Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment

Abstract
:1. Introduction
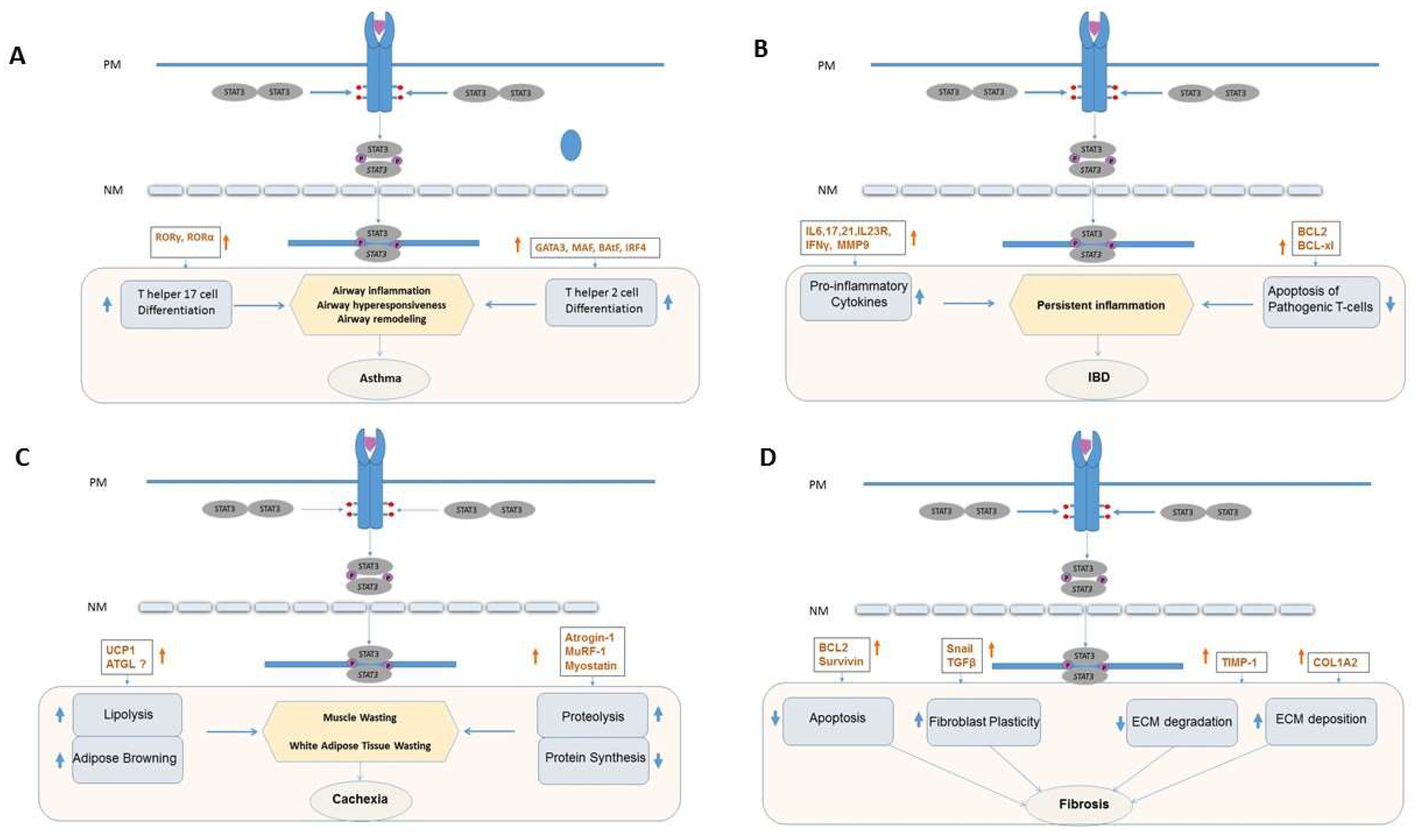
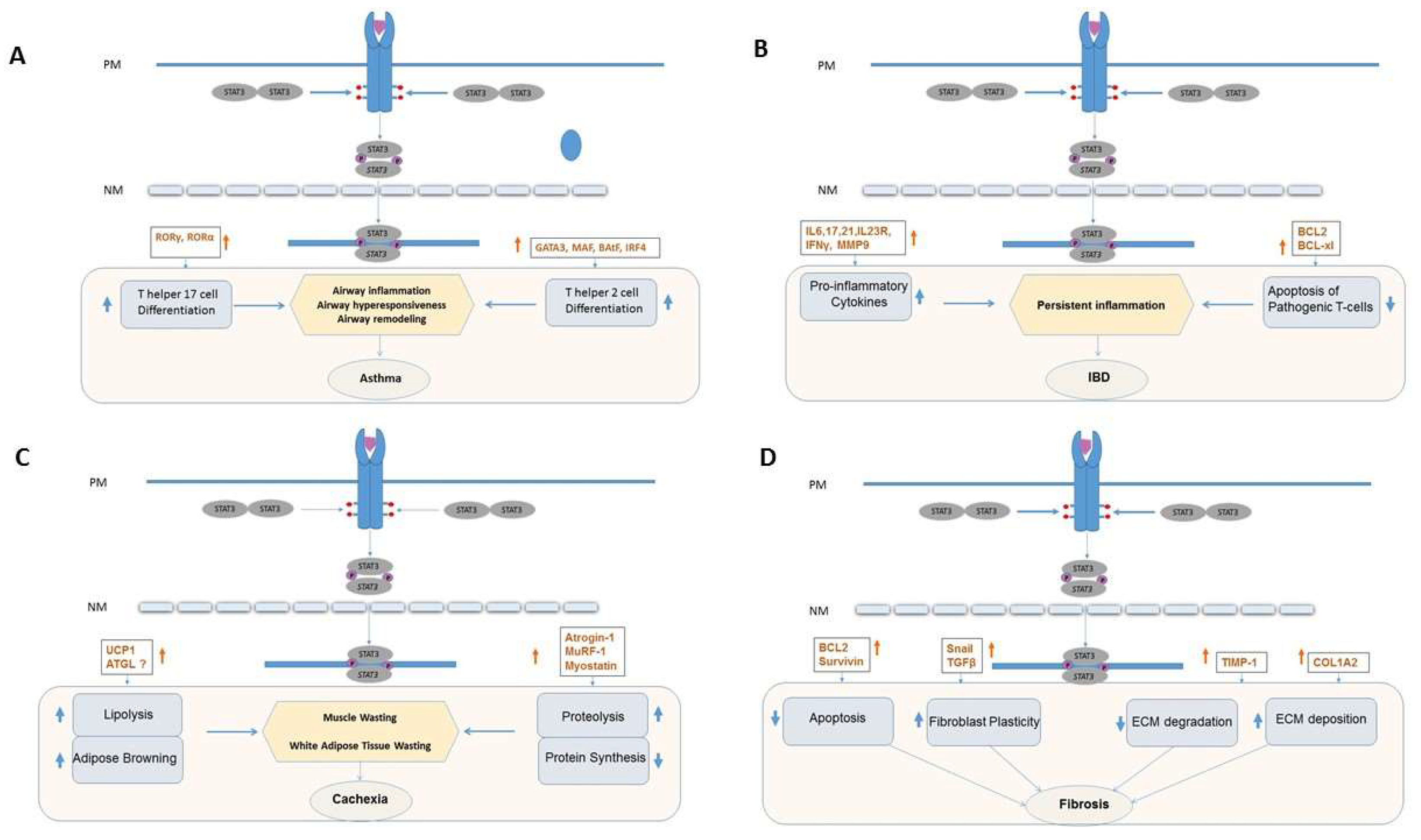
2. STAT3 and Asthma
2.1. Overview
2.2. Contribution of STAT3 and Its Targeting in Asthma
2.3. Drug Targeting of STAT3 in Asthma
3. STAT3 and Inflammatory Bowel Disease
3.1. Overview
3.2. Contribution of STAT3 to IBD
3.3. Targeting Cytokines and Cytokine Receptors in IBD
3.4. Drug Targeting of STAT3 in IBD
4. STAT3 and Cachexia
4.1. Overview of Cachexia
4.2. Inflammation and Cachexia
4.3. IL-6, JAK and STAT3 Signaling Axis in Cachexia
4.4. STAT3, Proteolysis, and Activation of the Ubiquitin Proteosome System (UPS)
4.5. STAT3 in Autophagy-Mediated Muscle Loss
4.6. STAT3 in Lipolysis and Adipose Tissue Browning
4.7. Drug Targeting of STAT3 in Cachexia
5. STAT3 and Fibrosis
5.1. Overview of Fibrosis
5.2. Contribution of STAT3 to Fibrosis
5.3. STAT3 and the ECM
5.4. STAT3 and Fibroblast Apoptosis
5.5. STAT3 and Fibroblast Plasticity
5.6. Interplay between STAT3, TGF-β1 and Other Signaling Networks
5.7. Drug Targeting STAT3 to Treat Fibrosis
6. STAT3 and Integration of Complex Signaling Networks
7. Outlook
Funding
Conflicts of Interest
References
- Darnell, J.E.J.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C.J.; Rose-John, S.; Lissilaa, R.; Ferlin, W.; Ernst, M.; Hertzog, P.J.; Mansell, A.; Jenkins, B.J. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Caiello, I.; Minnone, G.; Holzinger, D.; Vogl, T.; Prencipe, G.; Manzo, A.; De Benedetti, F.; Strippoli, R. IL-6 amplifies TLR mediated cytokine and chemokine production: Implications for the pathogenesis of rheumatic inflammatory diseases. PLoS ONE 2014, 9, e107886. [Google Scholar] [CrossRef] [PubMed]
- Moran, A.; Akcan Arikan, A.; Mastrangelo, M.-A.A.; Wu, Y.; Yu, B.; Poli, V.; Tweardy, D.J. Prevention of trauma and hemorrhagic shock-mediated liver apoptosis by activation of stat3α. Int. J. Clin. Exp. Med. 2008, 1, 213–247. [Google Scholar] [PubMed]
- Moran, A.; Tsimelzon, A.I.; Mastrangelo, M.-A.A.; Wu, Y.; Yu, B.; Hilsenbeck, S.G.; Poli, V.; Tweardy, D.J. Prevention of trauma/hemorrhagic shock-induced lung apoptosis by IL-6-mediated activation of Stat3. Clin. Transl. Sci. 2009, 2, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Thacker, S.A.; Robinson, P.; Abel, A.; Tweardy, D.J. Modulation of the unfolded protein response during hepatocyte and cardiomyocyte apoptosis in trauma/hemorrhagic shock. Sci. Rep. 2013, 3, 1187. [Google Scholar] [CrossRef] [PubMed]
- Akinbami, L.J.; Moorman, J.E.; Bailey, C.; Zahran, H.S.; King, M.; Johnson, C.A.; Liu, X. Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010. NCHS Data Brief 2012, 94, 1–8. [Google Scholar]
- Fahy, J.V. Eosinophilic and neutrophilic inflammation in asthma: Insights from clinical studies. Proc. Am. Thorac. Soc. 2009, 6, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E. Asthma: Defining of the persistent adult phenotypes. Lancet 2006, 368, 804–813. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Poon, A.H.; Hamid, Q. Asthma phenotypes and endotypes. Curr. Opin. Pulm. Med. 2013, 19, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Thoracic Society. Proceedings of the ATS workshop on refractory asthma: Current understanding, recommendations, and unanswered questions. Am. J. Respir. Crit. Care Med. 2000, 162, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Al-Ramli, W.; Prefontaine, D.; Chouiali, F.; Martin, J.G.; Olivenstein, R.; Lemiere, C.; Hamid, Q. TH17-associated cytokines (IL-17A and IL-17F) in severe asthma. J. Allergy Clin. Immunol. 2009, 123, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; Dupont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef] [PubMed]
- Simeone-Penney, M.C.; Severgnini, M.; Tu, P.; Homer, R.J.; Mariani, T.J.; Cohn, L.; Simon, A.R. Airway epithelial STAT3 is required for allergic inflammation in a murine model of asthma. J. Immunol. 2007, 178, 6191–6199. [Google Scholar] [CrossRef] [PubMed]
