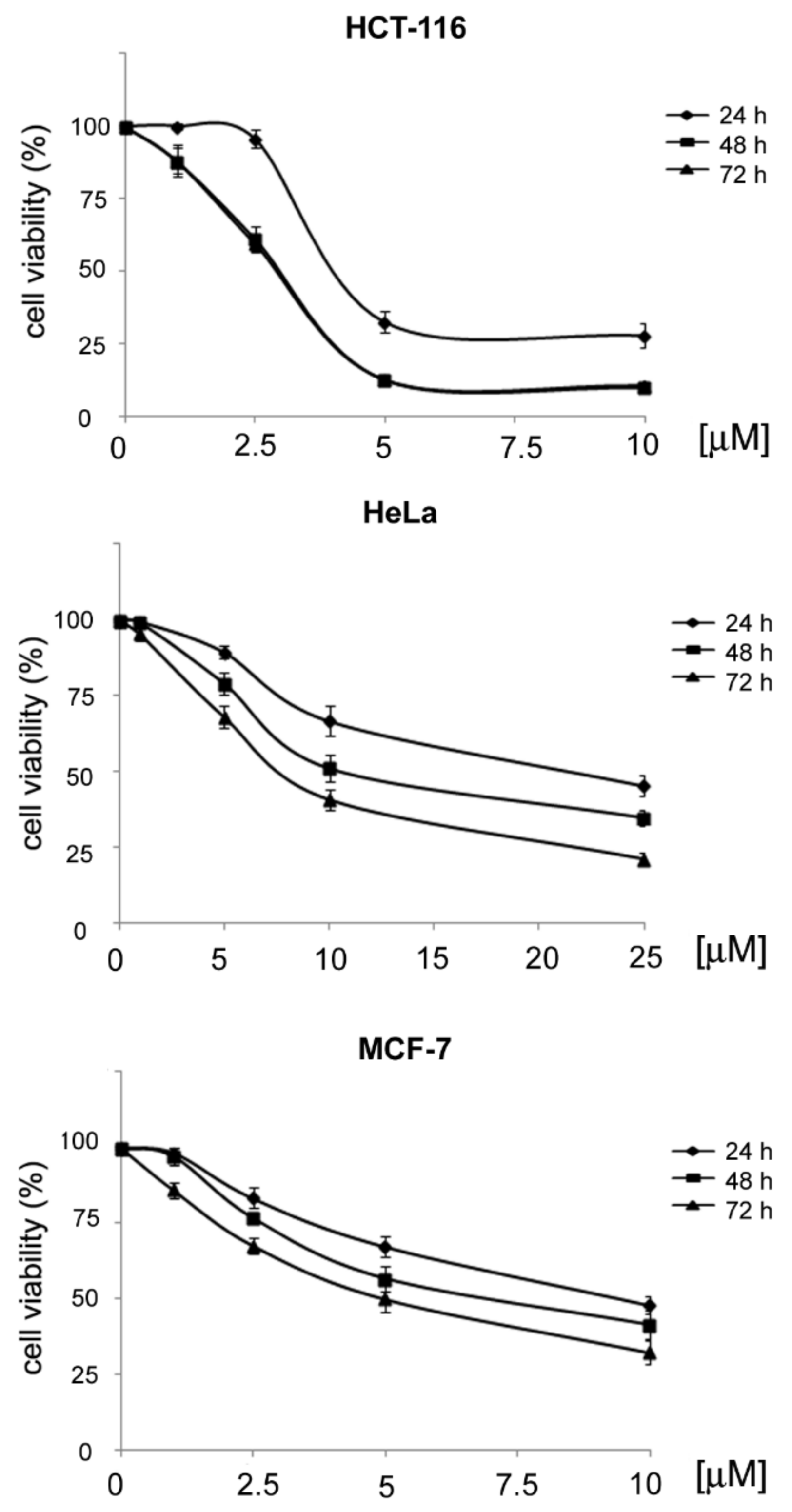
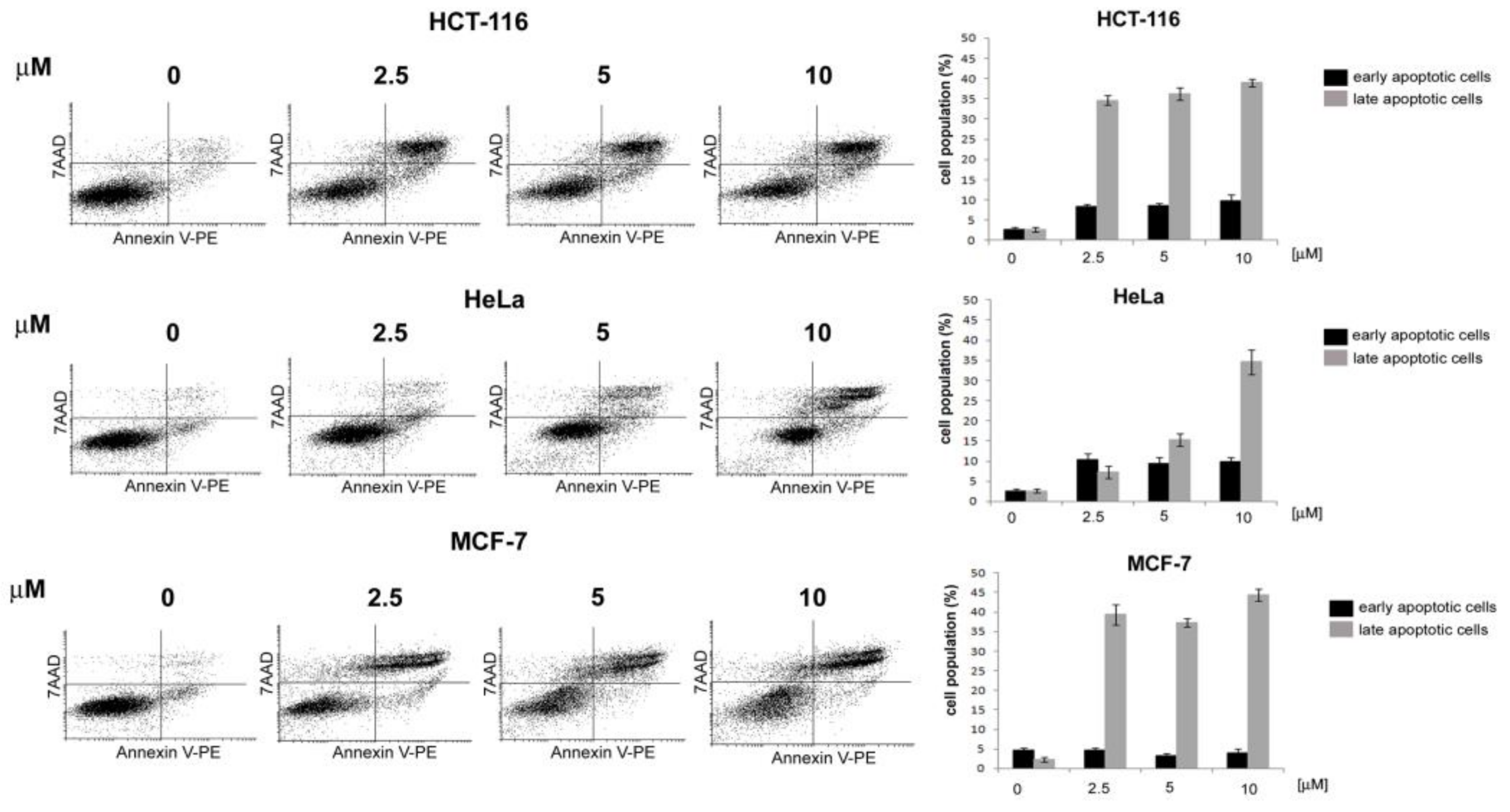
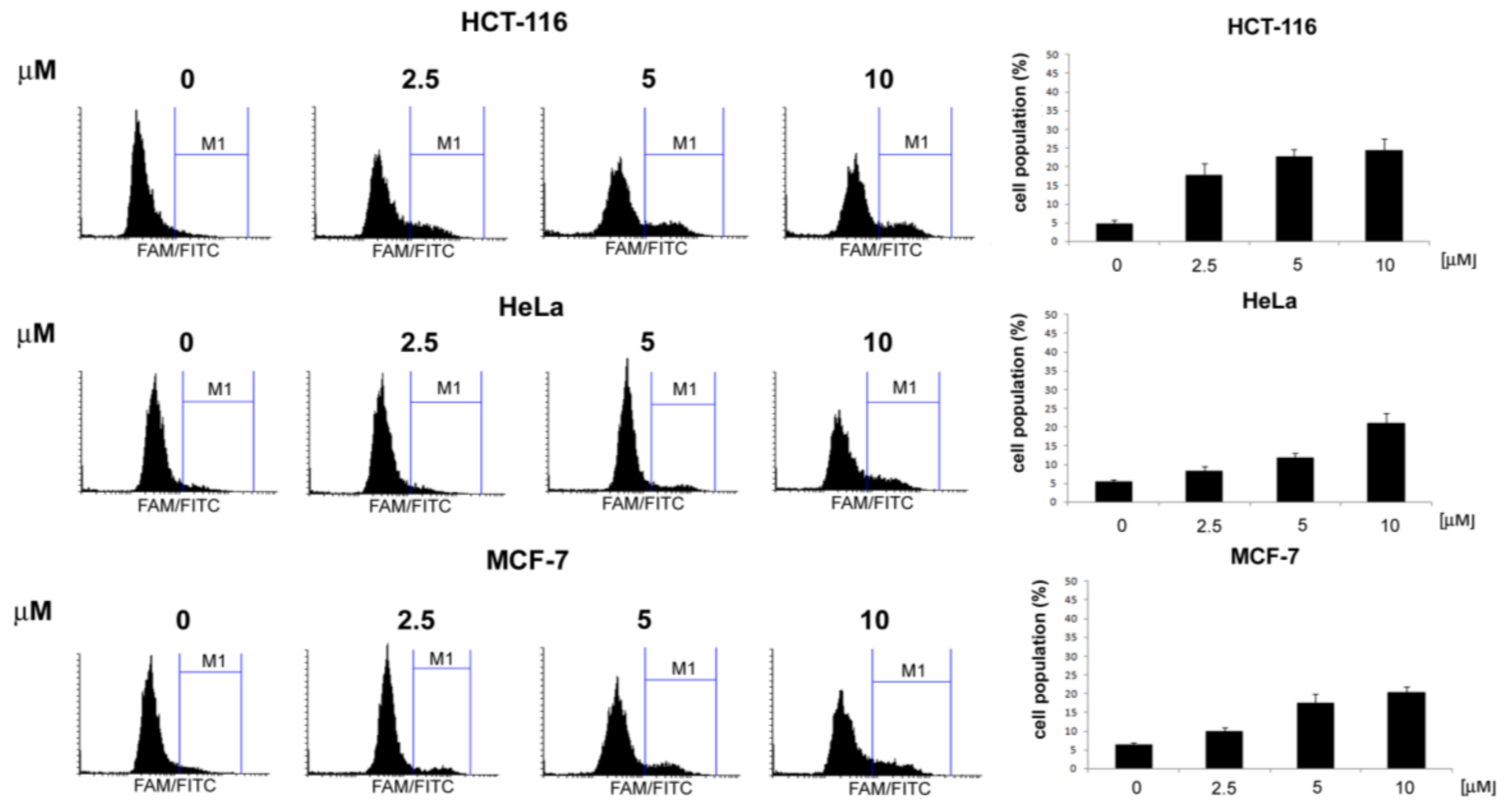
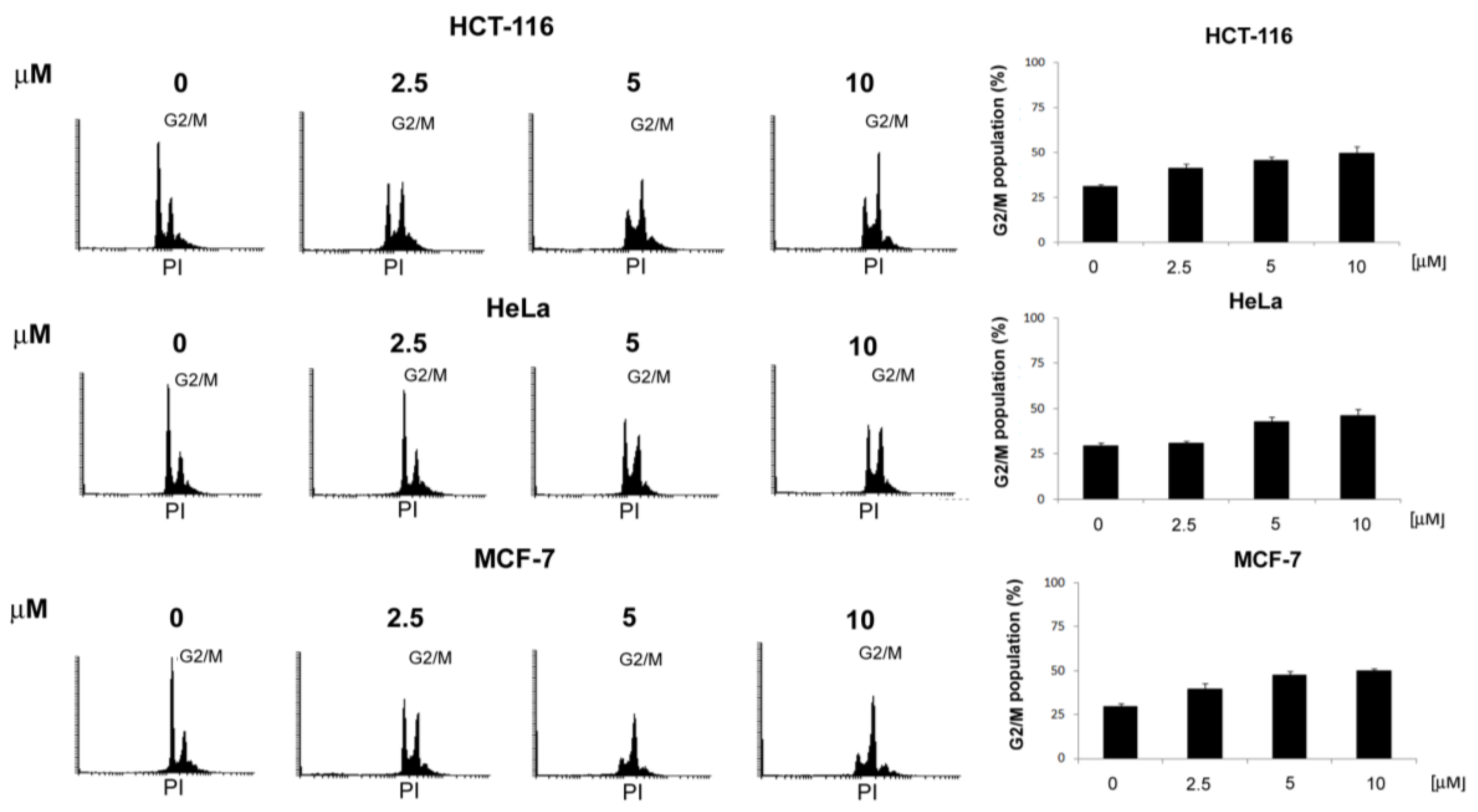
3.2.1. General Procedures for the Preparation of 4-Acetamido-N-(aryl/heteroaryl)Benzenesulfonamides
Method A. To a solution of aminocomponent (3 mmol) in dry pyridine (3 mL), 4-acetamidobenzene-1-sulfonyl chloride (3 mol, 701 mg) was added in portions over 5 min, and reaction mixture was stirred for 2–20 h at room temperature or 105 °C. The reaction mixture was poured into ice-cooled water and stirred for 30 min. The mixture was acidified with dilute hydrochloric acid (1 M) to pH ~ 3–3.5 and