Post-Receptor Inhibitors of the GHR-JAK2-STAT Pathway in the Growth Hormone Signal Transduction


Abstract
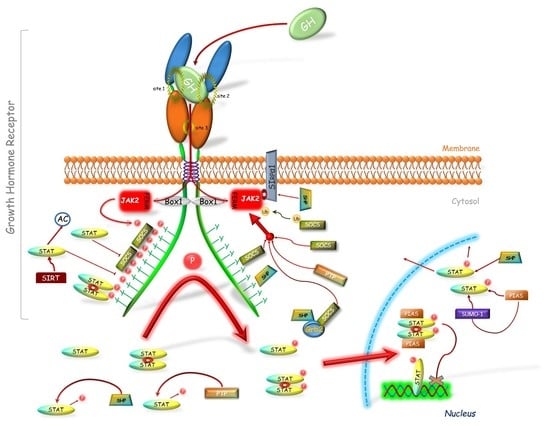
{kind=link}
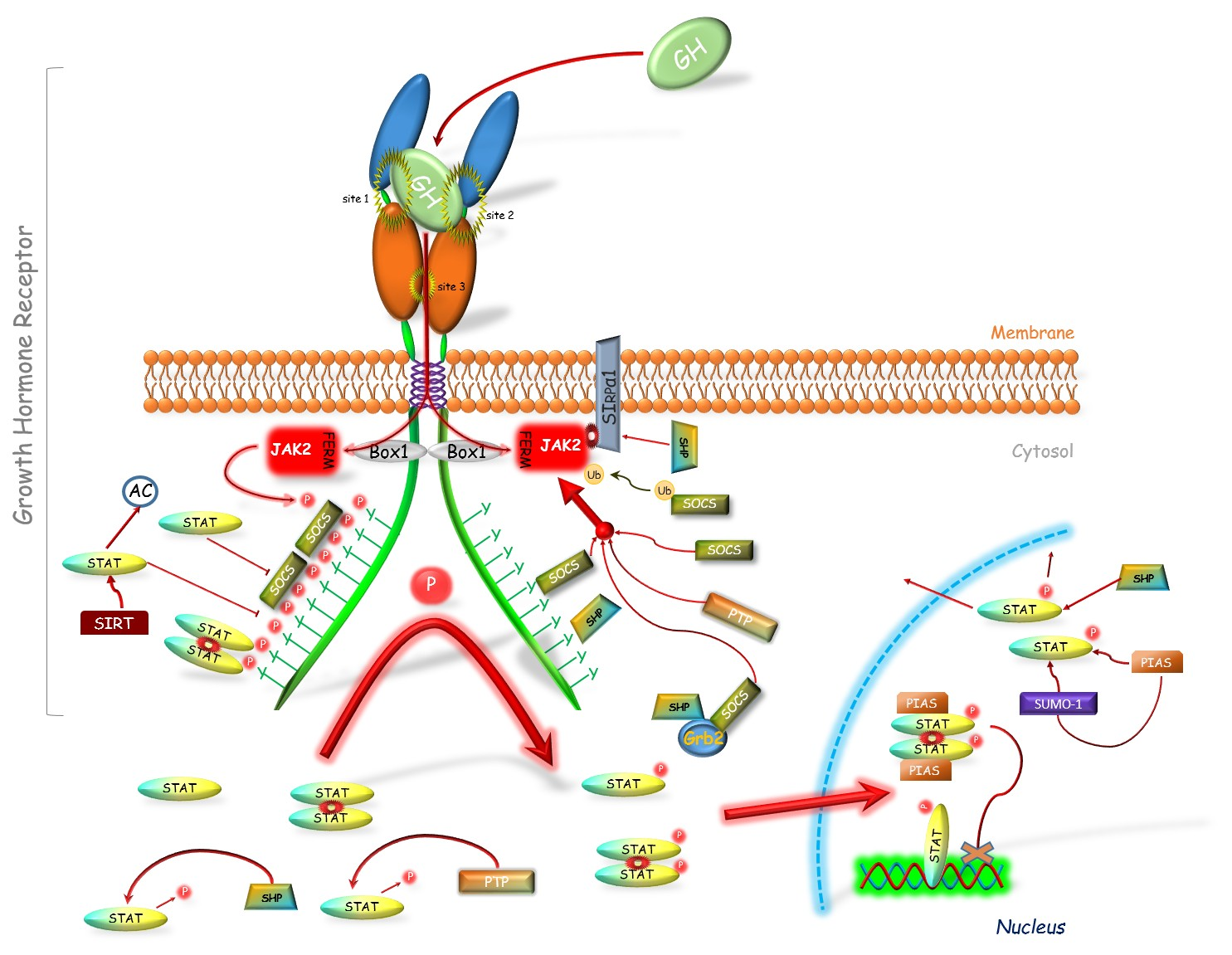
{kind=link}
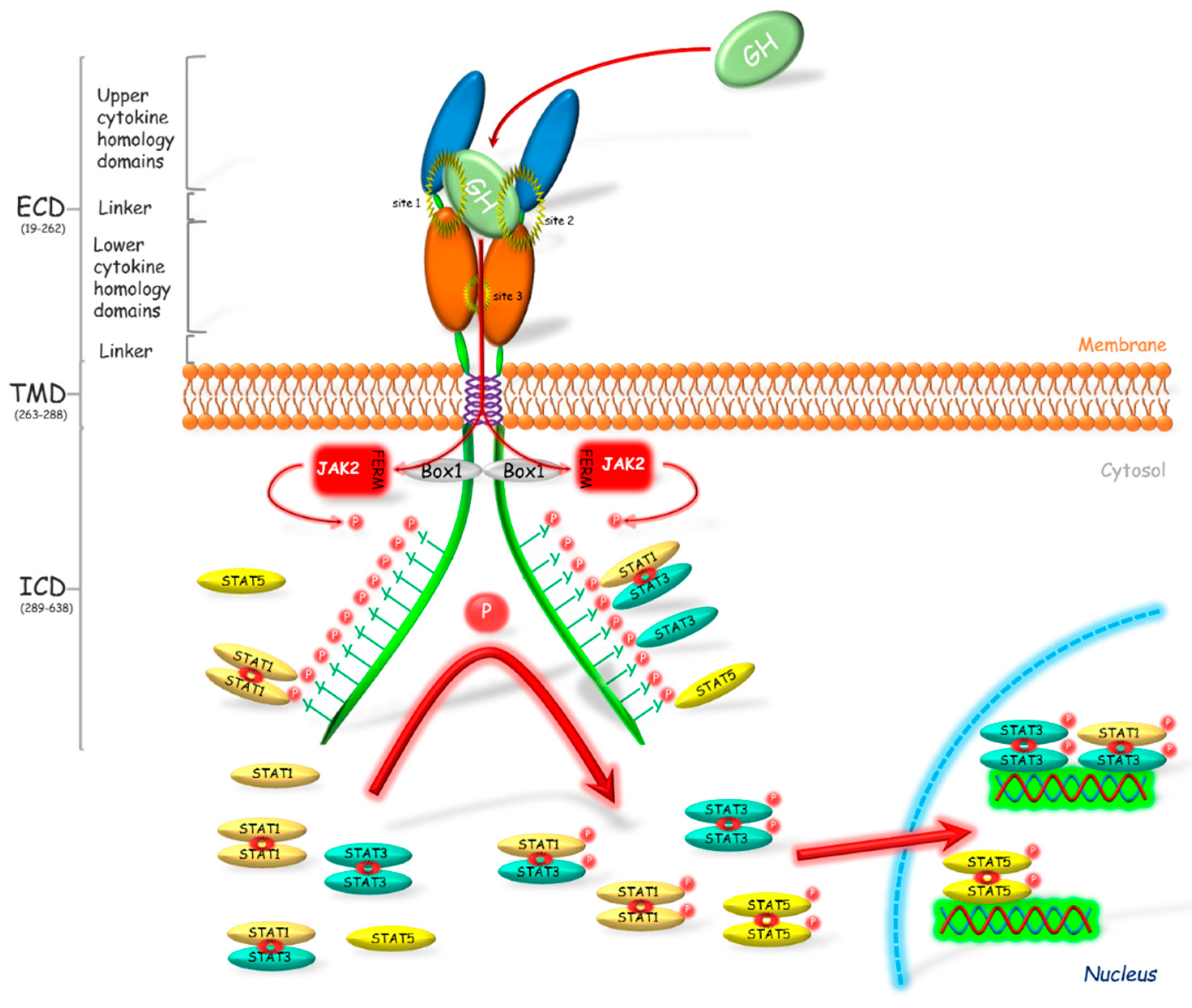
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Somatotropic Axis
1.2. GHR-JAK2-STAT Pathway
2. GHR-JAK2-STAT Inhibitors
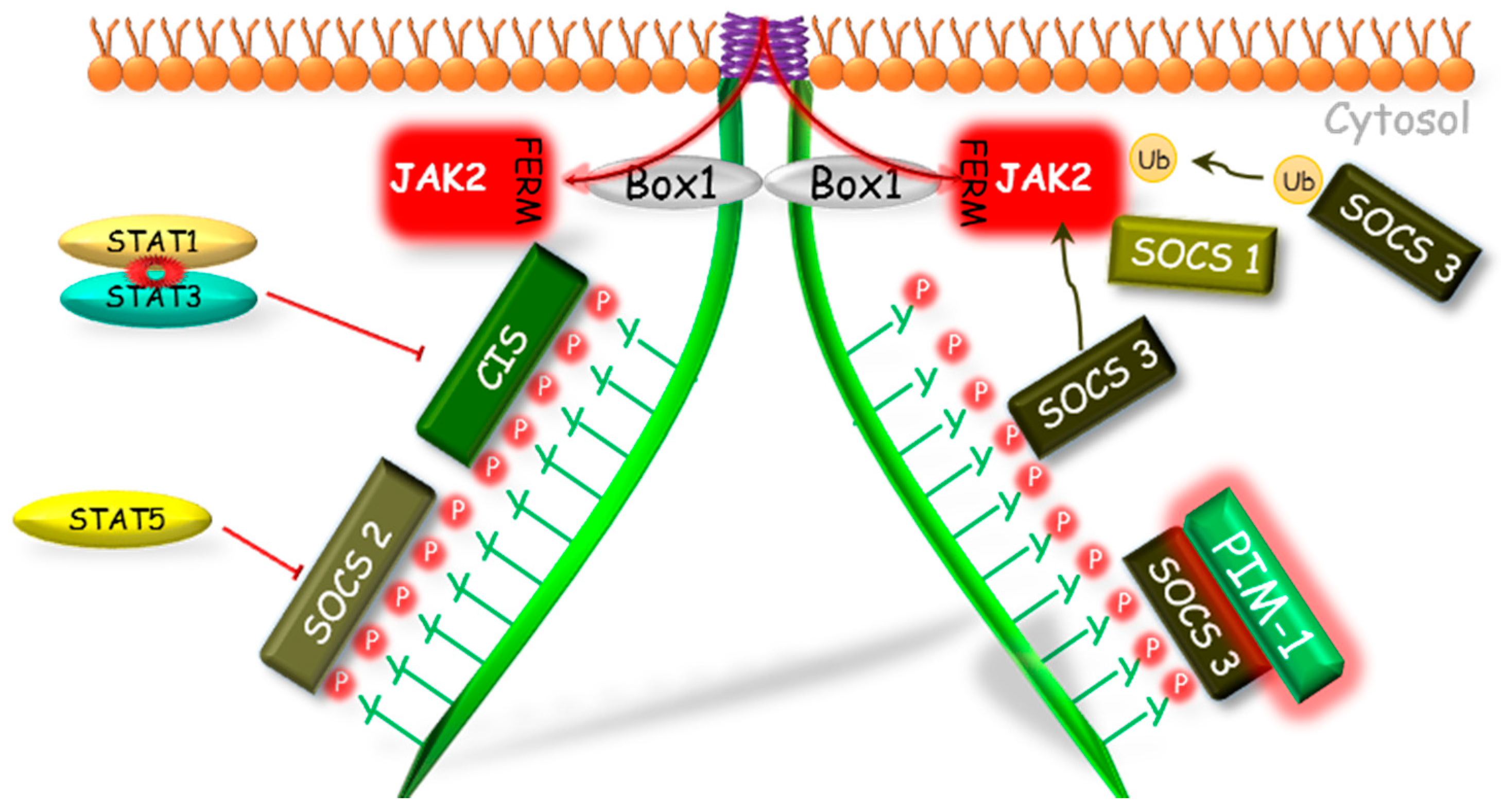
2.1. Suppressors of Cytokine Signaling (SOCS)
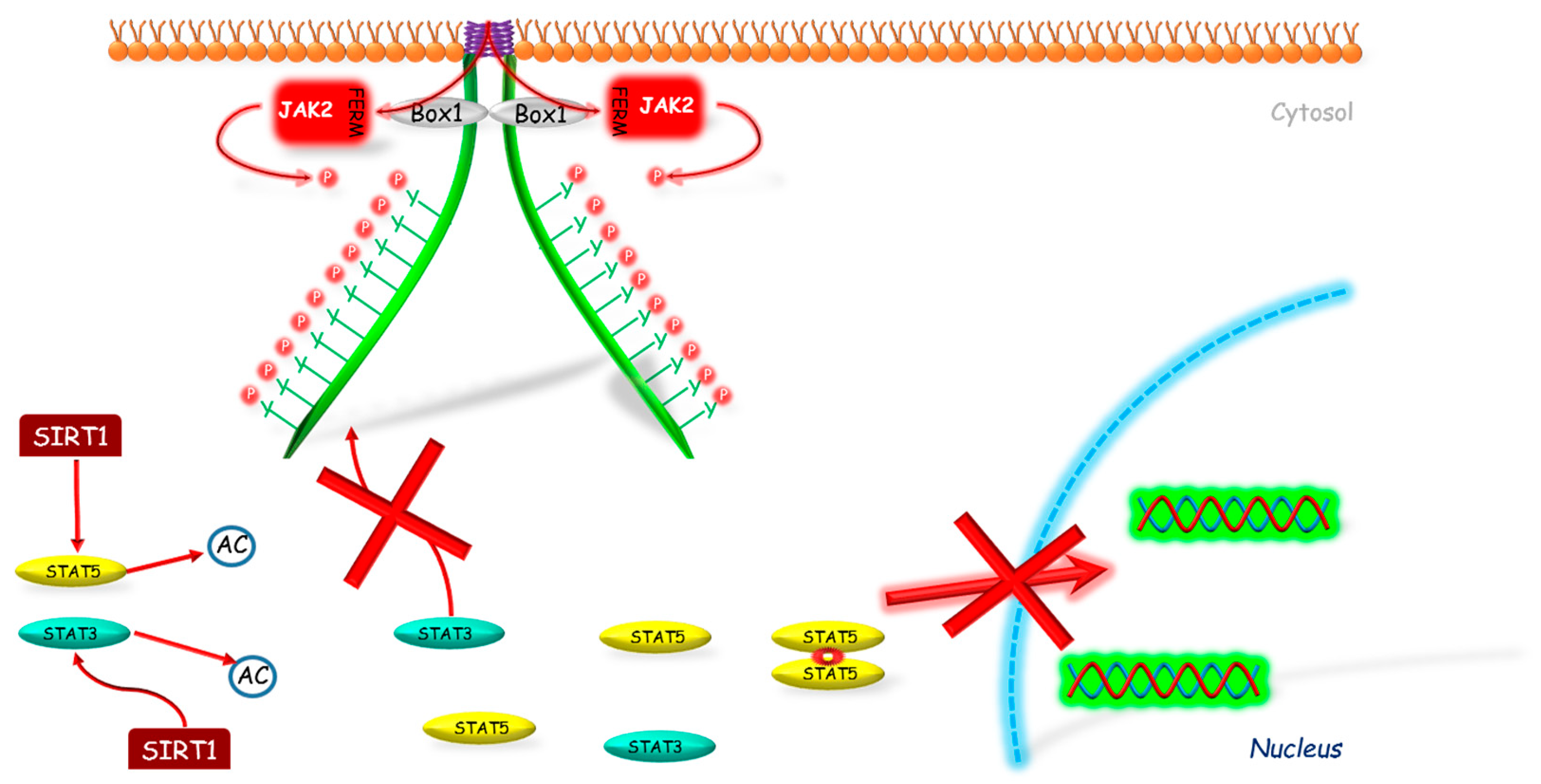
2.2. Sirtuin 1 (SIRT1)
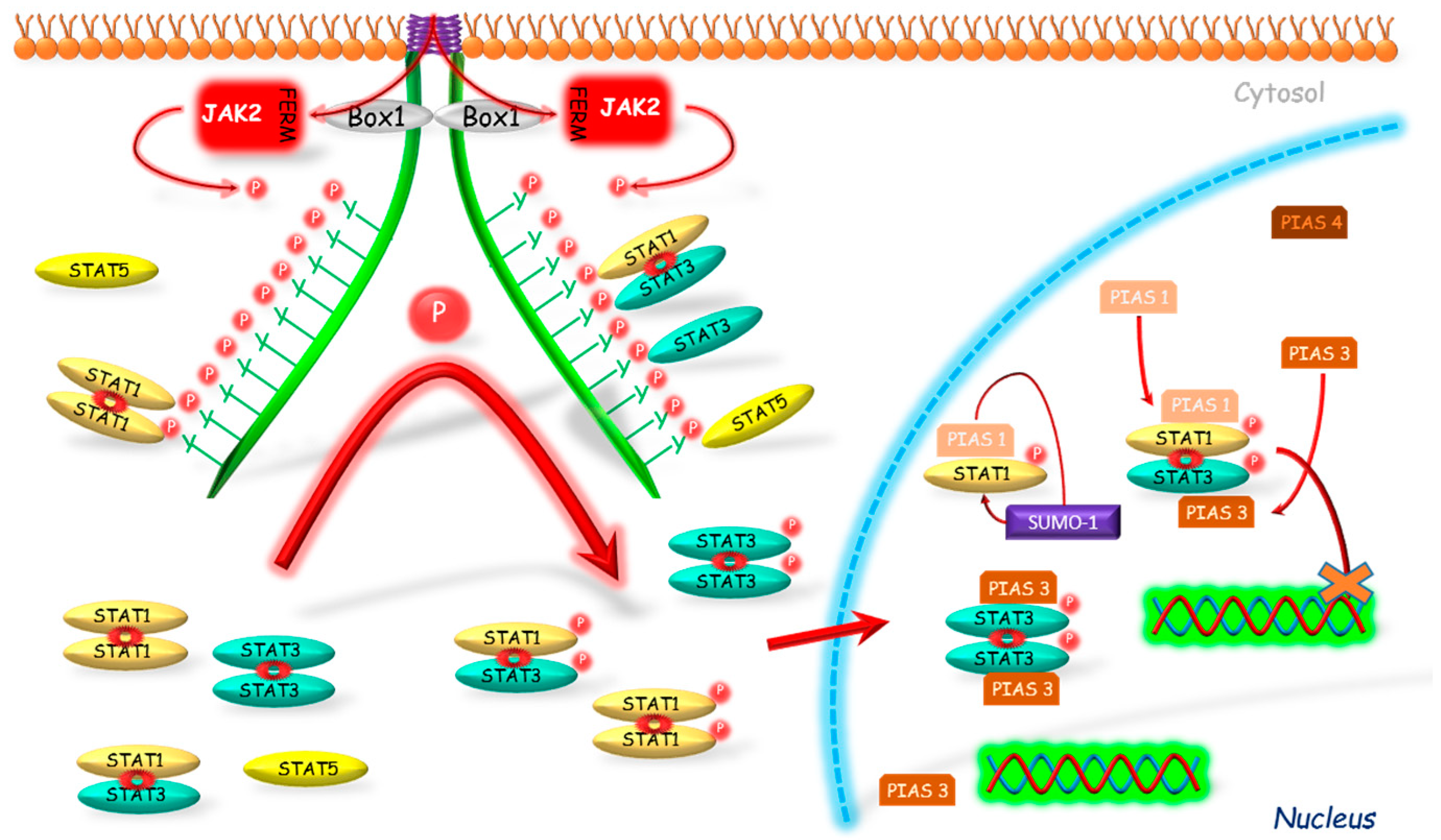
2.3. Protein Inhibitor of Activated STAT (PIAS)
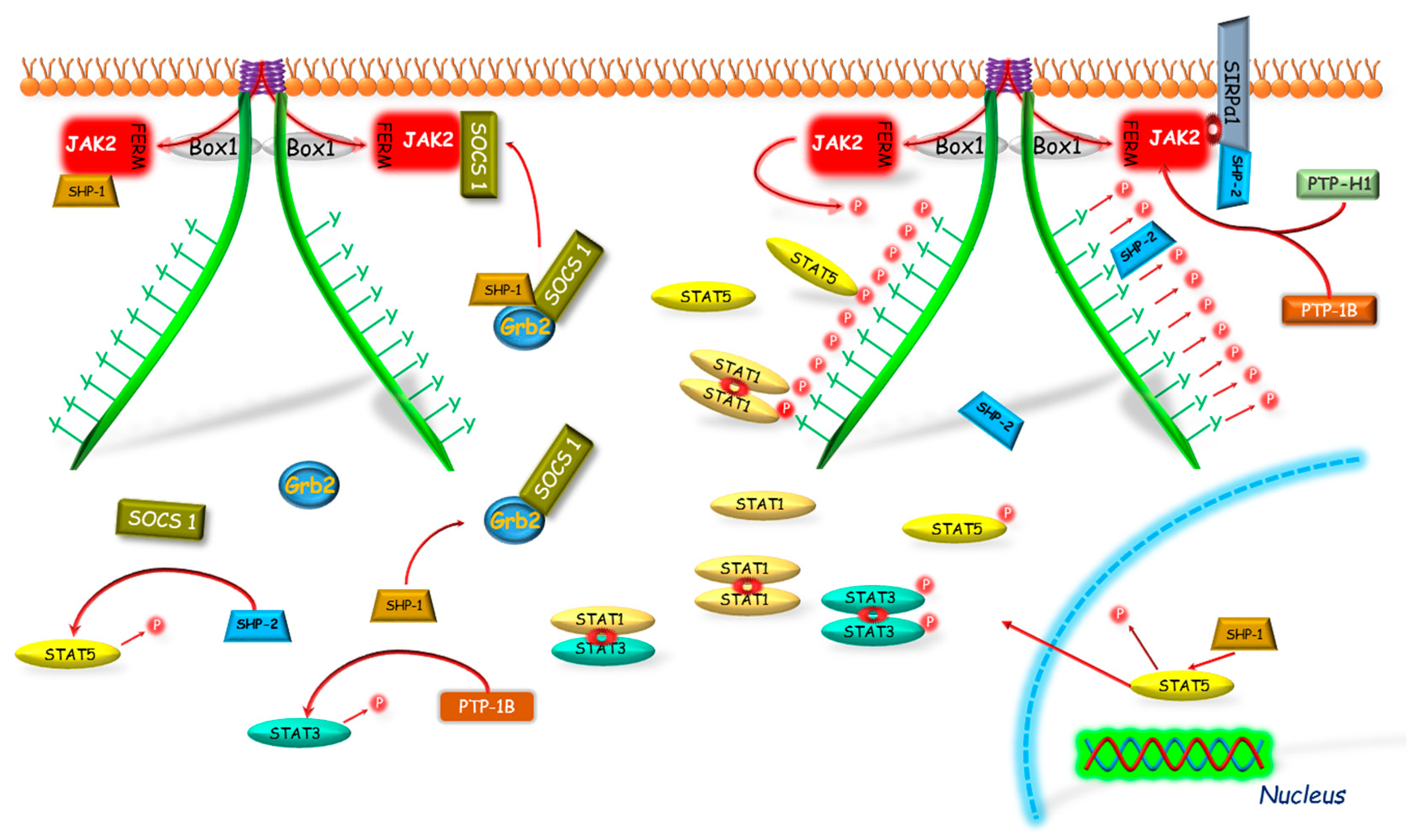
2.4. Phosphatases
2.4.1. Protein Tyrosine Phosphatase (PTP-1B and PTP-H1)
2.4.2. Src Homology 2 (SH2) Domain Containing Protein Tyrosine Phosphatase (SHP-1)
2.4.3. Src Homology 2 (SH2) Domain Containing Protein Tyrosine Phosphatase (SHP-2)
2.5. Signal Regulatory Proteins (SIRPs)
3. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Liu, Y.; Chen, J.; Tang, X.; Chen, P.; Zhang, M. The 20 kDa and 22 kDa forms of human growth hormone (hGH) exhibit different intracellular signalling profiles and properties. Gen. Comp. Endocrinol. 2017, 248, 49–54. [Google Scholar] [CrossRef]
- Hirt, H.; Kimelman, J.; Birnbaum, M.J.; Chen, E.Y.; Seeburg, P.H.; Eberhardt, N.L.; Barta, A. The human growth hormone gene locus: Structure, evolution, and allelic variations. DNA 1987, 6, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Velegrakis, A.; Sfakiotaki, M.; Sifakis, S. Human placental growth hormone in normal and abnormal fetal growth. Biomed. Rep. 2017, 7, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Devesa, J.; Almengló, C.; Devesa, P. Multiple Effects of Growth Hormone in the Body: Is it really the hormone for growth? Clin. Med. Insights Endocrinol. Diabetes 2016, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Soendergaard, C.; Kvist, P.H.; Thygesen, P.; Reslow, M.; Nielsen, O.H.; Kopchick, J.J.; Holm, T.L. Characterization of Growth Hormone Resistance in Experimental and Ulcerative Colitis. Int. J. Mol. Sci. 2017, 18, 2046. [Google Scholar] [CrossRef] [PubMed]
- Khatib, N.; Gaidhane, S.; Gaidhane, A.M.; Khatib, M.; Simkhada, P.; Gode, D.; Zahiruddin, Q.S. Ghrelin: Ghrelin as a Regulatory Peptide in Growth Hormone Secretion. J. Clin. Diagn. Res. 2014, 8, MC13–MC17. [Google Scholar] [CrossRef] [PubMed]
- Ceranowicz, P.; Warzecha, Z.; Dembinski, A. Peptidyl hormones of endocrine cells origin in the gut–their discovery and physiological relevance. J. Physiol. Pharmacol. 2015, 66, 11–27. [Google Scholar] [PubMed]
- Warzecha, Z.; Dembiński, A.; Ceranowicz, P.; Dembiński, M.; Cieszkowski, J.; Konturek, S.J.; Polus, A.; Pawlik, W.W.; Kuwahara, A.; Kato, I. Influence of ghrelin on gastric and duodenal growth and expression of digestive enzymes in young mature rats. J. Physiol. Pharmacol. 2006, 57, 425–437. [Google Scholar] [PubMed]
- Wang, Q.; Bing, C.; Al-Barazanji, K.; Mossakowaska, D.E.; Wang, X.M.; McBay, D.L.; Neville, W.A.; Taddayon, M.; Pickavance, L.; Dryden, S.; et al. Interactions between leptin and hypothalamic neuropeptide Y neurons in the control of food intake and energy homeostasis in the rat. Diabetes 1997, 46, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Watanobe, H.; Habu, S. Leptin regulates growth hormone-releasing factor, somatostatin, and α-melanocyte-stimulating hormone but not neuropeptide Y release in rat hypothalamus in vivo: Relation with growth hormone secretion. J. Neurosci. 2002, 22, 6265–6271. [Google Scholar] [CrossRef] [PubMed]
