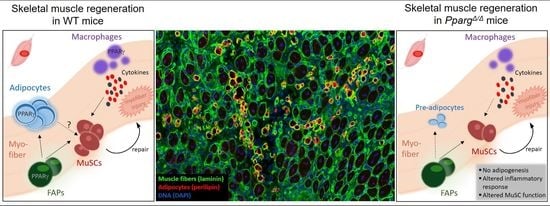
PPARγ Controls Ectopic Adipogenesis and Cross-Talks with Myogenesis During Skeletal Muscle Regeneration


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
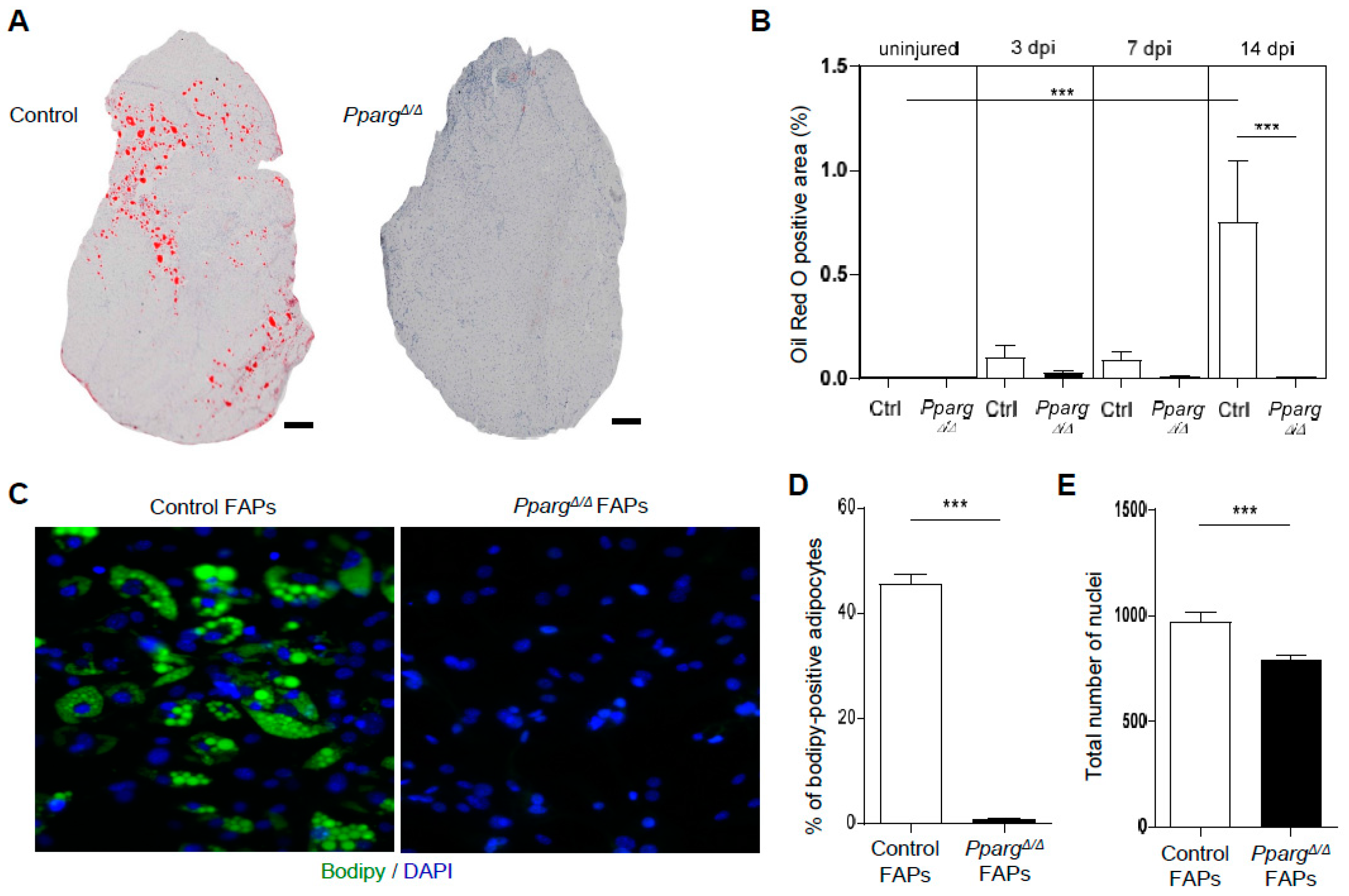
2.1. PPARγ Deletion Completely Abolishes Ectopic Adipogenesis During Skeletal Muscle Regeneration
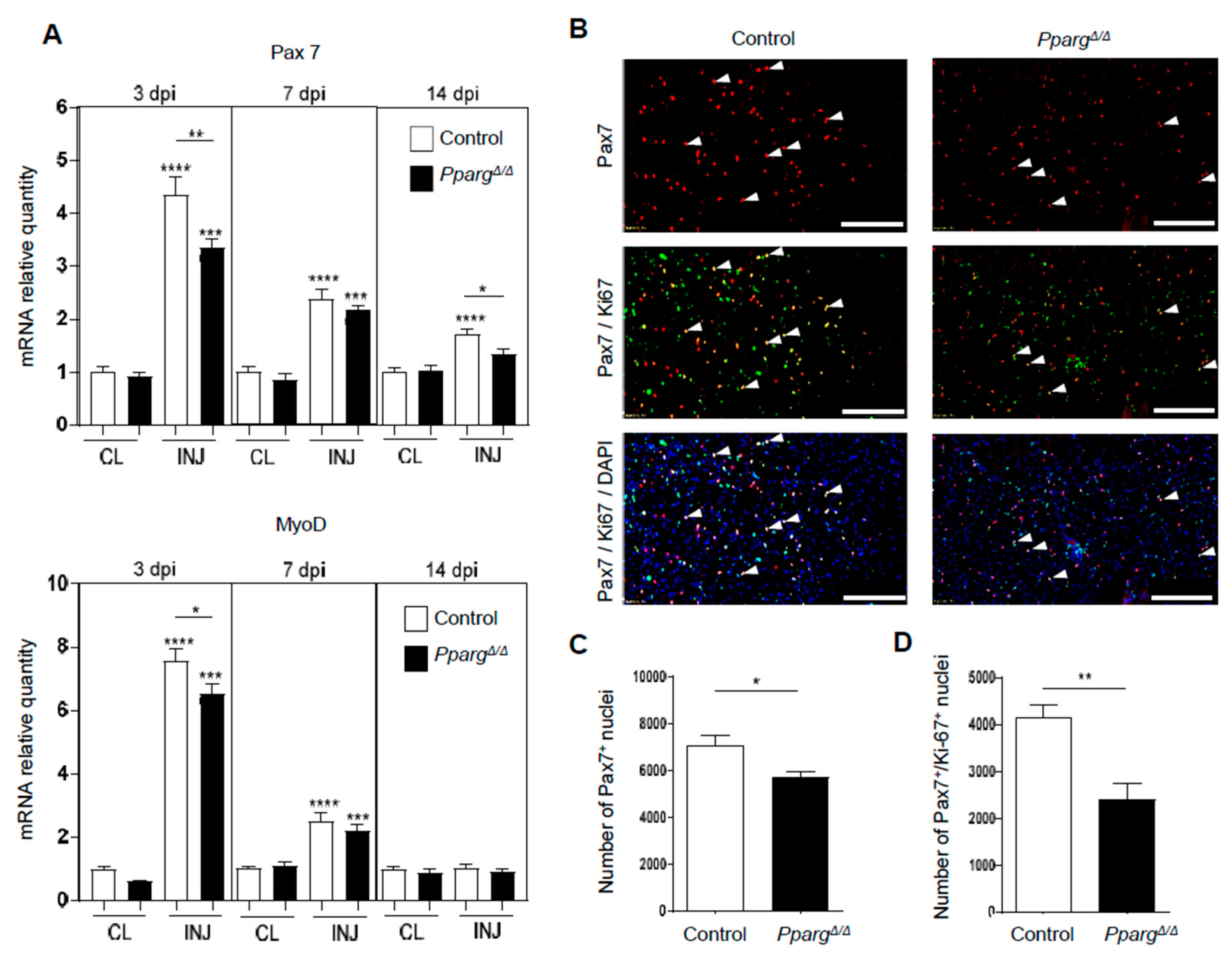
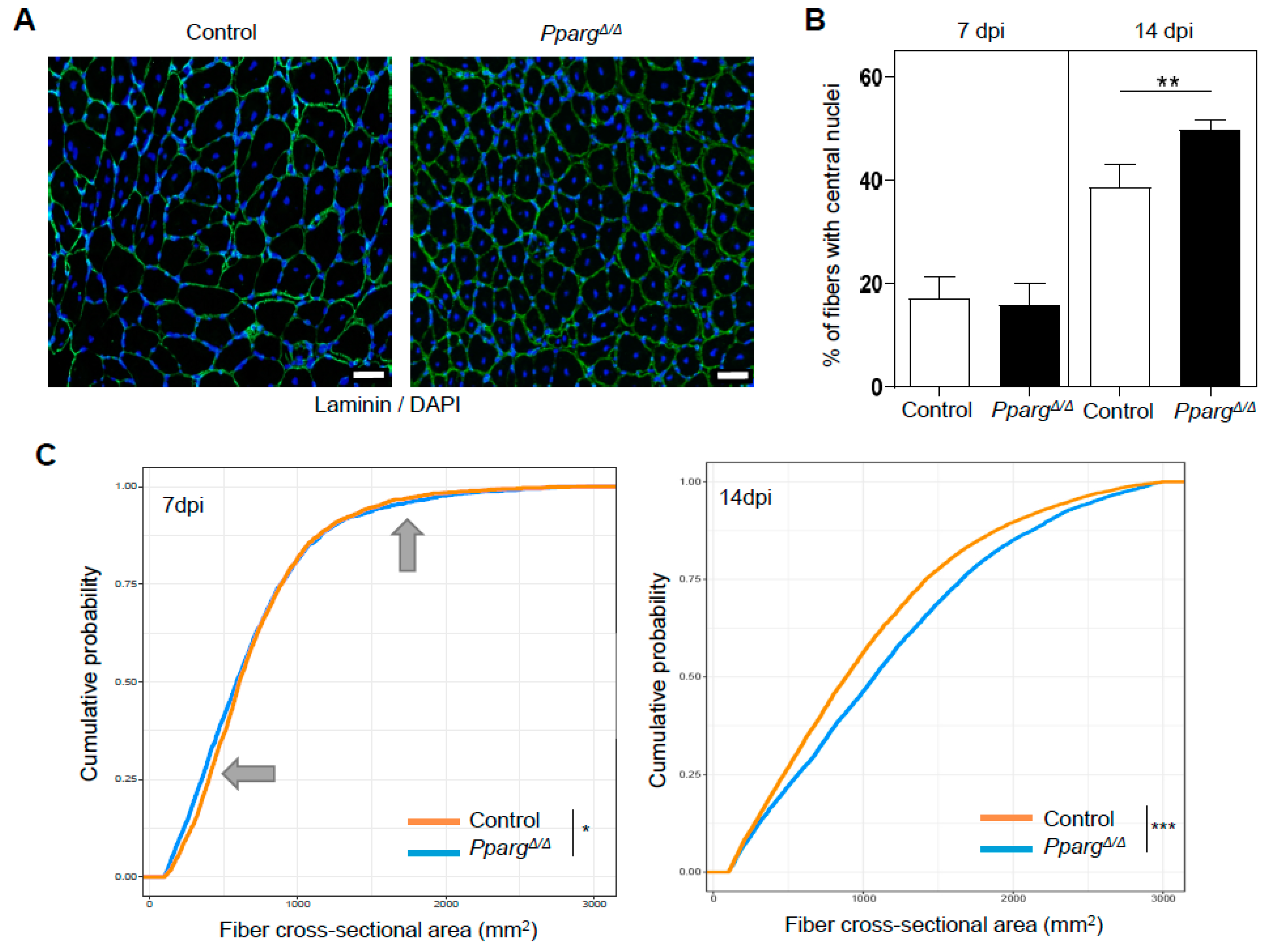
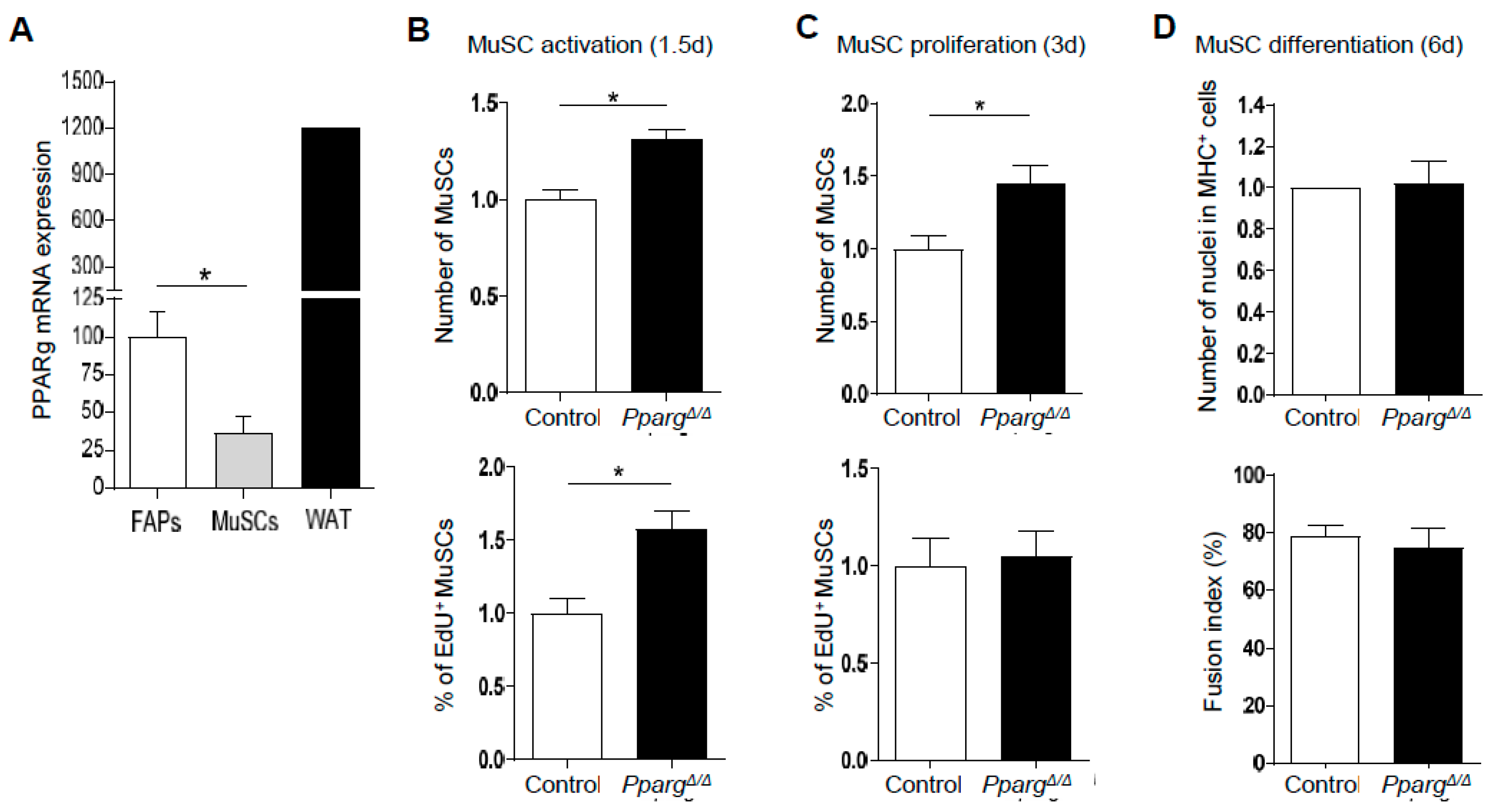
2.2. Loss of PPARγ Impairs Muscle Stem Cell Function and Cross-Talks with Myogenesis In Vivo
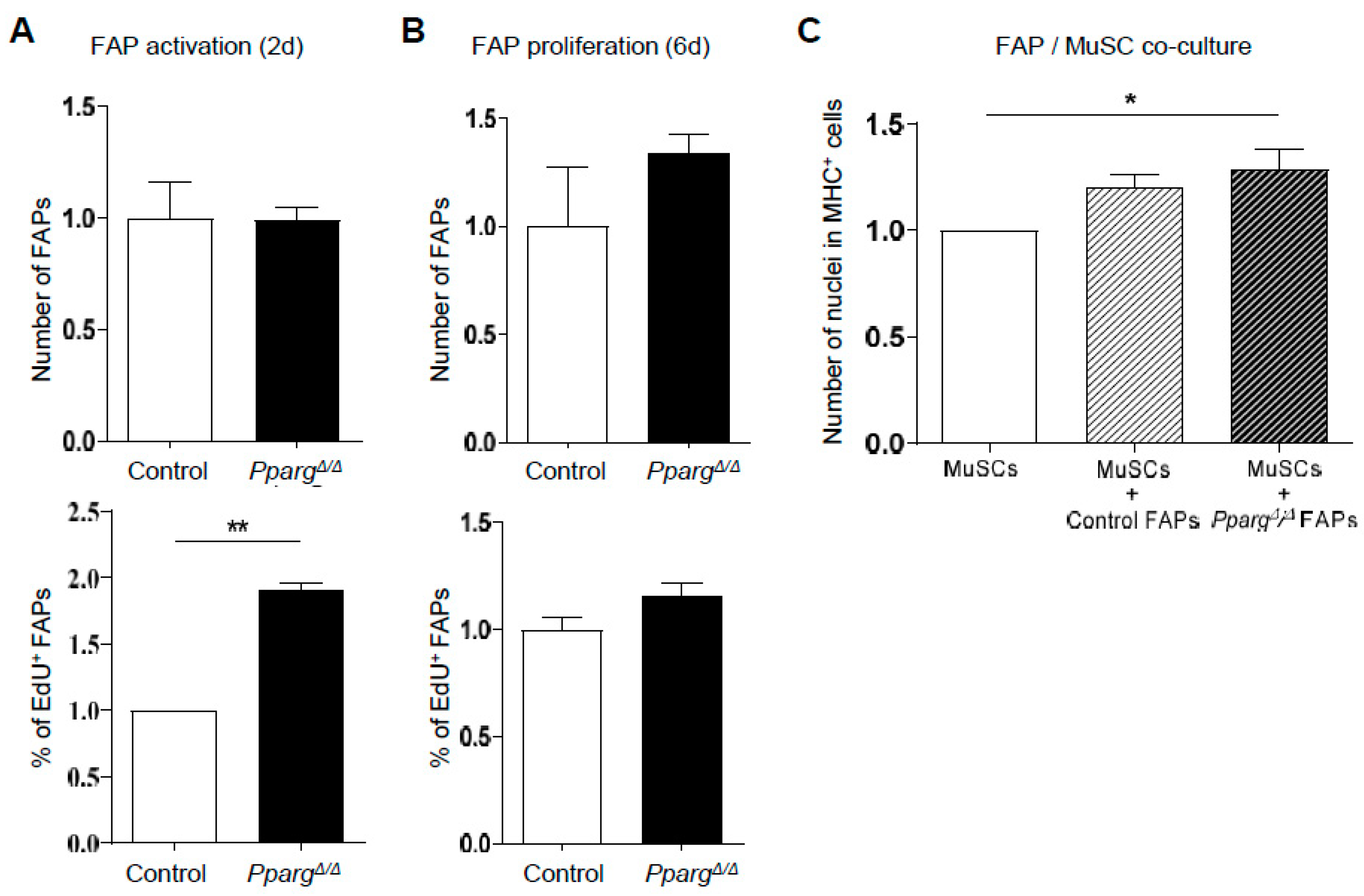
2.3. PPARγ KO Does Not Directly Impair the Intrinsic Function of MuSC Function and the FAP/MuSC Cross-Talk Ex Vivo
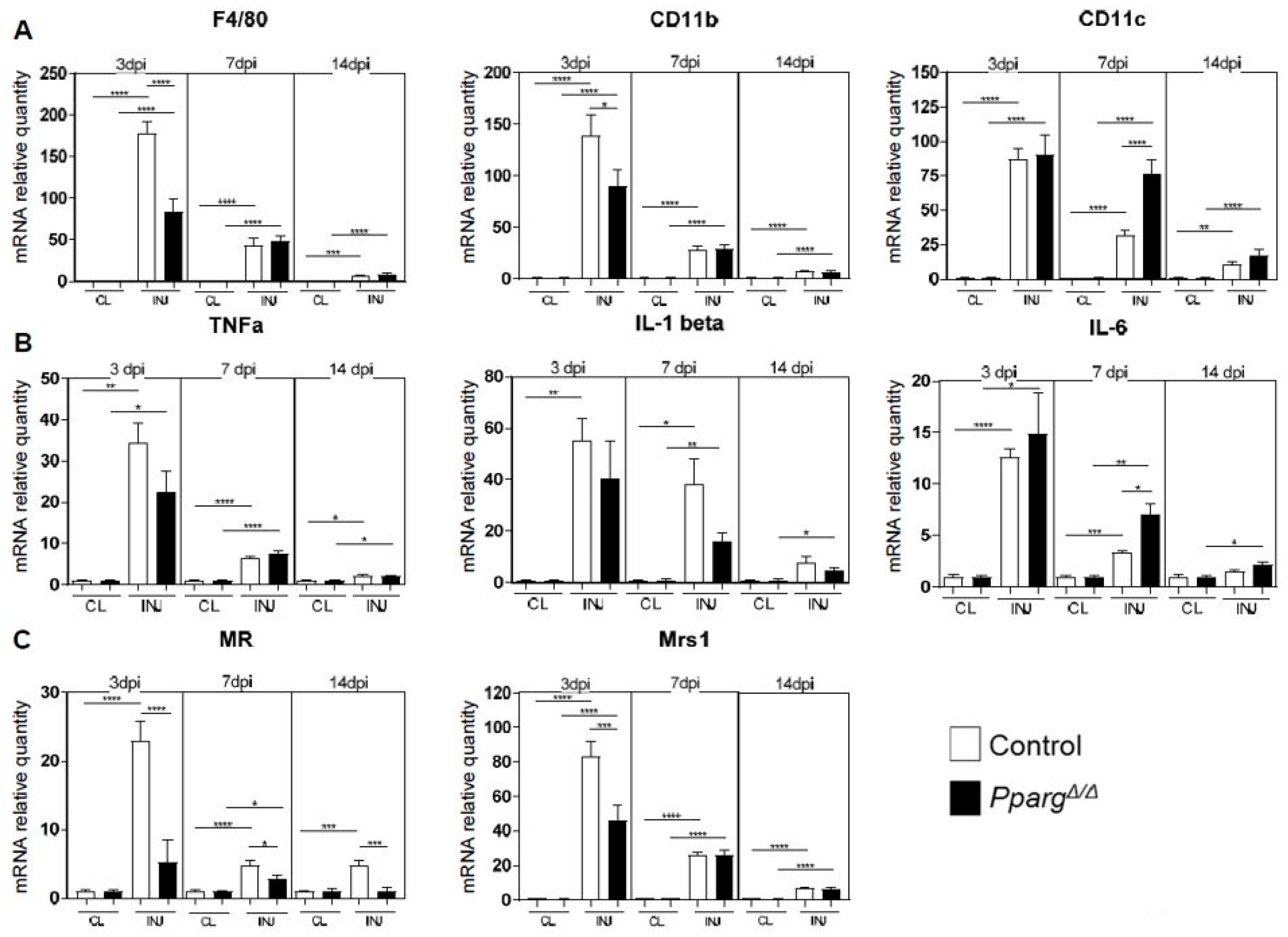
2.4. Altered Inflammatory Signatures in PPARγ-KO Mice During Muscle Regeneration
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Muscle Progenitor Cell Isolation
4.3. Cell Culture
4.4. Immunocytochemistry for In Vitro Studies
4.5. Immunohistochemistry for In Vivo Studies
4.6. Quantitative PCR
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Dumont, N.A.; Rudnicki, M.A. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 2013, 14, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abmayr, S.M.; Pavlath, G.K. Myoblast fusion: Lessons from flies and mice. Development 2012, 139, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Mounier, R.; Horvath, A.; Cuvellier, S.; Dumont, F.; Poliska, S.; Ardjoune, H.; Juban, G.; Nagy, L.; Chazaud, B. Highly dynamic transcriptional signature of distinct macrophage subsets during sterile inflammation, resolution, and tissue repair. J. Immunol. 2016, 196, 4771–4782. [Google Scholar] [CrossRef] [PubMed]
