or periphery. J. Clin. Investig. 2003, 112, 1310–1312. [Google Scholar] [CrossRef]
- Ulsenheimer, A.; Gerlach, J.T.; Gruener, N.H.; Jung, M.C.; Schirren, C.A.; Schraut, W.; Zachoval, R.; Pape, G.R.; Diepolder, H.M. Detection of functionally altered hepatitis C virus-specific CD4 T cells in acute and chronic hepatitis C. Hepatology 2003, 37, 1189–1198. [Google Scholar] [CrossRef]
- MacDonald, A.J.; Duffy, M.; Brady, M.T.; McKiernan, S.; Hall, W.; Hegarty, J.; Curry, M.; Mills, K.H. CD4 T helper type 1 and regulatory T cells induced against the same epitopes on the core protein in hepatitis C virus-infected persons. J. Infect. Dis. 2002, 185, 720–727. [Google Scholar] [CrossRef]
- Graham, C.S.; Wells, A.; Liu, T.; Sherman, K.E.; Peters, M.; Chung, R.T.; Bhan, A.K.; Andersen, J.; Koziel, M.J.; Team, A.S. Antigen-specific immune responses and liver histology in HIV and hepatitis C coinfection. AIDS 2005, 19, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, D.E.; Ikeda, F.; Li, Y.; Nakamoto, N.; Ganesan, S.; Valiga, M.E.; Nunes, F.A.; Rajender Reddy, K.; Chang, K.M. Peripheral virus-specific T-cell interleukin-10 responses develop early in acute hepatitis C infection and become dominant in chronic hepatitis. J. Hepatol. 2008, 48, 903–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, M.; Sene, D.; Pol, S.; Bourliere, M.; Poynard, T.; Charlotte, F.; Cacoub, P.; Caillat-Zucman, S. Intrahepatic virus-specific IL-10-producing CD8 T cells prevent liver damage during chronic hepatitis C virus infection. Hepatology 2006, 44, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accapezzato, D.; Francavilla, V.; Paroli, M.; Casciaro, M.; Chircu, L.V.; Cividini, A.; Abrignani, S.; Mondelli, M.U.; Barnaba, V. Hepatic expansion of a virus-specific regulatory CD8(+) T cell population in chronic hepatitis C virus infection. J. Clin. Investig. 2004, 113, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.W.; Oh, B.S.; Kwon, J.H.; You, C.R.; Chung, K.W.; Kay, C.S.; Jung, H.S. Serum interleukin-6 and C-reactive protein as a prognostic indicator in hepatocellular carcinoma. Cytokine 2012, 60, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, J.; Hu, X.; Liu, S.; He, B. Prognostic and Therapeutic Values of Tumor Necrosis Factor-Alpha in Hepatocellular Carcinoma. Med. Sci. Monit. 