- Stritesky, G.L.; Muthukrishnan, R.; Sehra, S.; Goswami, R.; Pham, D.; Travers, J.; Nguyen, E.T.; Levy, D.E.; Kaplan, M.H. The transcription factor STAT3 is required for T helper 2 cell development. Immunity 2011, 34, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Cho, M.; Choi, G.; Na, H.; Chung, Y. Dynamic control of Th2 cell responses by STAT3 during allergic lung inflammation in mice. Int. Immunopharmacol. 2015, 28, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Chew, G.Y.J.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired th17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Laurence, A.; O’Shea, J.J. Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Semin. Immunol. 2007, 19, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Dong, C. IL-17 cytokines in immunity and inflammation. Emerg. Microbes Infect. 2013, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, H.; Yao, Y.; Xia, D.; Zhou, J. The overexpression of heparin-binding epidermal growth factor is responsible for Th17-induced airway remodeling in an experimental asthma model. J. Immunol. 2010, 185, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Gavino, A.C.; Nahmod, K.; Bharadwaj, U.; Makedonas, G.; Tweardy, D.J. STAT3 inhibition prevents lung inflammation, remodeling, and accumulation of Th2 and Th17 cells in a murine asthma model. Allergy 2016, 71, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Fitzsimons, C.; Smith, P.A. JAK/STAT inhibitors and other small molecule cytokine antagonists for the treatment of allergic disease. Ann. Allergy Asthma Immunol. 2018, 120, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Bel, E.H.; Ortega, H.G.; Pavord, I.D. Glucocorticoids and mepolizumab in eosinophilic asthma. N. Engl. J. Med. 2014, 371, 2433–2434. [Google Scholar] [CrossRef] [PubMed]
- Bel, E.H.; Wenzel, S.E.; Thompson, P.J.; Prazma, C.M.; Keene, O.N.; Yancey, S.W.; Ortega, H.G.; Pavord, I.D. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N. Engl. J. Med. 2014, 371, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Ortega, H.G.; Liu, M.C.; Pavord, I.D.; Brusselle, G.G.; FitzGerald, J.M.; Chetta, A.; Humbert, M.; Katz, L.E.; Keene, O.N.; Yancey, S.W.; et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 2014, 371, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Pavord, I.D.; Korn, S.; Howarth, P.; Bleecker, E.R.; Buhl, R.; Keene, O.N.; Ortega, H.; Chanez, P. Mepolizumab for severe eosinophilic asthma (DREAM): A multicentre, double-blind, placebo-controlled trial. Lancet 2012, 380, 651–659. [Google Scholar] [CrossRef]
- Wiegman, C.H.; Russell, K.E.; Seiffert, J.; Rossios, C.; Adcock, I.M.; Rothaul, A.; Main, M.; Morgan, F. The Selective pan-Janus Kinase (JAK) Inhibitor VR588 Demonstrates Potent Anti-Inflammatory Activity in a Murine Chronic House Dust Mite (HDM) Model of Asthma. In Proceedings of the American Thoracic Society International Conference Abstracts, Denver, CO, USA, 15–20 May 2015; American Thoracic Society: New York, NY, USA, 2015; p. A6435. [Google Scholar]
- Oskeritzian, C.A.; Hait, N.C.; Wedman, P.; Chumanevich, A.; Kolawole, E.M.; Price, M.M.; Falanga, Y.T.; Harikumar, K.B.; Ryan, J.J.; Milstien, S.; et al. The sphingosine-1-phosphate/sphingosine-1-phosphate receptor 2 axis regulates early airway T-cell infiltration in murine mast cell-dependent acute allergic responses. J. Allergy Clin. Immunol. 2015, 135, 1008–1018.e1. [Google Scholar] [CrossRef] [PubMed]
- Simeone-Penney, M.C. STAT3: A Novel Regulator of Airway Inflammation and Remodeling in Asthma; Tufts University—Sackler School of Graduate Biomedical Sciences: Medford, MA, USA, 2008. [Google Scholar]
- Ito, H.; Takazoe, M.; Fukuda, Y.; Hibi, T.; Kusugami, K.; Andoh, A.; Matsumoto, T.; Yamamura, T.; Azuma, J.; Nishimoto, N.; et al. A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 2004, 126, 989–996, discussion 947. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Safety and Efficacy of TJ301 IV in Participants with Active Ulcerative Colitis; U.S. National Library of Medicine: Bethesda, MD, USA, 2017.