- Luque, R.M.; Peinado, J.R.; Gracia-Navarro, F.; Broglio, F.; Ghigo, E.; Kineman, R.D.; Malagón, M.M.; Castaño, J.P. Cortistatin mimics somatostatin by inducing a dual, dose-dependent stimulatory and inhibitory effect on growth hormone secretion in somatotropes. J. Mol. Endocrinol. 2006, 36, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Rubinek, T.; Modan-Moses, D. Klotho and the Growth Hormone/Insulin-Like Growth Factor 1 Axis: Novel Insights into Complex Interactions. Vitam. Horm. 2016, 101, 85–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, J.; McDonald, C.; Lupino, K.; Zhai, X.; Wilkins, B.J.; Hakonarson, H.; Pei, L. GDF15 is a heart-derived hormone that regulates body growth. EMBO Mol. Med. 2017, 9, 1150–1164. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, D.; Díaz, O.; Devesa, P.; Devesa, J. Growth Hormone (GH) and Cardiovascular System. Int. J. Mol. Sci. 2018, 19, 290. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Waters, M.J. The growth hormone receptor: Mechanism of activation and clinical implications. Nat. Rev. Endocrinol. 2010, 6, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Waters, M.J. The growth hormone receptor. Growth Hormon. IGF Res. 2016, 28, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Melmed, S. Acromegaly pathogenesis and treatment. J. Clin. Investig. 2009, 119, 3189–3202. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R. Metabolic Actions of IGF-I in Normal Physiology and Diabetes. Endocrinol. Metab. Clin. N. Am. 2012, 41, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Simpson, H.L.; Jackson, N.C.; Shojaee-Moradie, F.; Jones, R.H.; Russell-Jones, D.L.; Sönksen, P.H.; Dunger, D.B.; Umpleby, A.M. Insulin-like growth factor I has a direct effect on glucose and protein metabolism, but no effect on lipid metabolism in type 1 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Edwall, D.; Prisell, P.T.; Levinovitz, A.; Jennische, E.; Norstedt, G. Expression of insulin-like growth factor I messenger ribonucleic acid in regenerating bone after fracture: Influence of indomethacin. J. Bone Miner. Res. 1992, 7, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Giacinti, C.; Nardis, C.; Borsellino, G.; Rizzuto, E.; Nicoletti, C.; Wannenes, F.; Battistini, L.; Rosenthal, N.; Molinaro, M.; et al. Local expression of IGF-1 accelerates muscle regeneration by rapidly modulating inflammatory cytokines and chemokines. FASEB J. 2007, 21, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Tiangco, D.A.; Papakonstantinou, K.C.; Mullinax, K.A.; Terzis, J.K. IGF-I and end-to-side nerve repair: A dose-response study. J. Reconstr. Microsurg. 2001, 17, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Apel, P.J.; Jun, J.; Callahan, M.; Northam, C.N.; Alton, T.B.; Sonntag, W.E.; Li, Z. Effect of locally delivered IGF-1 on nerve regeneration during aging: An experimental study in rats. Muscle Nerve 2010, 41, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, Z.; Dembinski, A.; Ceranowicz, P.; Konturek, S.J.; Tomaszewska, R.; Stachura, J.; Konturek, P.C. IGF-1 stimulates production of interleukin-10 and inhibits development of caerulein-induced pancreatitis. J. Physiol. Pharmacol. 2003, 54, 575–590. [Google Scholar] [PubMed]
- Mallol, C.; Casana, E.; Jimenez, V.; Casellas, A.; Haurigot, V.; Jambrina, C.; Sacristan, V.; Morró, M.; Agudo, J.; Vilà, L. AAV-mediated pancreatic overexpression of Igf1 counteracts progression to autoimmune diabetes in mice. Mol. Metab. 2017, 6, 664–680. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, Z.; Ceranowicz, P.; Dembinski, A.; Cieszkowski, J.; Kusnierz-Cabala, B.; Tomaszewska, R.; Kuwahara, A.; Kato, I. Therapeutic effect of ghrelin in the course of cerulein-induced acute pancreatitis in rats. J. Physiol. Pharmacol. 2010, 61, 419–427. [Google Scholar] [PubMed]
- Matuszyk, A.; Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Ceranowicz, D.; Gałązka, K.; Bonior, J.; Jaworek, J.; Bartuś, K.; Gil, K. Exogenous Ghrelin Accelerates the Healing of Acetic Acid-Induced Colitis in Rats. Int. J. Mol. Sci. 2016, 17, 1455. [Google Scholar] [CrossRef] [PubMed]
- Ceranowicz, P.; Warzecha, Z.; Cieszkowski, J.; Ceranowicz, D.; Kuśnierz-Cabala, B.; Bonior, J.; Jaworek, J.; Ambroży, T.; Gil, K.; Olszanecki, R. Essential role of growth hormone and IGF-1 in therapeutic effect of ghrelin in the course of acetic acid-induced colitis. Int. J. Mol. Sci. 2017, 18, 1118. [Google Scholar] [CrossRef] [PubMed]