2016, 22, 3694–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrajoli, A.; Keating, M.J.; Manshouri, T.; Giles, F.J.; Dey, A.; Estrov, Z.; Koller, C.A.; Kurzrock, R.; Thomas, D.A.; Faderl, S.; et al. The clinical significance of tumor necrosis factor-alpha plasma level in patients having chronic lymphocytic leukemia. Blood 2002, 100, 1215–1219. [Google Scholar]
- Ahmed, M.I.; Salahy, E.E.; Fayed, S.T.; El-Hefnawy, N.G.; Khalifa, A. Human papillomavirus infection among Egyptian females with cervical carcinoma: Relationship to spontaneous apoptosis and TNF-alpha. Clin. Biochem. 2001, 34, 491–498. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Grimshaw, M.J.; Kulbe, H.; Wilson, J.L.; Wilbanks, G.D.; Burke, F.; Balkwill, F.R. Expression and regulation of tumor necrosis factor alpha in normal and malignant ovarian epithelium. Mol. Cancer Ther. 2006, 5, 382–390. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, M.; Kono, H.; Amemiya, H.; Matsuda, M.; Suzuki, T.; Maki, A.; Fujii, H. Role of interleukin-18 and its receptor in hepatocellular carcinoma associated with hepatitis C virus infection. Int. J. Cancer 2006, 118, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Okamura, H.; Tsutsi, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Stark, G.R. NFkappaB-dependent signaling pathways. Exp. Hematol. 2002, 30, 285–296. [Google Scholar] [CrossRef]
- Hassan, M.; Selimovic, D.; El-Khattouti, A.; Soell, M.; Ghozlan, H.; Haikel, Y.; Abdelkader, O.; Megahed, M. Hepatitis C virus-mediated angiogenesis: Molecular mechanisms and therapeutic strategies. World J. Gastroenterol. 2014, 20, 15467–15475. [Google Scholar] [CrossRef] [PubMed]
- Tandle, A.; Blazer, D.G., III; Libutti, S.K. Antiangiogenic gene therapy of cancer: Recent developments. J. Transl. Med. 2004, 2, 22. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Selimovic, D.; Ghozlan, H.; Abdel-kader, O. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology 2009, 49, 1469–1482. [Google Scholar] [CrossRef] [Green Version]
- Salcedo, X.; Medina, J.; Sanz-Cameno, P.; Garcia-Buey, L.; Martin-Vilchez, S.; Borque, M.J.; Lopez-Cabrera, M.; Moreno-Otero, R. The potential of angiogenesis soluble markers in chronic hepatitis C. Hepatology 2005, 42, 696–701. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef]