stirred for 1 h at room temperature (compd 1), or with concentrated hydrochloric acid (11.8 M) to pH ~ 5.6–6 and stirred for overnight at room temperature (compds 9, 10, 11), respectively. The precipitated solid was filtered, washed with water (3 × 1 mL), methanol (2 × 1 mL), and dried.
Method B. To a solution of aminocomponent (3 mmol) in acetone (3 mL) and pyridine (0.9 mL), 4-acetamidobenzene-1-sulfonyl chloride (3 mmol, 701 mg) was added, and reaction mixture was stirred for 18‒48 h at room temperature. Afterwards, reaction solvents were evaporated under reduced pressure, and the residue was suspended in ice slush (40 mL). Obtained mixture was acidified with dilute hydrochloric acid (1 M) to pH ~ 2 and stirred for 1 h at room temperature. The precipitated solid was filtered, washed with water, and dried. Pure compounds were obtained after crystallization from ethanol.
4-Acetamido-N-(4-chlorophenyl)benzenesulfonamide (
1).
Method A. Starting from 4-chloroaniline (383 mg), and after 2 h at 105 °C the title compound
1 was obtained (711 mg, 73%): m.p. 195‒196.5 °C (ref. 196‒197.5 °C [
26]); IR (KBr) v
max 3360, 3226 (NH), 2921 (CH
3), 1689 (C=O), 1592, 1530 (NH
def), 1490 (C=C), 1329 (SO
2 asym), 1150 (SO
2 sym) cm
−1; anal. C 51.77, H 4.03, N 8.63% calcd. for C
14H
13ClN
2O
3S, C 51.70, H 4.00, N 8.62%.