- Cieszkowski, J.; Warzecha, Z.; Ceranowicz, P.; Ceranowicz, D.; Kusnierz-Cabala, B.; Pedziwiatr, M.; Dembinski, M.; Ambrozy, T.; Kaczmarzyk, T.; Pihut, M. Therapeutic effect of exogenous ghrelin in the healing of gingival ulcers is mediated by the release of endogenous growth hormone and insulin-like growth factor-1. J. Physiol. Pharmacol. 2017, 68, 609–617. [Google Scholar] [PubMed]
- De Vos, A.M.; Ultsch, M.; Kossiakoff, A.A. Human growth hormone and extracellular domain of its receptor: Crystal structure of the complex. Science 1992, 255, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.A.; Le Roith, D. Control of growth by the somatropic axis: Growth hormone and the insulin-like growth factors have related and independent roles. Annu. Rev. Physiol. 2001, 63, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Gent, J.; van Kerkhof, P.; Roza, M.; Bu, G.; Strous, G.J. Ligand-independent growth hormone receptor dimerization occurs in the endoplasmic reticulum and is required for ubiquitin system-dependent endocytosis. Proc. Natl. Acad. Sci. USA 2002, 99, 9858–9863. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Wooh, J.W.; Tunny, K.A.; Waters, M.J. Growth hormone receptor; mechanism of action. Int. J. Biochem. Cell Biol. 2008, 40, 1984–1989. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; Adams, J.J.; Pelekanos, R.A.; Wan, Y.; McKinstry, W.J.; Palethorpe, K.; Seeber, R.M.; Monks, T.A.; Eidne, K.A.; Parker, M.W.; et al. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat. Struct. Mol. Biol. 2005, 12, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Argetsinger, L.S.; Campbell, G.S.; Yang, X.; Witthuhn, B.A.; Silvennoinen, O.; Ihle, J.N.; Carter-Su, C. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell 1993, 74, 237–244. [Google Scholar] [CrossRef]
- Zhu, T.; Goh, E.L.; Lobie, P.E. Growth hormone stimulates the tyrosine phosphorylation and association of p125 focal adhesion kinase (FAK) with JAK2. Fak is not required for stat-mediated transcription. J. Biol. Chem. 1998, 273, 10682–10689. [Google Scholar] [CrossRef] [PubMed]
- Alves dos Santos, C.M.; van Kerkhof, P.; Strous, G.J. The signal transduction of the growth hormone receptor is regulated by the ubiquitin/proteasome system and continues after endocytosis. J. Biol. Chem. 2001, 276, 10839–10846. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Herrington, J.; Smit, L.S.; Schwartz, J.; Carter-Su, C. The role of STAT proteins in growth hormone signaling. Oncogene 2000, 19, 2585–2597. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by statl and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Beuvink, I.; Hess, D.; Flotow, H.; Hofsteenge, J.; Groner, B.; Hynes, N.E. Stat5a Serine Phosphorylation. Serine 779 is constitutively phosphorylated in the mammary gland, and serine 725 phosphorylation influences prolactin-stimulatedin vitro DNA binding activity. J. Biol. Chem. 2000, 275, 10247–10255. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Xu, J.; Erwin, R.A.; Farrar, W.L.; Kirken, R.A.; Rui, H. Differential control of the phosphorylation state of proline-juxtaposed serine residues ser725 of stat5a and ser730 of STAT5β in prolactin-sensitive cells. J. Biol. Chem. 1998, 273, 30218–30224. [Google Scholar] [CrossRef] [PubMed]
- Gouilleux, F.; Wakao, H.; Mundt, M.; Groner, B. Prolactin induces phosphorylation of Tyr694 of Stat5 (MGF), a prerequisite for DNA binding and induction of transcription. EMBO J. 1994, 13, 4361–4369. [Google Scholar] [PubMed]
- Pircher, T.J.; Petersen, H.; Gustafsson, J.A.; Haldosén, L.A. Extracellular signal-regulated kinase (ERK) interacts with signal transducer and activator of transcription (STAT) 5a. Mol. Endocrinol. 1999, 13, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J. Role of Jak kinases and STATs in cytokine signal transduction. Int. J. Hematol. 2001, 73, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Ross, J.A.; Rodriguez, G.; Nagy, Z.S.; Wilson, H.L.; Kirken, R.A. Signal transducer and activator of transcription 5b (STAT5β) serine 193 is a novel cytokine-induced phospho-regulatory site that is constitutively activated in primary hematopoietic malignancies. J. Biol. Chem. 2012, 287, 16596–16608. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. JAK-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N. STATs: Signal transducers and activators of transcription. Cell 1996, 84, 331–334. [Google Scholar] [CrossRef]