- Elpek, G.O. Angiogenesis and liver fibrosis. World J. Hepatol. 2015, 7, 377–391. [Google Scholar] [CrossRef]
- Duffy, A.M.; Bouchier-Hayes, D.J.; Harmey, J.H. Vascular Endothelial Growth Factor (VEGF) and Its Role in Non-Endothelial Cells: Autocrine Signalling by VEGF. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Mise, M.; Arii, S.; Higashituji, H.; Furutani, M.; Niwano, M.; Harada, T.; Ishigami, S.; Toda, Y.; Nakayama, H.; Fukumoto, M.; et al. Clinical significance of vascular endothelial growth factor and basic fibroblast growth factor gene expression in liver tumor. Hepatology 1996, 23, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Horbach, A.; Kubitz, R.; Frilling, A.; Haussinger, D. Disruption of hepatocellular tight junctions by vascular endothelial growth factor (VEGF): A novel mechanism for tumor invasion. J. Hepatol. 2004, 41, 274–283. [Google Scholar] [CrossRef]
- Park, Y.N.; Kim, Y.B.; Yang, K.M.; Park, C. Increased expression of vascular endothelial growth factor and angiogenesis in the early stage of multistep hepatocarcinogenesis. Arch. Pathol. Lab. Med. 2000, 124, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Bupathi, M.; Kaseb, A.; Janku, F. Angiopoietin 2 as a therapeutic target in hepatocellular carcinoma treatment: Current perspectives. OncoTargets Ther. 2014, 7, 1927–1932. [Google Scholar] [CrossRef]
- Torimura, T.; Ueno, T.; Kin, M.; Harada, R.; Taniguchi, E.; Nakamura, T.; Sakata, R.; Hashimoto, O.; Sakamoto, M.; Kumashiro, R.; et al. Overexpression of angiopoietin-1 and angiopoietin-2 in hepatocellular carcinoma. J. Hepatol. 2004, 40, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Ker, C.G.; Chen, H.Y.; Juan, C.C.; Lo, H.W.; Shen, Y.Y.; Chen, J.S.; Lee, K.T.; Sheen, P.C. Role of angiogenesis in hepatitis and hepatocellular carcinoma. Hepatogastroenterology 1999, 46, 646–650. [Google Scholar] [PubMed]