4-Acetamido-N-[2-(phenylthio)phenyl]benzenesulfonamide (2). Method B. Starting from 2-(phenylthio)aniline (604 mg), and after 48 h the title compound 2 was obtained (997 mg, 83%): m.p. 141–144 °C; IR (KBr) vmax 3316, 3278, 3247, 3179 (NH), 2917, 2851 (CH3), 1662 (C=O), 1594, 1523 (NH def), 1474 (C=C), 1336 (SO2 asym), 1164 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.06 (s, 3H, CH3), 7.02 (d, J = 8.3 Hz, 1H, arom.), 7.07‒7.13 (m, 4H, arom.), 7.20 (t, 1H, arom.), 7.28‒7.29 (m, 3H, arom.), 7.61 (d, J = 8.8 Hz, 2H, arom.), 7.66 (d, J = 8.8 Hz, 2H, arom.), 9.61 (s, 1H, NH), 10.29 (s, 1H, NH) ppm; anal. C 60.28, H 4.55, N 7.03% calcd. for C20H18N2O3S2, C 60.38, H 4.57, N 7.06%.
4-Acetamido-N-(2-phenoxyphenyl)benzenesulfonamide (
3).
Method B. Starting from 2-(phenoxy)aniline (556 mg), and after 48 h the title compound
3 was obtained (966 mg, 84%): m.p. 161.5‒162.5 °C (ref. 162 °C [
27]); IR (KBr) v
max 3332, 3289 (NH), 2924 (CH
3), 1676 (C=O), 1592, 1510 (NH
def), 1491 (C=C), 1335 (SO
2 asym), 1160 (SO
2 sym) cm
−1;
1H NMR (500 MHz, DMSO-
d6) δ 2.07 (s, 3H, CH
3), 6.65 (d,
J = 7.9 Hz, 2H, arom.), 6.69 (d,
J = 7.8 Hz, 1H, arom.), 7.02‒7.10 (m, 3H, arom.), 7.27 (t, 2H, arom.), 7.35 (d,
J = 7.8 Hz, 1H, arom.), 7.62 (s, 4H, arom.), 9.77 (s, 1H, NH), 10.26 (s, 1H, NH) ppm;
13C NMR (125 MHz, DMSO-d
6) δ 24.8, 119.1, 119.2, 124.1, 124.2, 126.1, 127.1, 128.5, 128.6, 130.4, 134.5, 143.7, 150.2, 156.8, 169.6 ppm; anal. C 60.81, H 4.74, N 7.33% calcd. for C
20H
18N
2O
4S, C 60.52, H 4.74, N 7.30%.
4-Acetamido-N-[2-(phenylthio)pyridin-3-yl]benzenesulfonamide (4). Method B. Starting from 2-(phenylthio)pyridin-3-amine (607 mg), and after 48 h compound 4 was obtained (902 mg, 75%): m.p. 160‒161 °C; IR (KBr) vmax 3366, 3262 (NH), 2923, 2889 (CH3), 1692 (C=O), 1592, 1530 (NH def), 1441 (C=C), 1328 (SO2 asym), 1156 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.08 (s, 3H, CH3), 7.11 (dd, J = 4.9, 7.8 Hz, 1H, arom.), 7.23 (d, J = 5.4 Hz, 2H, arom.), 7.28‒7.39 (m, 4H, arom.), 7.66 (d, J = 8.3 Hz, 2H, arom.), 7.73 (d, J = 8.8 Hz, 2H, arom.), 8.14 (s, 1H, arom.), 10.00 (s, 1H, NH), 10.34 (s, 1H, NH) ppm; 13C NMR (125 MHz, DMSO-d6) δ 24.8, 119.2, 121.8, 128.8, 129.1, 129.7, 130.9, 131.4, 134.4, 135.2, 135.3, 144.0, 148.3, 156.9, 169.7 ppm; anal. C 57.12, H 4.29, N 10.52% calcd. for C19H17N3O3S2, C 57.00, H 4.21, N 10.49%.
4-Acetamido-N-(2-phenoxypyridin-3-yl)benzenesulfonamide (5). Method B. Starting from 2-phenoxypyridin-3-amine (559 mg), and after 30 h the title compound 5 was obtained (972 mg, 85%): m.p. 202.1–204.4 °C; IR (KBr) vmax 3316, 3275 (NH), 2924, 2853 (CH3), 1674 (C=O), 1592, 1512 (NH def), 1451 (C=C), 1334 (SO2 asym), 1161 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.08 (s, 3H, CH3), 6.73 (d, J = 7.8 Hz, 2H, arom.), 7.08 (dd, J = 7.8, 4.9 Hz, 1H, arom.), 7.15 (t, 1H, arom.), 7.30 (t, 2H, arom.), 7.66‒7.70 (m, 4H, arom.), 7.73 (dd, J = 7.9, 1.5 Hz, 1H, arom.), 7.83 (dd, J = 4.9, 1.5 Hz, 1H, arom.), 10.06 (s, 1H, NH), 10.32 (s, 1H, NH) ppm; 13C NMR (125 MHz, DMSO-d6) δ 24.6, 119.0, 119.6, 121.7, 121.9, 124.9, 128.4, 129.7, 134.0, 134.8, 143.7, 143.9, 153.7, 156.6, 169.5 ppm; anal. C 59.52, H 4.47, N 10.96% calcd. for C19H17N3O4S, C 59.37, H 4.49, N 10.93%.
4-Acetamido-N-(quinolin-8-yl)benzenesulfonamide (
6).
Method B. Starting from quinolin-8-amine (433 mg), and after 25 h the title compound
6 was obtained (800 mg, 78%): m.p. 193‒194 °C (ref. 194 °C [
28]); IR (KBr) v
max 3365, 3243 (NH), 2926, 2851 (CH
3), 1691 (C=O), 1597, 1533 (NH
def), 1504, 1470 (C=N, C=C), 1335 (SO
2 asym), 1170 (SO
2 sym) cm
−1;
1H NMR (500 MHz, DMSO-
d6) δ 2.00 (s, 3H, CH
3), 7.50 (t, 1H, arom.), 7.57 (dd,
J = 8.3, 3.9 Hz, 1H, arom.), 7.61‒7.68 (m, 4H, arom.), 7.83 (d,
J = 8.8 Hz, 2H, arom.), 8.34 (dd,
J = 8.3, 1.5 Hz, 1H, arom.), 8.84 (dd,
J = 3.9, 1.5 Hz, 1H, arom.), 9.79 (s, 1H, NH), 10.24 (s, 1H, NH) ppm; anal. C 59.81, H 4.43, N 12.31% calcd. for C
17H
15N
3O
3S, C 59.71, H 4.30, N 12.29%.