- U.S. National Library of Medicine. Phase 1 Study of Safety and Biological Effects of C326, an Inhibitor of IL-6, in Crohn’s Disease. 2006–2007; U.S. National Library of Medicine: Bethesda, MD, USA, 2006.
- Herrlinger, K.R.; Diculescu, M.; Fellermann, K.; Hartmann, H.; Howaldt, S.; Nikolov, R.; Petrov, A.; Reindl, W.; Otte, J.M.; Stoynov, S.; et al. Efficacy, safety and tolerability of vidofludimus in patients with inflammatory bowel disease: The ENTRANCE study. J. Crohn’s Colitis 2013, 7, 636–643. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Chen, Y.; Tato, C.M.; Laurence, A.; Joyce-Shaikh, B.; Blumenschein, W.M.; McClanahan, T.K.; O’Shea, J.J.; Cua, D.J. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat. Immunol. 2009, 10, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Fedorak, R.N.; Scherl, E.; Fleisher, M.R.; Katz, S.; Johanns, J.; Blank, M.; Rutgeerts, P. Ustekinumab Crohn’s Disease Study Group. A randomized trial of Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with moderate-to-severe Crohn’s disease. Gastroenterology 2008, 135, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Tibbles, H.; Ozer, Z.; Qazi, S.; Vassilev, A. Anti-inflammatory activity profile of JANEX-1 in preclinical animal models. Bioorg. Med. Chem. 2008, 16, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Vranic, I.; Su, C.; Rousell, S.; Niezychowski, W.; Study, A.I. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N. Engl. J. Med. 2012, 367, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Cottone, M.; Orlando, A.; Papi, C. Tofacitinib in active ulcerative colitis. N. Engl. J. Med. 2012, 367, 1959–1961. [Google Scholar]
- De Vries, L.C.S.; Wildenberg, M.E.; De Jonge, W.J.; D’Haens, G.R. The Future of Janus Kinase Inhibitors in Inflammatory Bowel Disease. J Crohn’s Colitis 2017, 11, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthakrishnan, A.N. Filgotinib for Crohn’s disease-expanding treatment options. Lancet 2017, 389, 228–229. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Panes, J.; D’Haens, G.R.; Colombel, J.F.; Zhou, Q.; Huang, B.; Enejosa, J.V.; Pangan, A.L.; Lacerda, A.P. Safety and Efficacy of ABT-494 (Upadacitinib), an Oral Jak1 Inhibitor, as Induction Therapy in Patients with Crohn’s Disease: Results from Celest. Gastroenterology 2017, 152, S1308–S1309. [Google Scholar] [CrossRef]
- Robinson, P.; Khalil, A.; Engineer, N.; Montoya, K.; Magness, E.; Rashid, A.; Eckols, T.; Ivan, C.; Yang, P.; Bharadwaj, B.; Tweardy, D. Genetic and small molecule modulation of STAT3 in a mouse model of Ulcerative colitis. University of Texas MD Anderson Cancer Center: Houston, TX, USA, 2018; unpublished. [Google Scholar]
- Robinson, P.; Legi, A.; Khalil, A.; Engineer, N.; Magness, E.; Montoya, K.; Rashid, A.; Eckols, T.; Tweardy, D. Genetic and small molecule modulation of STAT3 in a mouse model of Crohn’s disease. University of Texas MD Anderson Cancer Center: Houston, TX, USA, 2018; unpublished. [Google Scholar]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Dong, Y.; Garibotto, G.; Mitch, W.E. Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Suvannasankha, A.; Crean, C.D.; White, V.L.; Chen, C.-S.; Farag, S.S. The novel histone deacetylase inhibitor, AR-42, inhibits gp130/Stat3 pathway and induces apoptosis and cell cycle arrest in multiple myeloma cells. Int. J. Cancer 2011, 129, 204–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerchione, C.; Peluso, I.; Nappi, D.; Pareto, A.E.; Picardi, M.; Martinelli, V.; Pane, F. Ruxolitinib rechallenge in combination with hydroxyurea is effective in reverting cachexia and reducing blood transfusion demand and splenomegaly symptoms in a patient with primary myelofibrosis. Ann. Hematol. 2017, 96, 697–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jatoi, A. Anti-inflammatory therapy: Exploring exercise, serum-derived bovine immunoglobulin/protein isolates, and ruxolitinib for the cancer-associated weight loss syndrome. Curr. Opin. Support. Palliat. Care 2013, 7, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escriva, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milara, J.; Ballester, B.; Morell, A.; Ortiz, J.L.; Escriva, J.; Fernandez, E.; Perez-Vizcaino, F.; Cogolludo, A.; Pastor, E.; Artigues, E.; et al. JAK2 mediates lung fibrosis, pulmonary vascular remodelling and hypertension in idiopathic pulmonary fibrosis: An experimental study. Thorax 2018, 73, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Sallam, A.M.; Esmat, A.; Abdel-Naim, A.B. Cucurbitacin-B attenuates CCl4-induced hepatic fibrosis in mice through inhibition of STAT-3. Chem. Biol. Drug Des. 2018, 91, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, E.; Younes, A. Targeting the JAK-STAT pathway in lymphoma: A focus on pacritinib. Expert Opin. Investig. Drugs 2013, 22, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Komrokji, R.S.; Seymour, J.F.; Roberts, A.W.; Wadleigh, M.; To, L.B.; Scherber, R.; Turba, E.; Dorr, A.; Zhu, J.; Wang, L.; et al. Results of a phase 2 study of pacritinib (SB1518), a JAK2/JAK2(V617F) inhibitor, in patients with myelofibrosis. Blood 2015, 125, 2649–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mughal, T.I.; Vaddi, K.; Sarlis, N.J.; Verstovsek, S. Myelofibrosis-associated complications: Pathogenesis, clinical manifestations, and effects on outcomes. Int. J. Gen. Med. 2014, 7, 89–101. [Google Scholar] [PubMed]
- Vannucchi, A.M.; Kantarjian, H.M.; Kiladjian, J.-J.; Gotlib, J.; Cervantes, F.; Mesa, R.A.; Sarlis, N.J.; Peng, W.; Sandor, V.; Gopalakrishna, P.; et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica 2015, 100, 1139–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.N.; Miller, C.B.; Silver, R.T.; et al. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: Results of a median 3-year follow-up of COMFORT-I. Haematologica 2015, 100, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Ma, L.; Gong, R.; Tolbert, E.; Mao, H.; Ponnusamy, M.; Chin, Y.E.; Yan, H.; Dworkin, L.D.; Zhuang, S. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. 2010, 78, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Lu, B.; Zhang, X.; Zhang, J.; Lai, L.; Li, D.; Wu, Y.; Song, Y.; Luo, J.; Pang, X.; et al. Cucurbitacin E, a tetracyclic triterpenes compound from Chinese medicine, inhibits tumor angiogenesis through VEGFR2-mediated Jak2-STAT3 signaling pathway. Carcinogenesis 2010, 31, 2097–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, U.; Eckols, T.K.; Xu, X.; Kasembeli, M.M.; Chen, Y.; Adachi, M.; Song, Y.; Mo, Q.; Lai, S.Y.; Tweardy, D.J. Small-molecule inhibition of STAT3 in radioresistant head and neck squamous cell carcinoma. Oncotarget 2016, 7, 26307–26330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedroza, M.; Le, T.T.; Lewis, K.; Karmouty-Quintana, H.; To, S.; George, A.T.; Blackburn, M.R.; Tweardy, D.J.; Agarwal, S.K. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. 2016, 30, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedroza, M.; To, S.; Assassi, S.; Wu, M.; Tweardy, D.; Agarwal, S.K. Role of STAT3 in skin fibrosis and transforming growth factor beta signalling. Rheumatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Yoo, W.; Stevenson, H.L.; Deshpande, D.; Shen, H.; Gagea, M.; Yoo, S.-Y.; Wang, J.; Eckols, T.K.; Bharadwaj, U.; et al. Multifunctional Effects of a Small-Molecule STAT3 Inhibitor on NASH and Hepatocellular Carcinoma in Mice. Clin. Cancer Res. 2017, 23, 5537–5546. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Harikumar, K.B. Sphingosine 1-Phosphate: A Novel Target for Lung Disorders. Front. Immunol. 2017, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.-C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Kasembeli, M.M.; Tweardy, D.J. STAT3 Inhibitors in Cancer: A Comprehensive Update. In STAT Inhibitors in Cancer; Ward, A.C., Ed.; STAT Inhibitors in Cancer; Springer International Publishing: Cham, Switzerland, 2016; pp. 95–161. [Google Scholar]