- Saclier, M.; Cuvellier, S.; Magnan, M.; Mounier, R.; Chazaud, B. Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS J. 2013, 280, 4118–4130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, D.; Kuznia, P.; Heshka, S.; Albu, J.; Heymsfield, S.B.; Goodpaster, B.; Visser, M.; Harris, T.B. Adipose tissue in muscle: A novel depot similar in size to visceral adipose tissue. Am. J. Clin. Nutr. 2005, 81, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Vettor, R.; Milan, G.; Franzin, C.; Sanna, M.; De Coppi, P.; Rizzuto, R.; Federspil, G. The origin of intermuscular adipose tissue and its pathophysiological implications. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E987–E998. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, J.N.; Bishop, J.Y.; Lo, I.K.; Flatow, E.L. Fatty infiltration and atrophy of the rotator cuff do not improve after rotator cuff repair and correlate with poor functional outcome. Am. J. Sports Med. 2007, 35, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Bishop, C.A.; Auh, S.; Newbould, R.D.; Fischbeck, K.H.; Janiczek, R.L. Quantifying disease activity in fatty-infiltrated skeletal muscle by ideal-cpmg in duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Samagh, S.P.; Kramer, E.J.; Melkus, G.; Laron, D.; Bodendorfer, B.M.; Natsuhara, K.; Kim, H.T.; Liu, X.; Feeley, B.T. MRI quantification of fatty infiltration and muscle atrophy in a mouse