- Haan, S.; Kortylewski, M.; Behrmann, I.; Muller-Esterl, W.; Heinrich, P.C.; Schaper, F. Cytoplasmic STAT proteins associate prior to activation. Biochem. J. 2000, 345, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Ndubuisi, M.I.; Guo, G.G.; Fried, V.A.; Etlinger, J.D.; Sehgal, P.B. Cellular physiology of stat3: Where’s the cytoplasmic monomer? J. Biol. Chem. 1999, 274, 25499–25509. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, J.; Brutsaert, S.; Olson, R.; Schindle, C. STATs dimerize in the absence of phosphorylation. J. Biol. Chem. 2003, 278, 34133–34140. [Google Scholar] [CrossRef] [PubMed]
- Udy, G.B.; Towers, R.P.; Snell, R.G.; Wilkins, R.J.; Park, S.H.; Ram, P.A.; Waxman, D.J.; Davey, H.W. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 7239–7244. [Google Scholar] [CrossRef] [PubMed]
- Teglund, S.; McKay, C.; Schuetz, E.; van Deursen, J.M.; Stravopodis, D.; Wang, D.; Brown, M.; Bodner, S.; Grosveld, G.; Ihle, J.N. Stat5a and Stat5β proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 1998, 93, 841–850. [Google Scholar] [CrossRef]
- Agha, A.; Monson, J.P. Modulation of glucocorticoid metabolism by the growth hormone-IGF-1 axis. Clin. Endocrinol. 2007, 66, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Kofoed, E.M.; Hwa, V.; Little, B.; Woods, K.A.; Buckway, C.K.; Tsubaki, J.; Pratt, K.L.; Bezrodnik, L.; Jasper, H.; Tepper, A.; et al. Growth hormone insensitivity associated with a STAT5β mutation. N. Engl. J. Med. 2003, 349, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Riedlinger, G.; Miyoshi, K.; Tang, W.; Li, C.; Deng, C.X.; Robinson, G.W.; Hennighausen, L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival and differentiation. Mol. Cell. Biol. 2004, 24, 8037–8047. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Scott, C.D.; Baxter, R.C. Regulation of the acid-labile subunit of the insulin-like growth factor complex in cultured rat hepatocytes. Endocrinology 1994, 135, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Boisclair, Y.R.; Seto, D.; Hsieh, S.; Hurst, K.R.; Ooi, G.T. Organization and chromosomal localization of the gene encoding the mouse acid labile subunit of the insulin-like growth factor binding complex. Proc. Natl. Acad. Sci. USA 1996, 93, 10028–10033. [Google Scholar] [CrossRef] [PubMed]
- Trengove, M.C.; Ward, A.C. SOCS proteins in development and disease. Am. J. Clin. Exp. Immunol. 2013, 2, 1–29. [Google Scholar] [PubMed]
- Hilton, D.J.; Richardson, R.T.; Alexander, W.S.; Viney, E.M.; Willson, T.A.; Sprigg, N.S.; Starr, R.; Nicholson, S.E.; Metcalf, D.; Nicola, N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA 1998, 95, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Maenaka, K.; Kotoshiba, S.; Matsumoto, M.; Kohda, D.; Conaway, R.C.; Weliky Conaway, J.; Nakayama, K.I. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004, 18, 3055–3065. [Google Scholar] [CrossRef] [PubMed]
- Vuong, B.Q.; Arenzana, T.L.; Showalter, B.M.; Losman, J.; Chen, X.P.; Mostecki, J.; Banks, A.S.; Limnander, A.; Fernandez, N.; Rothman, P.B. SOCS-1 localizes to the microtubule organizing complex-associated 20S proteasome. Mol. Cell. Biol. 2004, 24, 9092–9101. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, C.J.; Rico-Bautista, E.; Lorentzon, M.; Thaus, A.L.; Morgan, P.O.; Willson, T.A.; Zervoudakis, P.; Metcalf, D.; Street, I.; Nicola, N.A.; et al. SOCS2 negatively regulates growth hormone action in vitro and in vivo. J. Clin. Investig. 2005, 115, 397–440. [Google Scholar] [CrossRef] [PubMed]
- Leroith, D.; Nissley, P. Knock your SOCS off! J. Clin. Investig. 2005, 115, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Adams, T.E.; Hansen, J.A.; Starr, R.; Nicola, N.A.; Hilton, D.J.; Billestrup, N. Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J. Biol. Chem. 1998, 273, 1285–1287. [Google Scholar] [CrossRef] [PubMed]