- Choy, D.F.; Hart, K.M.; Borthwick, L.A.; Shikotra, A.; Nagarkar, D.R.; Siddiqui, S.; Jia, G.; Ohri, C.M.; Doran, E.; Vannella, K.M.; et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci. Transl. Med. 2015, 7, 301ra129. [Google Scholar] [CrossRef] [PubMed]
- Dahlhamer, J.M.; Zammitti, E.P.; Ward, B.W.; Wheaton, A.G.; Croft, J.B. Prevalence of Inflammatory Bowel Disease among Adults Aged >/=18 Years—United States, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Ha, F.; Khalil, H. Crohn’s disease: A clinical update. Therap. Adv. Gastroenterol. 2015, 8, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Kalla, R.; Ventham, N.T.; Satsangi, J.; Arnott, I.D. Crohn’s disease. BMJ 2014, 349, g6670. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef]
- Juckett, G.; Trivedi, R. Evaluation of chronic diarrhea. Am. Fam. Phys. 2011, 84, 1119–1126. [Google Scholar]
- Sands, B.E. From symptom to diagnosis: Clinical distinctions among various forms of intestinal inflammation. Gastroenterology 2004, 126, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Castano-Milla, C.; Chaparro, M.; Gisbert, J.P. Systematic review with meta-analysis: The declining risk of colorectal cancer in ulcerative colitis. Aliment. Pharmacol. Ther. 2014, 39, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Freeman, H.J. Colorectal cancer risk in Crohn’s disease. World J. Gastroenterol. 2008, 14, 1810–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harpaz, N.; Ward, S.C.; Mescoli, C.; Itzkowitz, S.H.; Polydorides, A.D. Precancerous lesions in inflammatory bowel disease. Best Pract. Res. Clin. Gastroenterol. 2013, 27, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees, C.W.; Barrett, J.C.; Parkes, M.; Satsangi, J. New IBD genetics: Common pathways with other diseases. Gut 2011, 60, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Mudter, J.; Neurath, M.F. Il-6 signaling in inflammatory bowel disease: Pathophysiological role and clinical relevance. Inflamm. Bowel Dis. 2007, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Fuss, I.J.; Blumberg, R.S. The immunology of mucosal models of inflammation. Annu. Rev. Immunol. 2002, 20, 495–549. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E.J. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovato, P.; Brender, C.; Agnholt, J.; Kelsen, J.; Kaltoft, K.; Svejgaard, A.; Eriksen, K.W.; Woetmann, A.; Odum, N. Constitutive STAT3 activation in intestinal T cells from patients with Crohn’s disease. J. Biol. Chem. 2003, 278, 16777–16781. [Google Scholar] [CrossRef] [PubMed]
- Mudter, J.; Weigmann, B.; Bartsch, B.; Kiesslich, R.; Strand, D.; Galle, P.R.; Lehr, H.A.; Schmidt, J.; Neurath, M.F. Activation pattern of signal transducers and activators of transcription (STAT) factors in inflammatory bowel diseases. Am. J. Gastroenterol. 2005, 100, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Musso, A.; Dentelli, P.; Carlino, A.; Chiusa, L.; Repici, A.; Sturm, A.; Fiocchi, C.; Rizzetto, M.; Pegoraro, L.; Sategna-Guidetti, C.; et al. Signal transducers and activators of transcription 3 signaling pathway: An essential mediator of inflammatory bowel disease and other forms of intestinal inflammation. Inflamm. Bowel Dis. 2005, 11, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Mudter, J.; Finotto, S.; Mullberg, J.; Jostock, T.; Wirtz, S.; Schutz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in crohn disease and experimental colitis in vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, A.; Hanada, T.; Mitsuyama, K.; Yoshida, T.; Kamizono, S.; Hoshino, T.; Kubo, M.; Yamashita, A.; Okabe, M.; Takeda, K.; et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J. Exp. Med. 2001, 193, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Clausen, B.E.; Kaisho, T.; Tsujimura, T.; Terada, N.; Forster, I.; Akira, S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 1999, 10, 39–49. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yoshizaki, K.; Kishimoto, T.; Ito, H. IL-6 is required for the development of Th1 cell-mediated murine colitis. J. Immunol. 2000, 164, 4878–4882. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Neurath, M.F. Signaling molecules: The pathogenic role of the IL-6/STAT-3 trans signaling pathway in intestinal inflammation and in colonic cancer. Curr. Drug Targets 2008, 9, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Kaisho, T.; Yoshida, N.; Takeda, J.; Kishimoto, T.; Akira, S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: Generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 1998, 161, 4652–4660. [Google Scholar] [CrossRef] [PubMed]
- Maritano, D.; Sugrue, M.L.; Tininini, S.; Dewilde, S.; Strobl, B.; Fu, X.; Murray-Tait, V.; Chiarle, R.; Poli, V. The STAT3 isoforms alpha and beta have unique and specific functions. Nat. Immunol. 2004, 5, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Bilsborough, J.; Viney, J.L. From model to mechanism: Lessons of mice and men in the discovery of protein biologicals for the treatment of inflammatory bowel disease. Expert Opin. Drug Discov. 2006, 1, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Geboes, K.; Colpaert, S.; D’Haens, G.R.; Rutgeerts, P.; Ceuppens, J.L. IL-15 is highly expressed in inflammatory bowel disease and regulates local T cell-dependent cytokine production. J. Immunol. 2000, 164, 3608–3615. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.M.; Putoczki, T.L.; Ernst, M. STAT3-Activating Cytokines: A Therapeutic Opportunity for Inflammatory Bowel Disease? J. Interferon Cytokine Res. 2015, 35, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Pallone, F.; Fina, D.; Caruso, R.; Monteleone, G. Role of IL-21 in inflammatory bowel disease. Expert Rev. Clin. Immunol. 2010, 6, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Munoz, F.; Dominguez-Lopez, A.; Yamamoto-Furusho, J.K. Role of cytokines in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 4280–4288. [Google Scholar] [CrossRef] [PubMed]