model of rotator cuff tears. J. Orthop. Res. 2013, 31, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Sambasivan, R.; Yao, R.; Kissenpfennig, A.; Van Wittenberghe, L.; Paldi, A.; Gayraud-Morel, B.; Guenou, H.; Malissen, B.; Tajbakhsh, S.; Galy, A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 2011, 138, 3647–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brioche, T.; Pagano, A.F.; Py, G.; Chopard, A. Muscle wasting and aging: Experimental models, fatty infiltrations, and prevention. Mol. Asp. Med. 2016, 50, 56–87. [Google Scholar] [CrossRef] [PubMed]
- Von Maltzahn, J.; Jones, A.E.; Parks, R.J.; Rudnicki, M.A. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. 2013, 110, 16474–16479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, B.; Serrano, A.L.; Tjwa, M.; Suelves, M.; Ardite, E.; De Mori, R.; Baeza-Raja, B.; Martinez de Lagran, M.; Lafuste, P.; Ruiz-Bonilla, V.; et al. Fibrinogen drives dystrophic muscle fibrosis via a tgfbeta/alternative macrophage activation pathway. Genes Dev. 2008, 22, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.; Sousa-Victor, P.; Ruiz-Bonilla, V.; Jardi, M.; Caelles, C.; Serrano, A.L.; Munoz-Canoves, P. P38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J. Cell Biol. 2011, 195, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Ikemoto-Uezumi, M.; Nakatani, M.; Morita, M.; Yamaguchi, A.; Yamada, H.; Nishino, I.; Hamada, Y.; et al. Identification and characterization of pdgfralpha+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 2014, 5, e1186. [Google Scholar] [CrossRef] [PubMed]