- Tollet-Egnell, P.; Flores-Morales, A.; Stavréus-Evers, A.; Sahlin, L.; Norstedt, G. Growth hormone regulation of SOCS-2, SOCS-3, and CIS messenger ribonucleic acid expression in the rat. Endocrinology 1999, 140, 3693–3704. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Seiliez, I.; Thissen, J.P.; Le Cam, A. Regulation of expression of the rat SOCS-3 gene in hepatocytes by growth hormone, interleukin-6 and glucocorticoids mRNA analysis and promoter characterization. Eur. J. Biochem. 2000, 267, 5849–5857. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.A.; Lindberg, K.; Hilton, D.J.; Nielsen, J.H.; Billestrup, N. Mechanism of inhibition of growth hormone receptor signaling by suppressor of cytokine signaling proteins. Mol. Endocrinol. 1999, 13, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.A.; Waxman, D.J. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J. Biol. Chem. 1999, 274, 35553–35561. [Google Scholar] [CrossRef] [PubMed]
- Vidal, O.M.; Merino, R.; Rico-Bautista, E.; Fernandez-Perez, L.; Chia, D.J.; Woelfle, J.; Ono, M.; Lenhard, B.; Norstedt, G.; Rotwein, P.; Flores-Morales, A. In vivo transcript profiling and phylogenetic analysis identifies suppressor of cytokine signaling 2 as a direct signal transducer and activator of transcription 5b target in liver. Mol. Endocrinol. 2007, 21, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.; Greenhalgh, C.J.; Viney, E.; Willson, T.A.; Starr, R.; Nicola, N.A.; Hilton, D.J.; Alexander, W.S. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature 2000, 405, 1069–1073. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Yasukawa, H.; Suzuki, A.; Kamizono, S.; Syoda, T.; Kinjyo, I.; Sasaki, M.; Johnston, J.A.; Yoshimura, A. Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 1999, 4, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, H.; Misawa, H.; Sakamoto, H.; Masuhara, M.; Sasaki, A.; Wakioka, T.; Ohtsuka, S.; Imaizumi, T.; Matsuda, T.; Ihle, J.N.; et al. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J. 1999, 18, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Kershaw, N.J.; Murphy, J.M.; Varghese, L.N.; Laktyushin, A.; Young, S.N.; Lucet, I.S.; Norton, R.S.; Nicola, N.A. Suppression of cytokine signaling by SOCS3: Characterization of the mode of inhibition and the basis of its specificity. Immunity 2012, 36, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, N.; Li, J.S. Mechanism of growth hormone insensitivity induced by endotoxin. Acta Pharmacol. Sin. 2002, 23, 16–22. [Google Scholar] [PubMed]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 2008, 19, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Favre, H.; Benhamoua, A.; Finidoria, J.; Kellya, P.A.; Ederya, M. Dual effects of suppressor of cytokine signaling (SOCS-2) on growth hormone signal transduction. FEBS Lett. 1999, 453, 63–66. [Google Scholar] [CrossRef]
- Stofega, M.R.; Argetsinger, L.S.; Wang, H.; Ullrich, A.; Carter-Su, C. Negative regulation of growth hormone receptor/JAK2 signaling by signal regulatory protein α. J. Biol. Chem. 2000, 275, 28222–28229. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Elliott, J.; Barry, A.C.; Hibbert, L.; Cacalano, N.A.; Johnston, J.A. SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol. Cell. Biol. 2005, 25, 9115–9126. [Google Scholar] [CrossRef] [PubMed]
- Piessevaux, J.; Lavens, D.; Montoye, T.; Wauman, J.; Catteeuw, D.; Vandekerckhove, J.; Belsham, D.; Peelman, F.; Tavernier, J. Functional cross-modulation between SOCS proteins can stimulate cytokine signaling. J. Biol. Chem. 2006, 281, 32953–32966. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.A.; Waxman, D.J. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5β signaling by continuous growth hormone. J. Biol. Chem. 2000, 275, 39487–39496. [Google Scholar] [CrossRef] [PubMed]