- Rebe, C.; Vegran, F.; Berger, H.; Ghiringhelli, F. STAT3 activation: A key factor in tumor immunoescape. JAK-STAT 2013, 2, e23010. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Shin-Ya, M.; Kishida, T.; Urano, A.; Takada, R.; Sakagami, J.; Imanishi, J.; Kita, M.; Ueda, Y.; Iwakura, Y.; et al. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin. Exp. Immunol. 2006, 146, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.-W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piekkala, M.; Hagstrom, J.; Tanskanen, M.; Rintala, R.; Haglund, C.; Kolho, K.-L. Low trypsinogen-1 expression in pediatric ulcerative colitis patients who undergo surgery. World J. Gastroenterol. 2013, 19, 3272–3280. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Kita, M.; Shin-Ya, M.; Kishida, T.; Urano, A.; Takada, R.; Sakagami, J.; Imanishi, J.; Iwakura, Y.; Okanoue, T.; et al. Involvement of IL-17A in the pathogenesis of DSS-induced colitis in mice. Biochem. Biophys. Res. Commun. 2008, 377, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Kanai, T.; Watanabe, M.; Okazawa, A.; Sato, T.; Yamazaki, M.; Okamoto, S.; Ishii, H.; Totsuka, T.; Iiyama, R.; Okamoto, R.; et al. Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn’s disease. Gastroenterology 2001, 121, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Nishiwaki, T.; Ina, K.; Goto, H.; Watanabe, O.; Tsuzuki, T.; Furuta, R.; Ando, T.; Hibi, K.; Kusugami, K. Possible involvement of the interleukin-15 and interleukin-15 receptor system in a heightened state of lamina propria B cell activation and differentiation in patients with inflammatory bowel disease. J. Gastroenterol. 2005, 40, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Ohta, N.; Hiroi, T.; Kweon, M.N.; Kinoshita, N.; Jang, M.H.; Mashimo, T.; Miyazaki, J.; Kiyono, H. IL-15-dependent activation-induced cell death-resistant Th1 type CD8 alpha beta+NK1.1+ T cells for the development of small intestinal inflammation. J. Immunol. 2002, 169, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Pizarro, T.T.; Michie, M.H.; Bentz, M.; Woraratanadharm, J.; Smith, M.F.J.; Foley, E.; Moskaluk, C.A.; Bickston, S.J.; Cominelli, F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: Expression and localization in intestinal mucosal cells. J. Immunol. 1999, 162, 6829–6835. [Google Scholar] [PubMed]
- Sengupta, N.; MacDonald, T.T. The role of matrix metalloproteinases in stromal/epithelial interactions in the gut. Physiology 2007, 22, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, H.H.; McKenzie, B.S.; Hue, S.; Thompson, C.; Joyce-Shaikh, B.; Stepankova, R.; Robinson, N.; Buonocore, S.; Tlaskalova-Hogenova, H.; Cua, D.J.; et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 2006, 25, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zheng, M.; Bindas, J.; Schwarzenberger, P.; Kolls, J.K. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm. Bowel Dis. 2006, 12, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Herrlinger, K.R.; Witthoeft, T.; Raedler, A.; Bokemeyer, B.; Krummenerl, T.; Schulzke, J.D.; Boerner, N.; Kueppers, B.; Emmrich, J.; Mescheder, A.; et al. Randomized, double blind controlled trial of subcutaneous recombinant human interleukin-11 versus prednisolone in active Crohn’s disease. Am. J. Gastroenterol. 2006, 101, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Katakura, K.; Lee, J.; Rachmilewitz, D.; Li, G.; Eckmann, L.; Raz, E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J. Clin. Investig. 2005, 115, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Alli, R.; Vogel, P.; Geiger, T.L. IL-10 modulates DSS-induced colitis through a macrophage-ROS-NO axis. Mucosal Immunol. 2014, 7, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Musch, E.; Andus, T.; Kruis, W.; Raedler, A.; Spehlmann, M.; Schreiber, S.; Krakamp, B.; Malek, M.; Malchow, H.; Zavada, F.; et al. Interferon-beta-1a for the treatment of steroid-refractory ulcerative colitis: A randomized, double-blind, placebo-controlled trial. Clin. Gastroenterol. Hepatol. 2005, 3, 581–586. [Google Scholar] [CrossRef]
- Qiu, B.S.; Pfeiffer, C.J.; Keith, J.C.J. Protection by recombinant human interleukin-11 against experimental TNB-induced colitis in rats. Dig. Dis. Sci. 1996, 41, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Ulmer, H.; Kaser, A.; Weiss, G. Role of IL-10 for induction of anemia during inflammation. J. Immunol. 2002, 169, 2204–2209. [Google Scholar] [CrossRef] [PubMed]
- Zindl, C.L.; Lai, J.F.; Lee, Y.K.; Maynard, C.L.; Harbour, S.N.; Ouyang, W.; Chaplin, D.D.; Weaver, C.T. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 12768–12773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coskun, M.; Salem, M.; Pedersen, J.; Nielsen, O.H. Involvement of JAK/STAT signaling in the pathogenesis of inflammatory bowel disease. Pharmacol. Res. 2013, 76, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Anker, S.D. Prevalence, incidence and clinical impact of cachexia: Facts and numbers-update 2014. J. Cachexia Sarcopenia Muscle 2014, 5, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Porporato, P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef] [PubMed]
- Latres, E.; Amini, A.R.; Amini, A.A.; Griffiths, J.; Martin, F.J.; Wei, Y.; Lin, H.C.; Yancopoulos, G.D.; Glass, D.J. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J. Biol. Chem. 2005, 280, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr. Top. Microbiol. Immunol. 2010, 346, 267–278. [Google Scholar] [PubMed]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Chovatiya, R.; Medzhitov, R. Stress, inflammation, and defense of homeostasis. Mol. Cell 2014, 54, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Lopez-Soriano, F.J.; Busquets, S. Counteracting inflammation: A promising therapy in cachexia. Crit. Rev. Oncog. 2012, 17, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, G.; Maccio, A.; Mura, L.; Massa, E.; Mudu, M.C.; Mulas, C.; Lusso, M.R.; Madeddu, C.; Dessi, A. Serum levels of leptin and proinflammatory cytokines in patients with advanced-stage cancer at different sites. J. Mol. Med. 2000, 78, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Hanna, D.L.; Zhang, W.; Baba, H.; Lenz, H.-J. Molecular Pathways: Cachexia Signaling-A Targeted Approach to Cancer Treatment. Clin. Cancer Res. 2016, 22, 3999–4004. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Frantz, J.D.; Tawa, N.E.J.; Melendez, P.A.; Oh, B.-C.; Lidov, H.G.W.; Hasselgren, P.-O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.J.; Murphy, K.T.; Jenkinson, L.; Laine, D.; Emmrich, K.; Faou, P.; Weston, R.; Jayatilleke, K.M.; Schloegel, J.; Talbo, G.; et al. Targeting of Fn14 Prevents Cancer-Induced Cachexia and Prolongs Survival. Cell 2015, 162, 1365–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug. Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Canoves, P.; Scheele, C.; Pedersen, B.K.; Serrano, A.L. Interleukin-6 myokine signaling in skeletal muscle: A double-edged sword? FEBS J. 2013, 280, 4131–4148. [Google Scholar] [CrossRef] [PubMed]