- Uezumi, A.; Nakatani, M.; Ikemoto-Uezumi, M.; Yamamoto, N.; Morita, M.; Yamaguchi, A.; Yamada, H.; Kasai, T.; Masuda, S.; Narita, A.; et al. Cell-surface protein profiling identifies distinctive markers of progenitor cells in human skeletal muscle. Stem Cell Rep. 2016, 7, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Lemos, D.R.; Babaeijandaghi, F.; Low, M.; Chang, C.K.; Lee, S.T.; Fiore, D.; Zhang, R.H.; Natarajan, A.; Nedospasov, S.A.; Rossi, F.M. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting tnf-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 2015, 21, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Lukjanenko, L.; Brachat, S.; Pierrel, E.; Lach-Trifilieff, E.; Feige, J.N. Genomic profiling reveals that transient adipogenic activation is a hallmark of mouse models of skeletal muscle regeneration. PLoS ONE 2013, 8, e71084. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.F.; Demangel, R.; Brioche, T.; Jublanc, E.; Bertrand-Gaday, C.; Candau, R.; Dechesne, C.A.; Dani, C.; Bonnieu, A.; Py, G.; et al. Muscle regeneration with intermuscular adipose tissue (IMAT) accumulation is modulated by mechanical constraints. PLoS ONE 2015, 10, e0144230. [Google Scholar] [CrossRef] [PubMed]
- Pisani, D.F.; Bottema, C.D.; Butori, C.; Dani, C.; Dechesne, C.A. Mouse model of skeletal muscle adiposity: A glycerol treatment approach. Biochem. Biophys. Res. Commun. 2010, 396, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Wagatsuma, A. Adipogenic potential can be activated during muscle regeneration. Mol. Cell. Biochem. 2007, 304, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef]