- Irandoust, M.I.; Aarts, L.H.; Roovers, O.; Gits, J.; Erkeland, S.J.; Touw, I.P. Suppressor of cytokine signaling 3 controls lysosomal routing of G-CSF receptor. EMBO J. 2007, 26, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Sabo, J.K.; Zhang, J.G.; Nicola, N.A.; Norton, R.S. The SOCS box encodes a hierarchy of affinities for Cullin5: Implications for ubiquitin ligase formation and cytokine signalling suppression. J. Mol. Biol. 2009, 387, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Peltola, K.J.; Paukku, K.; Aho, T.L.; Ruuska, M.; Silvennoinen, O.; Koskinen, P.J. Pim-1 kinase inhibits STAT5-dependent transcription via its interactions with SOCS1 and SOCS3. Blood 2004, 103, 3744–3750. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Gorospe, M.; de Cabo, R. AsSIRTing the DNA damage response. Trends Cell Biol. 2008, 18, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, A.; Reinberg, D. Calorie restriction and the exercise of chromatin. Genes Dev. 2009, 23, 1849–1869. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Paul, R.K.; Martin-Montalvo, A.; Scheibye-Knudsen, M.; Song, S.; He, H.J.; Armour, S.M.; Hubbard, B.P.; Bohr, V.A.; Wang, L.; et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J. Biol. Chem. 2011, 286, 19270–19279. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Iguchi, G.; Fukuoka, H.; Suda, K.; Bando, H.; Takahashi, M.; Nishizawa, H.; Seino, S.; Takahashi, Y. SIRT1 regulates adaptive response of the growth hormone–insulin-like growth factor-I axis under fasting conditions in liver. Proc. Natl. Acad. Sci. USA 2013, 110, 14948–14953. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Sharrocks, A.D. PIAS proteins and transcriptional regulation—More than just SUMO E3 ligases? Genes Dev. 2006, 20, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Rytinki, M.M.; Kaikkonen, S.; Pehkonen, P.; Jaaskelainen, T.; Palvimo, J.J. PIAS proteins: Pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 2009, 66, 3029–3041. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.C.; Johannsson, G.; Leong, G.M.; Ho, K.K.Y. Estrogen regulation of growth hormone action. Endocr. Rev. 2004, 25, 693–721. [Google Scholar] [CrossRef] [PubMed]
- Pilecka, I.; Whatmore, A.; Hooft van Huijsduijnen, R.; Destenaves, B.; Clayton, P. Growth hormone signalling: Sprouting links between pathways, human genetics and therapeutic options. Trends Endocrinol. Metab. 2007, 18, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Jiang, X.; Wong, A.O.L. PIAS1 as a feedback repressor of JAK/STAT signaling coupled to growth hormone receptor in fish model. In Proceedings of the Endocrine Society’s 95th Annual Meeting and Expo, San Francisco, CA, USA, 15–18 June 2013. [Google Scholar]
- Liu, B.; Gross, M.; ten Hoeve, J.; Shuai, K. A transcriptional corepressor of Stat1 with an essential LXXLL signature motif. Proc. Natl. Acad. Sci. USA 2001, 98, 3203–3207. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liao, J.; Rao, X.; Kushner, S.A.; Chung, C.D.; Chang, D.D.; Shuai, K. Inhibition of Stat1-mediated gene activation by PIAS1. Proc. Natl. Acad. Sci. USA 1998, 95, 10626–10631. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, D.; Vanhatupa, S.; Kotaja, N.; Yang, J.; Aittomäki, S.; Jänne, O.A.; Palvimo, J.J.; Silvennoinen, O. PIAS proteins promote SUMO-1 conjugation to STAT1. Blood 2003, 102, 3311–3313. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K. Modulation of STAT signaling by STAT-interacting proteins. Oncogene 2000, 19, 2638–2644. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Mink, S.; Wong, K.A.; Stein, N.; Getman, C.; Dempsey, P.W.; Wu, H.; Shuai, K. PIAS1 selectively inhibits interferon-inducible genes and is important in innate immunity. Nat. Immunol. 2004, 5, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of Stat3 signal transduction by PIAS3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Rycyzyn, M.A.; Clevenger, C.V. The intranuclear prolactin/cyclophilin B complex as a transcriptional inducer. Proc. Natl. Acad. Sci. USA 2002, 99, 6790–6795. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, C.; Curchod, M.L.; Wälchli, S.; Espanel, X.; Guerrier, M.; Arigoni, F.; Strous, G.; van Huijsduijnen, R.H. Identification of protein tyrosine phosphatases with specificity for the ligand-activated growth hormone receptor. Mol. Endocrinol. 2003, 17, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Tonks, N.K. Protein tyrosine phosphatases in signal transduction. Curr. Opin. Cell Biol. 1997, 9, 193–204. [Google Scholar] [CrossRef]