- Tsujinaka, T.; Fujita, J.; Ebisui, C.; Yano, M.; Kominami, E.; Suzuki, K.; Tanaka, K.; Katsume, A.; Ohsugi, Y.; Shiozaki, H.; et al. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J. Clin. Investig. 1996, 97, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Baltgalvis, K.A.; Berger, F.G.; Pena, M.M.O.; Davis, J.M.; Muga, S.J.; Carson, J.A. Interleukin-6 and cachexia in ApcMin/+ mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R393–R401. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Baynes, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS ONE 2011, 6, e24650. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, T.J.; Smith, J.T.; Schuster, M.; Dragnev, K.H.; Rigas, J.R. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin. Biol. Ther. 2011, 11, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Silva, K.A.S.; Dong, J.; Dong, Y.; Dong, Y.; Schor, N.; Tweardy, D.J.; Zhang, L.; Mitch, W.E. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 2015, 290, 11177–11187. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Rossi Fanelli, F.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, K.; Andersen, S.; Degen, S.; Tadini, V.; Grosjean, J.; Hatakeyama, S.; Tesfahun, A.N.; Moestue, S.; Kim, J.; Nonstad, U.; et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 2017, 7, 2046. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Spiegelman, B.M. Cachexia & Brown Fat: A Burning Issue in Cancer. Trends Cancer 2016, 2, 461–463. [Google Scholar] [PubMed]
- Han, J.; Meng, Q.; Shen, L.; Wu, G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 2018, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Derecka, M.; Gornicka, A.; Koralov, S.B.; Szczepanek, K.; Morgan, M.; Raje, V.; Sisler, J.; Zhang, Q.; Otero, D.; Cichy, J.; et al. Tyk2 and Stat3 regulate brown adipose tissue differentiation and obesity. Cell Metab. 2012, 16, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Babaei, R.; Schuster, M.; Meln, I.; Lerch, S.; Ghandour, R.A.; Pisani, D.F.; Bayindir-Buchhalter, I.; Marx, J.; Wu, S.; Schoiswohl, G.; et al. Jak-TGFbeta cross-talk links transient adipose tissue inflammation to beige adipogenesis. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.Y.; Luk, C.T.; Brunt, J.J.; Sivasubramaniyam, T.; Lu, S.-Y.; Schroer, S.A.; Woo, M. Adipocyte-specific deficiency of Janus kinase (JAK) 2 in mice impairs lipolysis and increases body weight, and leads to insulin resistance with ageing. Diabetologia 2014, 57, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Tsoli, M.; Schweiger, M.; Vanniasinghe, A.S.; Painter, A.; Zechner, R.; Clarke, S.; Robertson, G. Depletion of white adipose tissue in cancer cachexia syndrome is associated with inflammatory signaling and disrupted circadian regulation. PLoS ONE 2014, 9, e92966. [Google Scholar] [CrossRef] [PubMed]
- Cernkovich, E.R.; Deng, J.; Bond, M.C.; Combs, T.P.; Harp, J.B. Adipose-specific disruption of signal transducer and activator of transcription 3 increases body weight and adiposity. Endocrinology 2008, 149, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yin, C.; Wang, S.; Xiao, Y. JAK-STAT in lipid metabolism of adipocytes. JAK-STAT 2013, 2, e27203. [Google Scholar] [CrossRef] [PubMed]
- Koltes, D.A.; Spurlock, M.E.; Spurlock, D.M. Adipose triglyceride lipase protein abundance and translocation to the lipid droplet increase during leptin-induced lipolysis in bovine adipocytes. Domest. Anim. Endocrinol. 2017, 61, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Koltes, D.; Spurlock, D. Leptin increases adipose triglyceride lipase in bovine primary adipocytes. FASEB J. 2013, 27, 373.8. [Google Scholar]
- Tseng, Y.-C.; Liva, S.G.; Dauki, A.M.; Henderson, S.E.; Kuo, Y.-C.; Benedict, J.A.; Kulp, S.K.; Bekaii-Saab, T.; Chen, C.-S.; Coss, C.C. Combined Androgen Administration and HDAC Inhibition in Experimental Cancer Cachexia. bioRxiv 2017, 214155. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr1. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, J.; Mendoza, F.A.; Jimenez, S.A. Strategies for anti-fibrotic therapies. Biochim. Biophys. Acta 2013, 1832, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agha, E.E.; Kramann, R.; Schneider, R.K.; Li, X.; Seeger, W.; Humphreys, B.D.; Bellusci, S. Mesenchymal Stem Cells in Fibrotic Disease. Cell Stem Cell 2017, 21, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Suwara, M.I.; Green, N.J.; Borthwick, L.A.; Mann, J.; Mayer-Barber, K.D.; Barron, L.; Corris, P.A.; Farrow, S.N.; Wynn, T.A.; Fisher, A.J.; et al. IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2014, 7, 684–693. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Pasche, B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat. Rev. Rheumatol. 2009, 5, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Dauer, D.J.; Ferraro, B.; Song, L.; Yu, B.; Mora, L.; Buettner, R.; Enkemann, S.; Jove, R.; Haura, E.B. Stat3 regulates genes common to both wound healing and cancer. Oncogene 2005, 24, 3397–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, S.; Itami, S.; Takeda, K.; Tarutani, M.; Yamaguchi, Y.; Miura, H.; Yoshikawa, K.; Akira, S.; Takeda, J. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999, 18, 4657–4668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, H.; Chinen, T.; Yoshida, T.; Kinjyo, I.; Takaesu, G.; Shiraishi, H.; Iida, M.; Kobayashi, T.; Yoshimura, A. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene 2006, 25, 2520–2530. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Xu, S.; Denton, C.P.; Abraham, D.J.; Ponticos, M. STAT3 controls COL1A2 enhancer activation cooperatively with JunB, regulates type I collagen synthesis posttranscriptionally, and is essential for lung myofibroblast differentiation. Mol. Biol. Cell 2018, 29, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Iness, A.; Yoon, J.; Grider, J.R.; Murthy, K.S.; Kellum, J.M.; Kuemmerle, J.F. Noncanonical STAT3 activation regulates excess TGF-beta1 and collagen I expression in muscle of stricturing Crohn’s disease. J. Immunol. 2015, 194, 3422–3431. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-Y.; Jeon, J.-H.; Jung, Y.-A.; Jung, G.-S.; Lee, E.J.; Choi, Y.-K.; Park, K.-G.; Choe, M.S.; Jang, B.K.; Kim, M.-K.; et al. Fyn deficiency attenuates renal fibrosis by inhibition of phospho-STAT3. Kidney Int. 2016, 90, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Giannandrea, M.; Parks, W.C. Diverse functions of matrix metalloproteinases during fibrosis. Dis. Models Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhou, Y.; Tan, R.; Xiong, M.; He, W.; Fang, L.; Wen, P.; Jiang, L.; Yang, J. Mice lacking the matrix metalloproteinase-9 gene reduce renal interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. Renal Physiol. 2010, 299, F973–F982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, F.; Babitz, S.A.; Rhee, A.; Hile, K.L.; Zhang, H.; Meldrum, K.K. Mesenchymal stem cells protect against obstruction-induced renal fibrosis by decreasing STAT3 activation and STAT3-dependent MMP-9 production. Am. J. Physiol. Renal Physiol. 2017, 312, F25–F32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J. Matrix metalloproteinases and tissue inhibitor of metalloproteinases are essential for the inflammatory response in cancer cells. J. Signal Transduct. 2010, 2010, 985132. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lafdil, F.; Wang, L.; Yin, S.; Feng, D.; Gao, B. Tissue inhibitor of metalloproteinase 1 (TIMP-1) deficiency exacerbates carbon tetrachloride-induced liver injury and fibrosis in mice: Involvement of hepatocyte STAT3 in TIMP-1 production. Cell Biosci. 2011, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moodley, Y.P.; Misso, N.L.A.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Habiel, D.M.; Hogaboam, C. Heterogeneity in fibroblast proliferation and survival in idiopathic pulmonary fibrosis. Front. Pharmacol. 2014, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-Y.; Hu, J.-J.; Shen, J.; Wang, M.-L.; Zhang, Q.-Q.; Qu, Y.; Lu, L.-G. Stat3 signaling activation crosslinking of TGF-beta1 in hepatic stellate cell exacerbates liver injury and fibrosis. Biochim. Biophys. Acta 2014, 1842, 2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef] [PubMed]