- Rosen, E.D.; Walkey, C.J.; Puigserver, P.; Spiegelman, B.M. Transcriptional regulation of adipogenesis. Genes Dev. 2000, 14, 1293–1307. [Google Scholar] [PubMed]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, B.M. PPAR-gamma: Adipogenic regulator and thiazolidinedione receptor. Diabetes 1998, 47, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Mounier, R.; Patsalos, A.; Gogolak, P.; Peloquin, M.; Horvath, A.; Pap, A.; Daniel, B.; Nagy, G.; Pintye, E.; et al. Macrophage PPARgamma, a lipid activated transcription factor controls the growth factor GDF3 and skeletal muscle regeneration. Immunity 2016, 45, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.W.; Chen, L.; Fisher, S.J.; Szanto, I.; Ristow, M.; Jozsi, A.C.; Hirshman, M.F.; Rosen, E.D.; Goodyear, L.J.; Gonzalez, F.J.; et al. Muscle-specific PPARγ-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J. Clin. Investig. 2003, 112, 608–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hevener, A.L.; He, W.; Barak, Y.; Le, J.; Bandyopadhyay, G.; Olson, P.; Wilkes, J.; Evans, R.M.; Olefsky, J. Muscle-specific Pparg deletion causes insulin resistance. Nat. Med. 2003, 9, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A. Control of macrophage activation and function by PPARS. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Li, A.C.; Binder, C.J.; Gutierrez, A.; Brown, K.K.; Plotkin, C.R.; Pattison, J.W.; Valledor, A.F.; Davis, R.A.; Willson, T.M.; Witztum, J.L.; et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J. Clin. Investig. 