- Garcia-Monzon, C.; Sanchez-Madrid, F.; Garcia-Buey, L.; Garcia-Arroyo, A.; Garcia-Sanchez, A.; Moreno-Otero, R. Vascular adhesion molecule expression in viral chronic hepatitis: Evidence of neoangiogenesis in portal tracts. Gastroenterology 1995, 108, 231–241. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Waris, G.; Mikolon, D.; Stupack, D.G.; Siddiqui, A. Hepatitis C virus stabilizes hypoxia-inducible factor 1alpha and stimulates the synthesis of vascular endothelial growth factor. J. Virol. 2007, 81, 10249–10257. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.K.; Brimacombe, C.L.; Rowe, I.A.; Reynolds, G.M.; Fletcher, N.F.; Stamataki, Z.; Bhogal, R.H.; Simoes, M.L.; Ashcroft, M.; Afford, S.C.; et al. A dual role for hypoxia inducible factor-1alpha in the hepatitis C virus lifecycle and hepatoma migration. J. Hepatol. 2012, 56, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.; Caveda, L.; Sanz-Cameno, P.; Arroyo, A.G.; Martin-Vilchez, S.; Majano, P.L.; Garcia-Buey, L.; Sanchez-Madrid, F.; Moreno-Otero, R. Hepatocyte growth factor activates endothelial proangiogenic mechanisms relevant in chronic hepatitis C-associated neoangiogenesis. J. Hepatol. 2003, 38, 660–667. [Google Scholar] [CrossRef]
- Mazzanti, R.; Messerini, L.; Monsacchi, L.; Buzzelli, G.; Zignego, A.L.; Foschi, M.; Monti, M.; Laffi, G.; Morbidelli, L.; Fantappie, O.; et al. Chronic viral hepatitis induced by hepatitis C but not hepatitis B virus infection correlates with increased liver angiogenesis. Hepatology 1997, 25, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mas, V.R.; Maluf, D.G.; Archer, K.J.; Yanek, K.C.; Fisher, R.A. Angiogenesis soluble factors as hepatocellular carcinoma noninvasive markers for monitoring hepatitis C virus cirrhotic patients awaiting liver transplantation. Transplantation 2007, 84, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
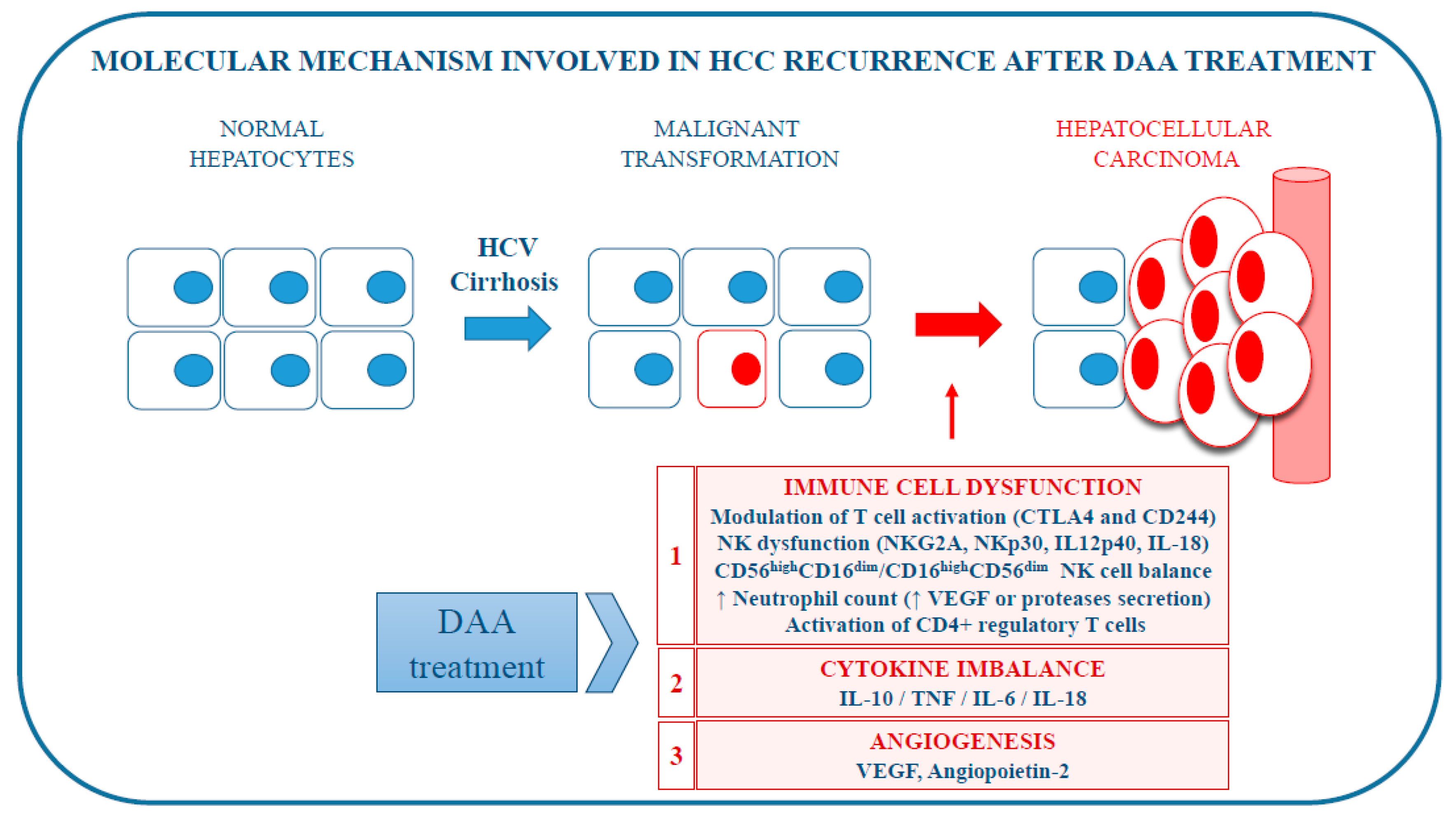
Figure 1.
Molecular mechanisms potentially involved in hepatocellular carcinoma (HCC) recurrence after direct-acting antiviral (DAA) treatment for chronic HCV infection.
Figure 1.
Molecular mechanisms potentially involved in hepatocellular carcinoma (HCC) recurrence after direct-acting antiviral (DAA) treatment for chronic HCV infection.

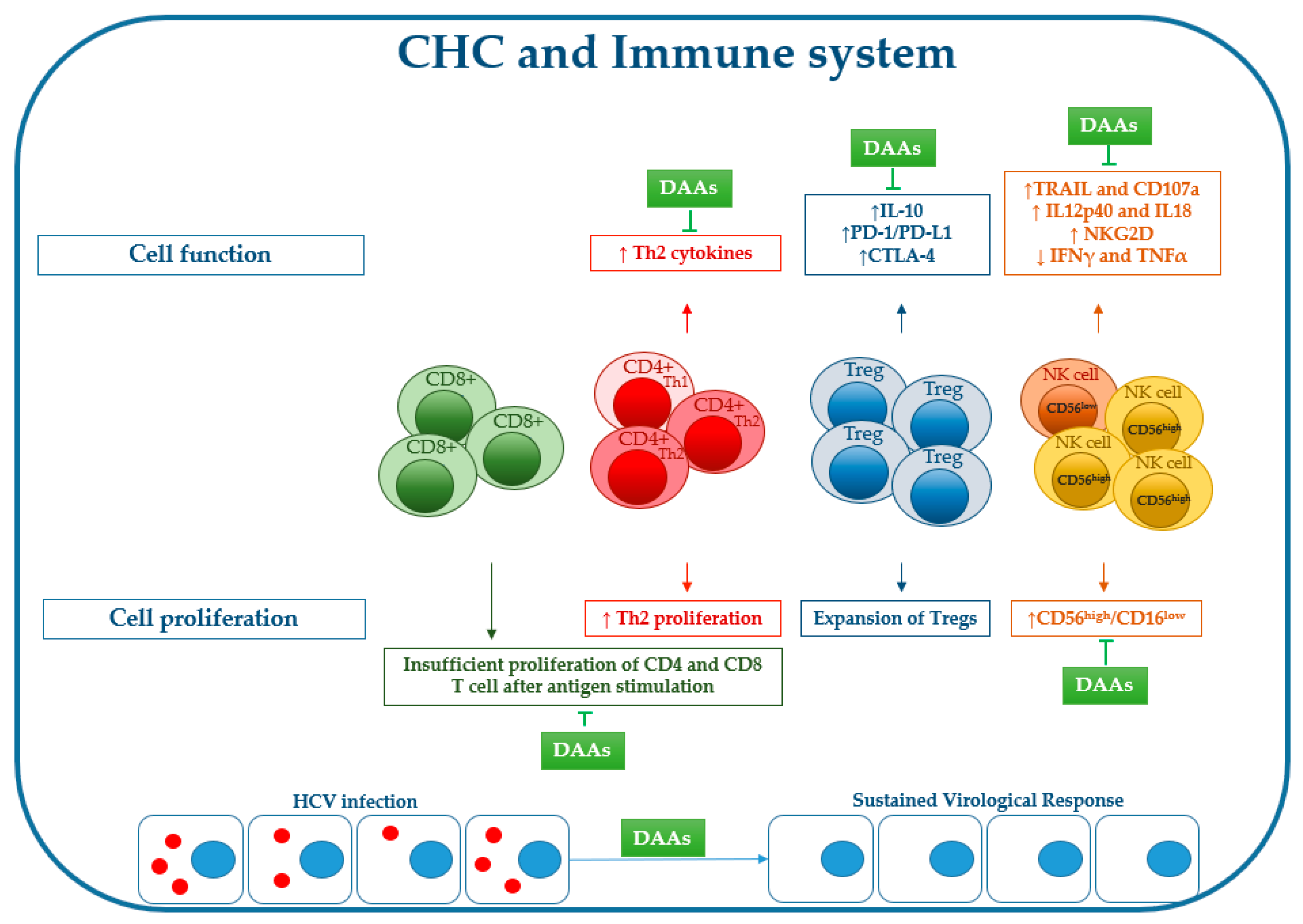
Figure 2.
Effects of chronic HCV infection on immune cell proliferation/function and effect of DAA treatment on recovery of virus-induced immune changes (for explanations, see the main text).
Figure 2.