4-Acetamido-N-(2-methylquinolin-8-yl)benzenesulfonamide (
7).
Method B. Starting from 2-methylquinolin-8-amine (475 mg), and after 18 h the title compound
7 was obtained (778 mg, 73%): m.p. 210‒213 °C (ref. 200 °C [
29]); IR (KBr) v
max 3352, 3243 (NH), 2925 (CH
3), 1694 (C=O), 1591, 1534 (NH
def), 1508, 1473 (C=N, C=C), 1342 (SO
2 asym), 1159 (SO
2 sym) cm
−1;
1H NMR (500 MHz, DMSO-
d6) δ 2.01 (s, 3H, CH
3), 2.67 (s, 3H, CH
3), 7.40‒7.46 (m, 2H, arom.), 7.57‒7.64 (m, 4H, arom.), 7.83 (d,
J = 8.8 Hz, 2H, arom.), 8.20 (d,
J = 8.3 Hz, 1H, arom.), 9.60 (s, 1H, NH), 10.24 (s, 1H, NH) ppm; anal. C 60.83, H 4.82, N 11.82% calcd. for C
18H
17N
3O
3S, C 60.49, H 5.16, N 11.53%.
4-Acetamido-N-(7-methylquinolin-8-yl)benzenesulfonamide (8). Method B. Starting from 7-methylquinolin-8-amine (475 mg), and after 22 h the title compound 8 (778 mg, 73%) was obtained: m.p. 232‒235 °C; IR (KBr) vmax 3361, 3224 (NH), 2928 (CH3), 1692 (C=O), 1598, 1541 (NH def), 1502, 1464 (C=N, C=C), 1333 (SO2 asym), 1170 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.04 (s, 3H, CH3), 2.55 (s, 3H, CH3), 7.32 (dd, J = 8.3, 4.4 Hz, 1H, arom.), 7.44 (d, J = 8.8 Hz, 2H, arom.), 7.48‒7.53 (m, 3H, arom.), 7.77 (d, J = 8.3 Hz, 1H, arom.), 8.22 (dd, J = 8.3, 1.5 Hz, 1H, arom.), 8.41 (dd, J = 3.9, 1.4 Hz, 1H, arom.), 9.49 (s, 1H, NH), 10.18 (s, 1H, NH) ppm; 13C NMR (125 MHz, DMSO-d6) δ 19.93, 24.6, 118.1, 121.1, 126.8, 127.1, 128.6, 130.3, 131.6, 134.8, 136.2, 138.5, 143.0, 144.4, 149.6, 169.3 ppm; anal. C 60.83, H 4.82, N 11.82% calcd. for C18H17N3O3S, C 60.66, H 4.71, N 11.82%.
4-Acetamido-N-(2-methylquinolin-6-yl)benzenesulfonamide (9). Method A. Starting from 2-methylquinolin-6-amine (475 mg), and after 20 h at room temperature the title compound 9 was obtained (938 mg, 88%): m.p. 265‒267 °C; IR (KBr) vmax 3353, 3236 (NH), 2921 (CH3), 1673 (C=O), 1611, 1591 (NH def), 1545, 1515, 1459 (C=N, C=C), 1320 (SO2 asym), 1155 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.02 (s, 3H, CH3), 2.58 (s, 3H, CH3), 7.33 (d, 1H, arom.), 7.43 (dd, 1H, arom.), 7.53 (s, 1H, arom.), 7.66 (d, J = 8.8 Hz, 2H, arom.), 7.72 (d, J = 8.8 Hz, 2H, arom.), 7.77 (d, 1H, arom.), 8.12 (d, 1H, arom.), 10.26 (s, 1H, NH), 10.47 (s, 1H, NH) ppm; anal. C 60.83, H 4.82, N 11.82% calcd. for C18H17N3O3S, C 60.53, H 4.02, N 11.45%.
4-Acetamido-N-(4H-1,2,4-triazol-4-yl)benzenesulfonamide (
10).
Method A. Starting from 4H-1,2,4-triazol-4-amine (475 mg), and after 3 h at 105 °C the title compound
10 was obtained (472 mg, 56%): m.p. 227–228 °C (205 °C (decomp.) [
30] ; IR (KBr) v
max 3296, 3251 (NH), 2931, 2861 (CH
3), 1673 (C=O), 1604, 1590 (NH
def), 1536, 1497 (C=N, C=C), 1334 (SO
2 asym), 1163 (SO
2 sym) cm
−1;
1H NMR (500 MHz, DMSO-d
6) δ 2.09 (s, 3H, CH
3), 7.61 (d,
J = 8.8 Hz, 2H, arom.), 7.81 (d,
J = 8.8 Hz, 2H, arom.), 8.27 (s, 2H, arom.), 10.45 (s, 1H, NH), 11.80 (s, 1H, NH) ppm;
13C NMR (125 MHz, DMSO-
d6) δ 24.9, 119.6, 129.1, 129.9, 143.5, 145.4, 170.0 ppm; anal. C 42.70, H 3.94, N 24.90% calcd. for C
10H
11N
5O
3S, C 42.38, H 3.71, N 24.50%.