2004, 114, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Nadra, K.; Quignodon, L.; Sardella, C.; Joye, E.; Mucciolo, A.; Chrast, R.; Desvergne, B. PPARgamma in placental angiogenesis. Endocrinology 2010, 151, 4969–4981. [Google Scholar] [CrossRef] [PubMed]
- Sardella, C.; Winkler, C.; Quignodon, L.; Hardman, J.A.; Toffoli, B.; Giordano Attianese, G.M.P.; Hundt, J.E.; Michalik, L.; Vinson, C.R.; Paus, R.; et al. Delayed hair follicle morphogenesis and hair follicle dystrophy in a lipoatrophy mouse model of pparg total deletion. J. Investig. Dermatol. 2018, 138, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Bassil, M.S.; Gougeon, R. Muscle protein anabolism in type 2 diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, D.B. Mouse models of inherited lipodystrophy. Dis. Model Mech. 2009, 2, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajas, L.; Landsberg, R.L.; Huss-Garcia, Y.; Sardet, C.; Lees, J.A.; Auwerx, J. E2Fs regulate adipocyte differentiation. Dev. Cell 2002, 3, 39–49. [Google Scholar] [PubMed]
- Angione, A.R.; Jiang, C.; Pan, D.; Wang, Y.X.; Kuang, S. PPARdelta regulates satellite cell proliferation and skeletal muscle regeneration. Skelet. Muscle 2011, 1, 33. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, P.; Manickam, R.; Ge, X.; Bonala, S.; McFarlane, C.; Sharma, M.; Wahli, W.; Kambadur, R. Inactivation of PPARbeta/delta adversely affects satellite cells and reduces postnatal myogenesis. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E122–E131. [Google Scholar] [CrossRef] [PubMed]