- Gu, F.; Dubé, N.; Kim, J.W.; Cheng, A.; de Jesus Ibarra-Sanchez, M.; Tremblay, M.L.; Boisclair, Y.R. Protein tyrosine phosphatase 1B attenuates growth hormone-mediated JAK2-STAT signaling. Mol. Cell. Biol. 2003, 11, 3753–3762. [Google Scholar] [CrossRef]
- Escrivá, F.; González-Rodriguez, Á.; Fernández-Millán, E.; Rondinone, C.M.; Álvarez, C.; Valverde, A.M. PTP1B deficiency enhances liver growth during suckling by increasing the expression of insulin-like growth factor-I. J. Cell. Physiol. 2010, 225, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.; Lees, E.K.; Mody, N.; Delibegovic, M. Regulation of growth hormone induced JAK2 and mTOR signalling by hepatic protein tyrosine phosphatase 1B. Diabetes Metab. 2015, 41, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Pilecka, I.; Patrignani, C.; Pescini, R.; Curchod, M.L.; Perrin, D.; Xue, Y.; Yasenchak, J.; Clark, A.; Magnone, M.C.; Zaratin, P.; et al. Protein-tyrosine phosphatase H1 controls growth hormone receptor signaling and systemic growth. J. Biol. Chem. 2007, 282, 35405–35415. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.L.; Cleveland, J.L.; Ihle, J.N. Protein tyrosine phosphatase containing SH2 domains: Characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12–p13. Mol. Cell. Biol. 1992, 12, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zheng, C.; Shou, P.; Li, N.; Cao, G.; Chen, Q.; Xu, C.; Du, L.; Yang, Q.; Cao, J.; et al. SHP1 Regulates Bone Mass by Directing Mesenchymal Stem Cell Differentiation. Cell. Rep. 2016, 16, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Horvat, A.; Schwaiger, F.; Hager, G.; Brocker, F.; Streif, R.; Knyazev, P.; Ullrich, A.; Kreutzberg, G.W. A novel role for protein tyrosine phosphatase shp1 in controlling glial activation in the normal and injured nervous system. J. Neurosci. 2001, 21, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Massa, P.T.; Saha, S.; Wu, C.; Jarosinski, K.W. Expression and function of the protein tyrosine phosphatase SHP-1 in oligodendrocytes. Glia 2000, 29, 376–385. [Google Scholar] [CrossRef]
- Minoo, P.; Zadeh, M.M.; Rottapel, R.; Lebrun, J.J.; Ali, S. A novel SHP-1/Grb2–dependent mechanism of negative regulation of cytokine-receptor signaling: Contribution of SHP-1 C-terminal tyrosines in cytokine signaling. Blood 2004, 103, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Hackett, R.H.; Wang, Y.D.; Sweitzer, S.; Feldman, G.; Wood, W.I.; Larner, A.C. Mapping of a cytoplasmic domain of the human growth hormone receptor that regulates rates of inactivation of Jak2 and Stat proteins. J. Biol. Chem. 1997, 272, 11128–11132. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.A.; Waxman, D.J. Interaction of growth hormone-activated STATs with SH2-containing phosphotyrosine phosphatase SHP-1 and nuclear JAK2 tyrosine kinase. J. Biol. Chem. 1997, 272, 17694–17702. [Google Scholar] [CrossRef] [PubMed]
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal structure of the tyrosine phosphatase SHP-2. Cell 1998, 92, 441–450. [Google Scholar] [CrossRef]
- Qu, C.K. The SHP-2 tyrosine phosphatase: Signaling mechanisms and biological functions. Cell Res. 2000, 10, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.L.; Jin, Y.J.; Burakoff, S.J. Cytosolic tyrosine dephosphorylation of STAT5. J. Biol. Chem. 2000, 275, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, L.; He, D.; Song, X.; Liang, X.; Zhao, Z.J.; Zhou, G.W. Crystal structure of human protein-tyrosine phosphatase SHP-1. J. Biol. Chem. 2003, 278, 6516–6520. [Google Scholar] [CrossRef] [PubMed]