4-Acetamido-N-(benzo[c][1,2,5]thiadiazol-4-yl)benzenesulfonamide (11). Method A. Starting from benzo[c][1,2,5]thiadiazol-4-amine (454 mg), and after 18 h at room temperature the title compound 11 was obtained (562 mg, 54%): m.p. 206‒207 °C; IR (KBr) vmax 3376, 3222 (NH), 2921 (CH3), 1676 (C=O), 1612, 1587 (NH def), 1543, 1493 (C=N, C=C), 1347 (SO2 asym), 1158 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.04 (s, 3H, CH3), 7.40 (d, 1H, arom.), 7.61 (t, 1H, arom.), 7.67 (d, J = 8.8 Hz, 2H, arom.), 7.75 (d, 1H, arom.), 7.81 (d, J = 8.8 Hz, 2H, arom.), 10.29 (s, 1H, NH), 10.87 (s, 1H, NH) ppm; anal. C 48.26, H 3.47, N 16.08% calcd. for C14H12N4O3S2, C 48.58, H 3.21, N 16.30%.
3.2.2. General Procedures for the Synthesis of 4-Amino-N-(aryl/heteroaryl)Benzenesulfonamides (12–22)
Method A: The 4-acetamido-N-(aryl/heteroaryl)benzenesulfonamide (2.5 mmol) was heated at 100 °C with a solution of NaOH (25 mmol) in water (11.5 mL) for 1 h; then, the solution was cooled and acidified to pH ~ 5 with 50% acetic acid. The solid was filtered off, washed with water (3 × 5 mL), 50% methanol (2 × 2.5 mL), and methanol (2.5 mL), and dried.
Method B: The 4-acetamido-N-(aryl/heteroaryl)benzenesulfonamide (2.5 mmol) was heated at 100 °C with hydrochloric acid (4 mL, 36%) in ethanol (10 mL) for 1 h, than the solution was cooled, treated with cold water (20 mL), and basified with ammonia to pH ~ 8–9. The solid was filtered off, washed with water (3 × 10 mL), and purified as indicated below.
4-Amino-N-(4-chlorophenyl)benzenesulfonamide (
12). Hydrolysis according to
Method A. Starting from
1 (812 mg), compound
12 (664 mg, 94%) was obtained: m.p. 195.5‒196 °C (ref. 194‒195 °C [
26]); IR (KBr) v
max 3412, 3346 (NH), 2923, 2855 (CH
3), 1635, 1597 (NH
def), 1493 (C=C), 1314 (SO
2 asym), 1152 (SO
2 sym) cm
−1; anal. C 50.97, H 3.92, N 9.91% calcd. for C
12H
11ClN
2O
2S, C 60.00, H 3.93, N 9.94%.
4-Amino-N-[2-(phenylthio)phenyl]benzenesulfonamide (13). Hydrolysis according to Method B. Starting from 2 (996 mg) and after purification on silica gel using benzene as the eluent, the title compound 13 (838 mg, 94%) was obtained as an oil; IR (NaCl plates) vmax 3479, 3377, 3304, 3228 (NH), 1626, 1594 (NH def), 1478 (C=C), 1316 (SO2 asym), 1151 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.01 (s, 2H, NH2), 6.51 (d, J = 7.8 Hz, 2H, arom.), 7.06‒7.12 (m, 4H, arom.), 7.16‒7.24 (m, 2H, arom.), 7.28‒7.34 (m, 5H, arom.), 9.03 (s, 1H, NH) ppm; 13C NMR (125 MHz, DMSO-d6) δ 113.2, 124.8, 125.1, 126.9, 128.0, 129.1, 129.6, 130.2, 130.5, 131.3, 133.7, 135.6, 137.7, 153.7 ppm.
4-Amino-N-(2-phenoxyphenyl)benzenesulfonamide (
14). Hydrolysis according to
Method B. Starting from
3 (956 mg) and after crystallization from ethanol, the title compound
14 (817 mg, 96%) was obtained: m.p. 142 °C (ref. 149 °C [
27]; IR (KBr) v
max 3498, 3394, 3305 (NH), 1630, 1596 (NH
def), 1490 (C=C), 1315 (SO
2 asym), 1147 (SO
2 sym) cm
−1;
1H NMR (500 MHz, DMSO-
d6) δ 6.00 (br. s, 2H, NH
2), 6.49 (d,
J = 8.3 Hz, 2H, arom.), 6.68‒6.74 (m, 3H, arom.), 7.00‒7.04 (m, 2H, arom.), 7.10 (t, 1H, arom.), 7.28‒7.38 (m, 5H, arom.), 9.36 (s, 1H, NH) ppm; anal. C 63.51, H 4.74, N 8.23% calcd. for C
18H
16N
2O
3S, C 63.57, H 4.60, N 8.21%.
4-Amino-N-[2-(phenylthio)pyridin-3-y]benzenesulfonamide (15). Hydrolysis according to Method B. Starting from 4 (999 mg) and after crystallization from ethanol, the title compound 15 (742 mg, 83%) was obtained: m.p. 146.8–147.6 °C dec.; IR (KBr) vmax 3451, 3362, 3268 (NH), 1635, 1593 (NH def), 1500, 1444 (C=N, C=C), 1312 (SO2 asym), 1156 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.00 (br. s, 2H, NH2), 6.57 (d, J = 8.3 Hz, 2H, arom.), 7.10 (dd, J = 8.1, 4.7 Hz, 1H, arom.), 7.27‒7.31 (m, 3H, arom.), 7.35‒7.37 (m, 5H, arom.), 8.12 (dd, J = 4.4, 1.5 Hz, 1H, arom.), 9.53 (s, 1H, NH) ppm; anal. C 57.12, H 4.23, N 11.76% calcd. for C17H15N3O2S2, C 56.89, H 4.07, N 11.60%.