- Balanis, N.; Wendt, M.K.; Schiemann, B.J.; Wang, Z.; Schiemann, W.P.; Carlin, C.R. Epithelial to mesenchymal transition promotes breast cancer progression via a fibronectin-dependent STAT3 signaling pathway. J. Biol. Chem. 2013, 288, 17954–17967. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Endo, K.; Furuya, S.; Minami, M.; Fukasawa, A.; Imamura, T.; Miyazawa, K. STAT3 integrates cooperative Ras and TGF-beta signals that induce Snail expression. Oncogene 2016, 35, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhong, Y.; Liu, G.; Zhang, X.; Xiao, B.; Huang, S.; Liu, H.; He, L. Role of Stat3 Signaling in Control of EMT of Tubular Epithelial Cells During Renal Fibrosis. Cell. Physiol. Biochem. 2017, 42, 2552–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Sumova, B.; Mallano, T.; Chen, C.-W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; van Laar, J.M. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-beta (TGF-beta) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yu, Y.; Sun, C.; Liu, T.; Liang, T.; Zhan, L.; Lin, X.; Feng, X.-H. STAT3 selectively interacts with Smad3 to antagonize TGF-beta. Oncogene 2016, 35, 4388–4398. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef]
- Wang, L.H.; Yang, X.Y.; Zhang, X.; Huang, J.; Hou, J.; Li, J.; Xiong, H.; Mihalic, K.; Zhu, H.; Xiao, W.; et al. Transcriptional inactivation of STAT3 by PPARgamma suppresses IL-6-responsive multiple myeloma cells. Immunity 2004, 20, 205–218. [Google Scholar] [CrossRef]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hoffmann, N.; Geldner, J.; Borchert, P.; Jonigk, D.; Mozes, M.M.; Hansmann, G. PPARgamma Links BMP2 and TGFbeta1 Pathways in Vascular Smooth Muscle Cells, Regulating Cell Proliferation and Glucose Metabolism. Cell Metab. 2017, 25, 1118–1134.e7. [Google Scholar] [CrossRef] [PubMed]
- Agha El, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Nunez Lopez, O.; Bohanon, F.J.; Wang, X.; Ye, N.; Corsello, T.; Rojas-Khalil, Y.; Chen, H.; Chen, H.; Zhou, J.; Radhakrishnan, R.S. STAT3 Inhibition Suppresses Hepatic Stellate Cell Fibrogenesis: HJC0123, a Potential Therapeutic Agent for Liver Fibrosis. RSC Adv. 2016, 6, 100652–100663. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Chandrasekharan, U.M.; Willard, B.; Tee, T.L.; Hsieh, J.K.; Przybycin, C.G.; Rini, B.I.; Dicorleto, P.E. Signal integration and gene induction by a functionally distinct STAT3 phosphoform. Mol. Cell. Biol. 2014, 34, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Waitkus, M.S.; Chandrasekharan, U.M.; Willard, B.; Haque, S.J.; Dicorleto, P.E. STAT3-mediated coincidence detection regulates noncanonical immediate early gene induction. J. Biol. Chem. 2013, 288, 11988–12003. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.Y.; Wilson, H.L.; Voltzke, K.J.; Williams, L.A.; Lee, H.J.; Wobker, S.E.; Kim, W.Y. Sav1 Loss Induces Senescence and Stat3 Activation Coinciding with Tubulointerstitial Fibrosis. Mol. Cell. Biol. 2017, 37, e00565-16. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.F.; Sanchez, B.J.; Hall, D.T.; Tremblay, A.-M.K.; Di Marco, S.; Gallouzi, I.-E. STAT3 promotes IFNgamma/TNFalpha-induced muscle wasting in an NF-kappaB-dependent and IL-6-independent manner. EMBO Mol. Med. 2017, 9, 622–637. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.-J.; Sepuri, N.B.V.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenni, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4924–E4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Inhibitor | Target | Preclinical/Clinical Model | Goals/Results | Ref. |
|---|---|---|---|---|
| Asthma | ||||
| TyrA1 | Jak | HDM-induced STAT3-mediated mice model of asthma | Blocked HDM-induced STAT3 activation and airway eosinophilia in mice | [14] |
| Vr588 | Jak | HDM-induced STAT3-mediated mice model of asthma. Intra-nasal VR588 (1.5 and 50 mg/kg) vs oral 15 mg/kg PK. HDM extract (25 μg) intranasally 5 days/week for 3 weeks with multiple intranasal doses (1.5 to7.5 mg/kg) given 1 hour prior to each HDM exposure; a separate group administered a VR588 7.5 mg/kg intranasal dose only during the last week of HDM treatment (n = 8). Comparison was made with oral tofacitinib (15 mg/kg) and fluticasone propionate (1.5 mg/kg). | VR588 resulted in significant reduction of AHR at least comparable to that achieved by FP. All VR588 doses significantly reduced BAL total cell count with a variety of doses inhibiting macrophage, neutrophil, lymphocyte and eosinophil counts. VR588 attenuated the induction of numerous cytokines (IL-4, IL-5, IL-17) compared with saline control, As well as HDM induced pSTAT3 | [27,28] |
| Mepolizumab | IL5 antagonist | Rhinovirus-induced Allergic Asthma Exacerbations; multicenter, double-blind, placebo-controlled DREAM trial | Mepolizumab is an effective and well tolerated treatment that reduces the risk of asthma exacerbations in patients with severe eosinophilic asthma | [24,25,26,27] |
| JTE-013 | S1PR2 | Dinitrophenyl (DNP) induced asthma model | Suppressed STAT3 activation, reduced chemokine secretion and prevented early T-cell recruitment in mice lungs after antigen challenge | [29] |
| STAT1/3dODN | STAT1/3 | DM-induced STAT3-mediated mice model of asthma | reduce airway inflammation and AHR in lungs of mice challenged with HDM | [30] |
| C188-9 | STAT3 | HDM-induced STAT3-mediated mice model of asthma | Normalization of IL-4, IL-5, IL-13, and IL-17A cytokine levels, as well as prevention of HDM-induced increases in Th2 cells, Th17 cells, and IL-4- and IL-17A-producing non-T cells | [22] |
| IBD | ||||
| Tocilizumab (MRA) | anti-IL6R MAb | Phase II Clinical: 36 patients with active Crohn’s disease (Crohn’s Disease Activity Index [CDAI] >150) randomly assigned to receive IV infusion of placebo/MRA/alternate MRA-placebo 12 weeks at 8 mg/kg | 80% of the patients (8 of 10) given biweekly MRA had a clinical response as compared with 31% of the placebo-treated patients (4 of 13; P = 0.019). | [31] |
| TJ301 (Olamkicept) | IL6R antagonist | Safety and Efficacy of intravenous TJ301 in Participants With Active Ulcerative Colitis | Ongoing Study | [32] |
| C326 | IL-6 Inhibitory Avimer protein | Placebo-Controlled, Phase 1, Single and Multiple IV Dose Escalation Study of the Safety, in Adults With Crohn’s Disease | Pharmacokinetics, Pharmacodynamics, and Immunogenicity of C326 in Adults With Crohn’s Disease | [33] |