Effects of chronic HCV infection on immune cell proliferation/function and effect of DAA treatment on recovery of virus-induced immune changes (for explanations, see the main text).


{kind=link}
{kind=link}
Table 1.
Summary of potential mechanisms involved in HCC recurrence.
| Hypothesis | Molecular Mechanism | Author | Ref. |
|---|---|---|---|
| Immune cell dysfunction | Modulation of T cell activation | Meissner et al., 2014 | [41] |
| Reduced NK cell activation | Burchill et al., 2015 | [42] | |
| Downregulation of NKG2A receptor | Spaan et al., 2016 | [43] | |
| Declined IL12p40, IL-18 serum level | Spaan et al., 2016 | [43] | |
| Modulation of differential white blood cell count | Casadei Gardini et al., 2018 | [44] | |
| Imbalance in NK cell subgroups | Chu et al., 2017 | [45] | |
| Decreased NKG2D | Chu et al., 2017 | [45] | |
| Decreased frequencies of NK cells | Ning et al., 2017; | [46] | |
| Meissner et al., 2017 | [47] | ||
| Immunosuppressive Tregs function | Langhans et al., 2017 | [48] | |
| Change in immune cytokine network | Rapid reduction of IL-10 serum level | Villani et al., 2016 | [49] |
| Increased TNFα secretion | Debes et al., 2018 | [50] | |
| Change in IL-6 serum level | Debes et al., 2018 | [50] | |
| Change in IL-18 serum level | Spaan et al., 2016; | [43] | |
| Ning et al., 2017; | [46] | ||
| Carlin et al., 2015 | [51] | ||
| Activation of angiogenesis | Increase of VEGF serum level | Villani et al., 2016; | [49] |
| Faillaci et al., 2018 | [52] | ||
| Increase of angiopoietin-2 serum level | Faillaci et al., 2018 | [52] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Villani, R.; Vendemiale, G.; Serviddio, G. Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy. Int. J. Mol. Sci. 2019, 20, 49. https://doi.org/10.3390/ijms20010049
AMA Style
Villani R, Vendemiale G, Serviddio G. Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy. International Journal of Molecular Sciences. 2019; 20(1):49. https://doi.org/10.3390/ijms20010049
Chicago/Turabian StyleVillani, Rosanna, Gianluigi Vendemiale, and Gaetano Serviddio. 2019. "Molecular Mechanisms Involved in HCC Recurrence after Direct-Acting Antiviral Therapy" International Journal of Molecular Sciences 20, no. 1: 49. https://doi.org/10.3390/ijms20010049
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.