4-Amino-N-(2-phenoxypyridin-3-yl)benzenesulfonamide (16). Hydrolysis according to Method B. Starting from 5 (959 mg) and after crystallization from ethanol, the title compound 16 (725 mg, 85%) was obtained: m.p. 163.1–164.7 °C; IR (KBr) vmax 3474, 3350, 3237 (NH), 1635, 1594 (NH def), 1492, 1448 (C=N, C=C), 1325 (SO2 asym), 1157 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.03 (s, 2H, NH2), 6.54 (d, J = 8.3 Hz, 2H, arom.), 6.82 (d, J = 7.9 Hz, 2H, arom.), 7.05 (dd, J = 7.9, 4.9 Hz, 1H, arom.), 7.16 (t, 1H, arom.), 7.34 (t, 2H, arom.), 7.38 (d, J = 8.8 Hz, 2H, arom.), 7.71 (dd, J = 7.8, 1.5 Hz, 1H, arom.), 7.79 (dd, J = 4.4, 1.5 Hz, 1H, arom.), 9.66 (s, 1H, NH) ppm; 13C NMR (125 MHz, DMSO-d6) δ 113.0, 119.5, 121.9, 122.7, 124.8, 125.1, 129.2, 129.7, 133.2, 143.0, 153.6, 153.9, 156.1 ppm; anal. C 59.81, H 4.43, N 12.31% calcd. for C17H15N3O3S, C 59.58, H 4.41, N 12.28%.
4-Amino-N-(quinolin-8-yl)benzenesulfonamide (
17). Hydrolysis according to
Method B. Starting from
6 (853 mg) the title compound
17 (696 mg, 93%) was obtained: m.p. 193‒193.3 °C (ref. 193 °C [
31]); IR (KBr) v
max 3468, 3375, 3252 (NH), 1628, 1593 (NH
def), 1504, 1472 (C=N, C=C), 1326 (SO
2 asym), 1153 (SO
2 sym) cm
−1;
1H NMR (500 MHz, DMSO-
d6) δ 6.00 (s, 2H, NH
2), 6.46 (d,
J = 8.8 Hz, 2H, arom.), 7.49‒7.53 (m, 3H, arom.), 7.58‒7.61 (m, 2H, arom.), 7.64 (d,
J = 7.8 Hz, 1H, arom.), 8.35 (d,
J = 8.3 Hz, 1H, arom.), 8.86 (d,
J = 4.4 Hz, 1H, arom.), 9.36 (s, 1H, NH) ppm; anal. C 60.18, H 4.38, N 14.04% calcd. for C
15H
13N
3O
2S, C 60.12, H 4.41, N 13.98%.
4-Amino-N-(2-methylquinolin-8-yl)benzenesulfonamide (
18). Hydrolysis according to
Method B. Starting from
7 (888 mg) and after crystallization from ethanol, the title compound
18 (689 mg, 88%) was obtained: m.p. 156‒158 °C (ref. m.p. was not determined [
29]); IR (KBr) v
max 3482, 3382, 3265 (NH), 1620, 1593 (NH
def), 1506, 1472 (C=N, C=C), 1345 (SO
2 asym), 1155 (SO
2 sym) cm
−1;
1H NMR (500 MHz, DMSO-
d6) δ 2.68 (s, 3H, CH
3), 6.00 (s, 2H, NH
2), 6.47 (d,
J = 8.8 Hz, 2H, arom.), 7.41 (t, 1H, arom.), 7.46 (d,
J = 8.3 Hz, 1H, arom.), 7.51‒7.57 (m, 4H, arom.), 8.21 (d,
J = 8.3 Hz, 1H, arom.), 9.23 (s, 1H, NH) ppm; anal. C 61.32, H 4.82, N 13.41% calcd. for C
16H
15N
3O
2S, C 61.25, H 4.89, N 13.21%.
4-Amino-N-(7-methylquinolin-8-yl)benzenesulfonamide (19). Hydrolysis according to Method B. Starting from 8 (888 mg) and after crystallization from ethanol, the title compound 19 (705 mg, 90%) was obtained: m.p. 202‒203 °C; IR (KBr) vmax 3477, 3378, 3239 (NH), 1630, 1595 (NH def), 1503, 1465 (C=N, C=C), 1316 (SO2 asym), 1153 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.55 (s, 3H, CH3), 5.79 (s, 2H, NH2), 6.31 (d, J = 8.8 Hz, 2H, arom.), 7.12 (d, J = 8.7 Hz, 2H, arom.), 7.36 (dd, J = 8.1, 4.2 Hz, 1H, arom.), 7.48 (d, J = 8.3 Hz, 1H, arom.), 7.73 (d, J = 8.8 Hz, 1H, arom.), 8.22 (d, J = 7.8 Hz, 1H, arom.), 8.55 (d, J = 3.9 Hz, 1H, arom.), 9.01 (s, 1H, NH) ppm; 13C NMR (125 MHz, DMSO-d6) δ 20.0, 112.2, 121.1, 125.6, 126.2, 126.9, 129.4, 130.3, 132.1, 136.2, 137.4, 144.1, 149.6, 152.9 ppm; anal. C 61.32, H 4.82, N 13.41% calcd. for C16H15N3O2S, C 61.24, H 4.76, N 13.40%.
4-Amino-N-(2-methylquinolin-6-yl)benzenesulfonamide (
20). Hydrolysis according to
Method A. Starting from
9 (888 mg) and after crystallization from acetone, the title compound
20 (548 mg, 70%) was obtained: m.p. 249‒252 °C (ref. 252 °C [
32]); IR (KBr) v
max 3474, 3308, 3235, 3176 (NH), 1638, 1597 (NH
def), 1507, 1429 (C=N, C=C), 1335 (SO
2 asym), 1149 (SO
2 sym) cm
−1; anal. C 61.32, H 4.82, N 13.41% calcd. for C
16H
15N
3O
2S, C 61.56, H 4.98, N 13.52%.
4-Amino-N-(4H-1,2,4-triazol-4-yl)benzenesulfonamide (
21). Hydrolysis according to
Method A. Starting from
10 (703 mg) the title compound
21 (532 mg, 89%) was obtained: m.p. 231‒232 °C (ref. 237 °C [
33]); IR (KBr) v
max 3415, 3337, 3213, 3148 (NH), 1658, 1595 (NH
def), 1529, 1457 (C=N, C=C), 1348 (SO
2 asym), 1164 (SO
2 sym) cm
−1; anal. C 40.16, H 3.79, N 29.27% calcd. for C
8H
9N5O
2S, C 40.48, H 3.96, N 28.77%.