- Mothe-Satney, I.; Piquet, J.; Murdaca, J.; Sibille, B.; Grimaldi, P.A.; Neels, J.G.; Rousseau, A.S. Peroxisome proliferator activated receptor beta (PPARbeta) activity increases the immune response and shortens the early phases of skeletal muscle regeneration. Biochimie 2017, 136, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Proserpio, V.; Lichtenberger, B.M.; Natsuga, K.; Sinclair, R.; Fujiwara, H.; Watt, F.M. Epidermal wnt/beta-catenin signaling regulates adipocyte differentiation via secretion of adipogenic factors. Proc. Natl. Acad. Sci. USA 2014, 111, E1501–E1509. [Google Scholar] [CrossRef] [PubMed]
- Festa, E.; Fretz, J.; Berry, R.; Schmidt, B.; Rodeheffer, M.; Horowitz, M.; Horsley, V. Adipocyte lineage cells contribute to the skin stem cell niche to drive hair cycling. Cell 2011, 146, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.A.; Horsley, V. Intradermal adipocytes mediate fibroblast recruitment during skin wound healing. Development 2013, 140, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, J.; Guo, B.; Xie, J.; Deng, S.; Fu, N.; Lin, S.; Li, G.; Lin, Y.; Cai, X. Crosstalk between adipose-derived stem cells and chondrocytes: When growth factors matter. Bone Res. 2016, 4, 15036. [Google Scholar] [CrossRef] [PubMed]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory t cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Fiore, D.; Judson, R.N.; Low, M.; Lee, S.; Zhang, E.; Hopkins, C.; Xu, P.; Lenzi, A.; Rossi, F.M.; Lemos, D.R. Pharmacological blockage