| Vidofludimus | IL17A, IL17F, and IFN-γ | Open-label uncontrolled entrance study of patients with IBD conducted at 13 study centers in Germany, Bulgaria and Romania | 12 weeks treatment phase; 8 out of 14 (57.1%) patients with CD and 6 out of 12 (50.0%) patients with UC were in steroid-free remission (complete responders). Another 4 (28.6%) patients in CD and 5 (41.7%) patients in UC were partial responders. Vidofludimus was well tolerated, with no drug-related serious adverse events. | [34] |
| Ustekinumab | anti-IL12p40 MAb | Double-blind, cross-over trial of the clinical effects of ustekinumab in 104 patients with moderate-to-severe Crohn’s disease (Population 1). Open label trial evaluated the effects of 4 weekly subcutaneous injections or 1 intravenous infusion of ustekinumab in 27 patients who were primary or secondary nonresponders to infliximab (population 2). | In population 1, clinical response rates for the combined groups given ustekinumab and placebo were 53% and 30% (p 0.02), respectively at weeks 4 and 6, and 49% and 40% (p 0.34), respectively at week 8. In a subgroup of 49 patients who were previously given infliximab (neither primary nor secondary nonresponders), clinical response to ustekinumab was significantly greater than the group given placebo (p < 0.05) through week 8. In population 2, the clinical responses at week 8 to subcutaneous and intravenous ustekinumab were 43% and 54%, respectively. | [35,36] |
| Janex-1 | Jak3 | Murine TNBS-induced colitis model | Attenuation of disease manifestations | [37,38,39] |
| Tofacitinib GLPG0634/GS-6034/CP-690550 | Jak3 | Phase II Clinical: moderate-to-severe UC | inducing clinical responses and remissions and has been FDA approved as the only nonsteroidal oral treatment that induces remission for moderate-to-severe UC | [40,41] |
| Filgotinib | Jak1 | Phase II Clinical: Crohn’s Disease | [42,43] | |
| Upadacitinib (ABT-494, AbbVie) | Jak1 | Phase II Clinical: Crohn’s Disease, Adult patients with active CD, with a CDAI 220-450, an average daily liquid/soft stool frequency (SF) ≥2.5 or daily abdominal pain (AP) score ≥2.0, and Simplified Endoscopic Score for CD (SES-CD) ≥6 (or ≥4 for those with isolated ileal disease), were randomized 1:1:1:1:1:1 to doubleblind induction therapy with placebo (PBO) or ABT-494 at 3, 6, 12, 24 mg twice daily (BID) or 24 mg once daily (QD) for 16 weeks, followed by blinded extension therapy for 36 weeks | This dose-ranging study demonstrated endoscopic improvement and clinical benefit of ABT-494 as induction therapy in patients with moderate-to-severe refractory CD, and a safety profile as expected with a JAK inhibitor in this population. | [44] |
| C188-9 | STAT3 | Murine models of DSS-induced UC and TNBS-induced CD | All manifestations of DSS-induced UC and TNBS-induced CD in mice were prevented by C188-9 treatment. C188-9 treatment also induced increased apoptosis of pathogenic CD4+ T-cells, and reduced colon levels of IL-17-positive cells in both models. | [45,46] |
| Cachexia | ||||
| STAT3 SH2 domain mimetic peptide (SIP) | STAT3 | C2C12 cell culture model of muscle differentiation | 48h treatment resulted in modest myofiber hypertrophy and prevented IL-6-induced fiber atrophy | [47] |
| C188-9 | STAT3 | CDK cachexia mouse model | C188-9 treatment antagonized catabolic signaling by decreasing myostatin expression and the activation of its downstream signaling mediators, p-Smad2 and p-Smad3. In addition, C188-9 increased muscle mass in tumor-bearing mice by augmenting muscle protein synthesis and suppressing protein degradation | [48] |
| AR-42 | HDAC inhibitor | C26 cachexia mouse model | anabolic androgen therapy in combination with HDAC Inhibitor AR-42 was shown to block STAT3 mediated muscle atrophy. | [49] |
| Ruxolitinib (INCB018424) | Jak1/2, STAT3 | Incyte, 127 patient, randomized phase II, Cancer associated weight loss trial focused on exocrinemetastatic pancreas cancer patients who had failed first-line chemotherapy, who typically suffer an inexorable decline in weight. Patients received capecitabine and, in addition, were randomly assigned to ruxolitinib versus placebo | The trial’s primary endpoint focused on survival, justified based on the negative prognostic effect of cancer-associated weight loss [11]. The hazard ratio for survival was 0.79 (one-sided P ¼ 0.12), but in an a priori subgroup analysis which was intended to identify patients most likely to benefit from JAK inhibition, the hazard ratio for survival was 0.47 (one-sided P ¼ 0.005). In this same subgroup, the 6-month survival rate with ruxolitinib was 42% compared to 11% with placebo. Importantly, ruxolitinib-treated patients manifested a significant improvement in body weight compared with placebo. Ruxolitinib was also relatively well tolerated | [50,51] |
| Fibrosis | ||||
| JSI-124 | Jak2 | bleomycin-induced lung fibrosis in rats | In rats administered JSI-124, a Jak2 inhibitor that targets STAT3 indirectly, several markers of bleomycin-induced lung fibrosis were reduced | [52,53] |
| cucurbitacin-B | Jak2 | carbon tetrachloride (CCl4) model of fibrosis | Decreased fibrosis and diminished levels of hydroxyproline in liver tissue as well as expression of collagen-1α, α-SMA and TGF-β | [54] |
| Pacritinib (SB1518) | Jak2 | Myelofobrosis, AML (Combined With Decitabine/Cytarabine) | Active drug in myelofibrosis. Going in the AML patients for safety efficacy as a STAT3 inhibitor in combination with Decitabine/Cytarabine | [55,56] |
| Ruxolitinib (INCB018424) | Jak1/2 | COMFORT (COntrolled MyeloFibrosis Study with ORal JAK Inhibitor Therapy)-I Trial | Ruxolitinib provided significant reductions in splenomegaly, improvements in myelofibrosis (MF)-related symptoms including cachexia, and a survival advantage relative to placebo in patients with intermediate-2 or high-risk MF. Ruxolitinib treatment was associated with increased weight (mean change: 3.9 kg vs. −1.9 kg), total cholesterol (mean percentage change: 26.4% vs. −3.3%), and albumin levels (mean percentage change: 5.8% vs. −1.7%) at week 24; sustained improvements were observed with longer-term ruxolitinib therapy. | [57,58,59] |
| S3I-201 | STAT3 | preclinical animal mouse model of renal interstitial fibrosis induced by unilateral ureteral obstruction | Attenuated interstitial fibrosis and showed a fibrotic suppression profile similar to other inhibitors | [60,61] |
| C188-9 | STAT3 |
|
| [62,63,64,65] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasembeli, M.M.; Bharadwaj, U.; Robinson, P.; Tweardy, D.J. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int. J. Mol. Sci. 2018, 19, 2299. https://doi.org/10.3390/ijms19082299
Kasembeli MM, Bharadwaj U, Robinson P, Tweardy DJ. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. International Journal of Molecular Sciences. 2018; 19(8):2299. https://doi.org/10.3390/ijms19082299
Chicago/Turabian StyleKasembeli, Moses M., Uddalak Bharadwaj, Prema Robinson, and David J. Tweardy. 2018. "Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment" International Journal of Molecular Sciences 19, no. 8: 2299. https://doi.org/10.3390/ijms19082299