- Nardozza, A.P.; D’Orazio, M.; Trapannone, R.; Corallino, S.; Filomeni, G.; Tartaglia, M.; Battistoni, A.; Cesareni, G.; Castagnoli, L. ROS and EGF are antagonistic cues controlling SHP-2 dimerization. Mol. Cell. Biol. 2012, 32, 1998–2009. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Kalidas, K.; Shaw, A.; Song, X.; Musat, D.L.; van der Burgt, I.; Brunner, H.G.; Bertola, D.R.; Crosby, A.; Ion, A.; et al. PTPN11 mutations in Noonan syndrome: Molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 2002, 70, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Yoshida, R. PTPN11 mutations and genotype-phenotype correlations in Noonan and LEOPARD syndromes. Pediatr. Endocrinol. Rev. 2005, 2, 669–674. [Google Scholar] [PubMed]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Raynal, P. Growth hormone and noonan syndrome: Update in dysfunctional signaling aspects and in therapy for short stature. Horm. Stud. 2014, 2. [Google Scholar] [CrossRef]
- Marin, T.M.; Keith, K.; Davies, B.; Conner, D.A.; Guha, P.; Kalaitzidis, D.; Wu, X.; Lauriol, J.; Wang, B.; Bauer, M.; et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome–associated PTPN11 mutation. J. Clin. Investig. 2011, 121, 1026–1043. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, E.M.; Cochrane, F.; Barclay, A.N.; van den Berg, T.K. Signal regulatory proteins in the immune system. J. Immunol. 2005, 175, 7781–7787. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, Y.; Matozaki, T.; Noguchi, T.; Iwamatsu, A.; Yamao, T.; Takahashi, N.; Tsuda, M.; Takada, T.; Kasuga, M. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol. Cell. Biol. 1996, 16, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Chen, Z.; Sures, I.; Wang, H.; Schilling, J.; Ullrich, A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997, 386, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Stofega, M.R.; Wang, H.; Ullrich, A.; Carter-Su, C. Growth hormone regulation of sirp and shp-2 tyrosyl phosphorylation and association. J. Biol. Chem. 1998, 273, 7112–7117. [Google Scholar] [CrossRef]
- Subramani, R.; Lopez-Valdez, R.; Salcido, A.; Boopalan, T.; Arumugam, A.; Nandy, S.; Lakshmanaswamy, R. Growth hormone receptor inhibition decreases the growth and metastasis of pancreatic ductal adenocarcinoma. Exp. Mol. Med. 2014, 46, e117. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, Y.; Waters, M.J.; Brooks, A.J. Role of the growth hormone–IGF-1 axis in cancer. Expert Rev. Endocrinol. Metab. 2011, 6, 71–84. [Google Scholar] [CrossRef]
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interferon Cytokine Res. 2016, 36, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, S.; Su, B.; Zhou, F.; Zhang, R.; Xu, T.; Zhang, R.; Leventaki, V.; Drakos, E.; Liu, W.; et al. Stat3 Contributes to Cancer Progression by Regulating Jab1/Csn5 Expression. Oncogene 2017, 36, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Vendrely, V.; Peuchant, E.; Buscail, E.; Moranvillier, I.; Rousseau, B.; Bedel, A.; Brillac, A.; de Verneuil, H.; Moreau-Gaudry, F.; Dabernat, S. Resveratrol and capsaicin used together as food complements reduce tumor growth and rescue full efficiency of low dose gemcitabine in a pancreatic cancer model. Cancer Lett. 2017, 390, 91–102. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT Pathway: Impact on Human Disease and Therapeutic Intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wójcik, M.; Krawczyńska, A.; Antushevich, H.; Herman, A.P. Post-Receptor Inhibitors of the GHR-JAK2-STAT Pathway in the Growth Hormone Signal Transduction. Int. J. Mol. Sci. 2018, 19, 1843. https://doi.org/10.3390/ijms19071843
Wójcik M, Krawczyńska A, Antushevich H, Herman AP. Post-Receptor Inhibitors of the GHR-JAK2-STAT Pathway in the Growth Hormone Signal Transduction. International Journal of Molecular Sciences. 2018; 19(7):1843. https://doi.org/10.3390/ijms19071843
Chicago/Turabian StyleWójcik, Maciej, Agata Krawczyńska, Hanna Antushevich, and Andrzej Przemysław Herman. 2018. "Post-Receptor Inhibitors of the GHR-JAK2-STAT Pathway in the Growth Hormone Signal Transduction" International Journal of Molecular Sciences 19, no. 7: 1843. https://doi.org/10.3390/ijms19071843
APA StyleWójcik, M., Krawczyńska, A., Antushevich, H., & Herman, A. P. (2018). Post-Receptor Inhibitors of the GHR-JAK2-STAT Pathway in the Growth Hormone Signal Transduction. International Journal of Molecular Sciences, 19(7), 1843. https://doi.org/10.3390/ijms19071843