of fibro/adipogenic progenitor expansion and suppression of regenerative fibrogenesis is associated with impaired skeletal muscle regeneration. Stem Cell Res. 2016, 17, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Takakuwa, R.; Marchand, S.; Dentz, E.; Bornert, J.M.; Messaddeq, N.; Wendling, O.; Mark, M.; Desvergne, B.; Wahli, W.; et al. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc. Natl. Acad. Sci. USA 2004, 101, 4543–4547. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dammone, G.; Karaz, S.; Lukjanenko, L.; Winkler, C.; Sizzano, F.; Jacot, G.; Migliavacca, E.; Palini, A.; Desvergne, B.; Gilardi, F.; et al. PPARγ Controls Ectopic Adipogenesis and Cross-Talks with Myogenesis During Skeletal Muscle Regeneration. Int. J. Mol. Sci. 2018, 19, 2044. https://doi.org/10.3390/ijms19072044
Dammone G, Karaz S, Lukjanenko L, Winkler C, Sizzano F, Jacot G, Migliavacca E, Palini A, Desvergne B, Gilardi F, et al. PPARγ Controls Ectopic Adipogenesis and Cross-Talks with Myogenesis During Skeletal Muscle Regeneration. International Journal of Molecular Sciences. 2018; 19(7):2044. https://doi.org/10.3390/ijms19072044
Chicago/Turabian StyleDammone, Gabriele, Sonia Karaz, Laura Lukjanenko, Carine Winkler, Federico Sizzano, Guillaume Jacot, Eugenia Migliavacca, Alessio Palini, Béatrice Desvergne, Federica Gilardi, and et al. 2018. "PPARγ Controls Ectopic Adipogenesis and Cross-Talks with Myogenesis During Skeletal Muscle Regeneration" International Journal of Molecular Sciences 19, no. 7: 2044. https://doi.org/10.3390/ijms19072044