4-Amino-N-(benzo[c][1,2,5]thiadiazol-4-yl)benzenesulfonamide (
22). Hydrolysis according to
Method A. Starting from
11 (871 mg) the title compound
22 (659 mg, 86%) was obtained: m.p. 206‒207 °C (ref. m.p. was not determined [
34]); IR (KBr) v
max 3468, 3379, 3329, 3201 (NH), 1626, 1591 (NH
def), 1541, 1496, 1449 (C=N, C=C), 1342 (SO
2 asym), 1158 (SO
2 sym) cm
−1; anal. C 47.04, H 3.29, N 18.29% calcd. for C
12H
10N
4O
2S
2, C 46.77, H 2.96, N 17.91%.
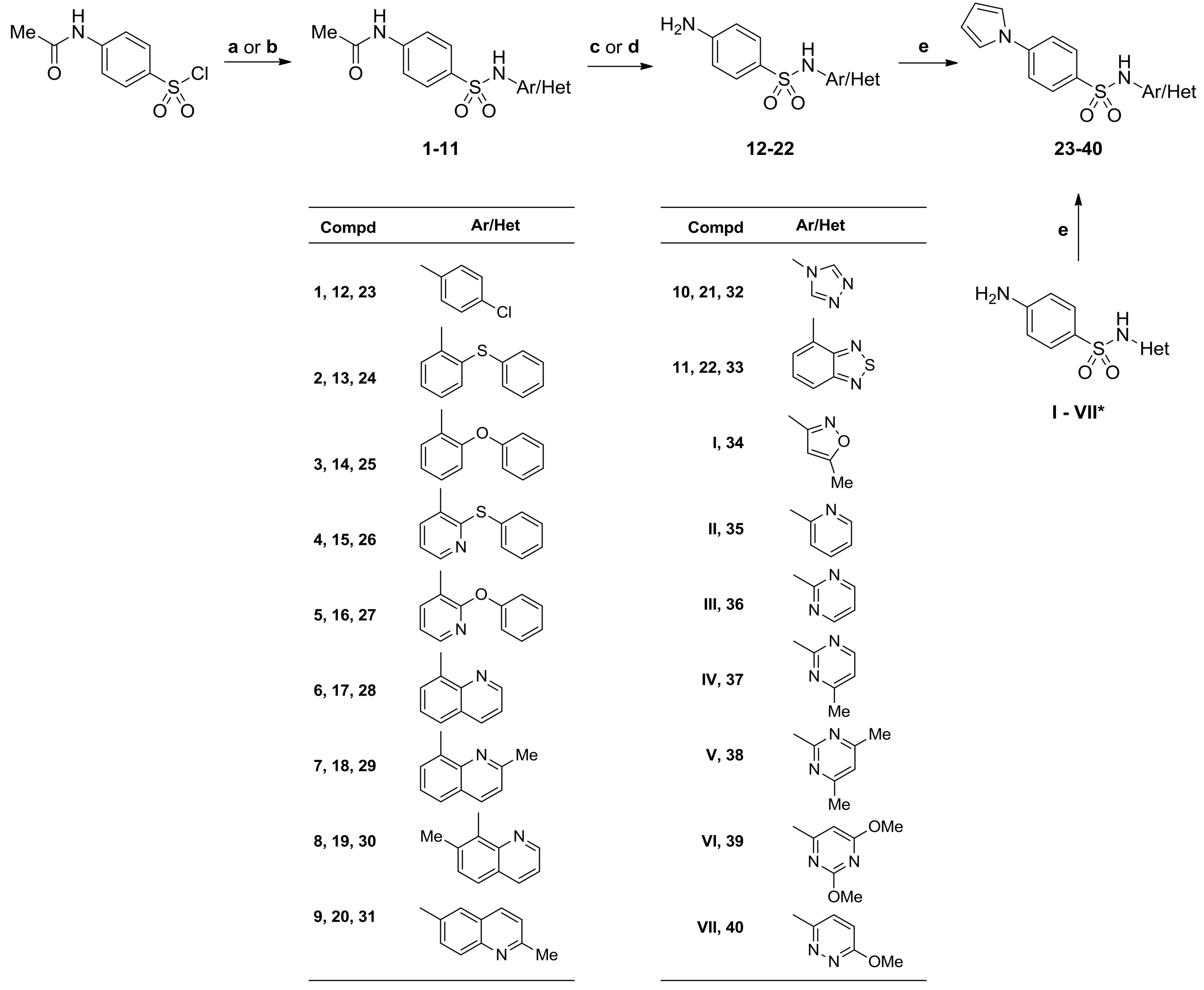
3.2.3. General Procedures for the Synthesis of N-(aryl/heteroaryl)-4-(1-H-pyrrol-1-yl)Benzenesulfonamides (23–40)
Method A. A mixture of 2,5-dimethoxytetrahydrofuran (198 mg, 1.5 mmol), p-dioxane (1.5 mL), glacial acetic acid (0.75 mL), and the corresponding 4-amino-N-(aryl/heteroaryl)benzenesulfonamide (13–19, I, II) (1.45 mmol) was stirred at reflux for 24 h. After cooling to room temperature, the mixture was evaporated in vacuum to dryness, and a residue was dissolved in boiling ethanol. After standing overnight, the precipitate was collected by filtration, washed with ethanol (2 × 2 mL), and dried at 105 °C.
Method B. A mixture of 2,5-dimethoxytetrahydrofuran (198 mg, 1.5 mmol), p-dioxane (1 mL), glacial acetic acid (2 mL), and the corresponding 4-amino-N-(heteroaryl)benzenesulfonamide (20, 21, VII) (1.4 mmol) was stirred at reflux for 24 h. After cooling to room temperature and standing overnight, the precipitate was collected by filtration, washed successively with p-dioxane (3 × 1.5 mL), water (4 × 2 mL) and ethanol (3 × 1.5 mL), and dried at temperatures gradually increasing to 105 °C.
Method C. A mixture of 2,5-dimethoxytetrahydrofuran (203 mg, 1.54 mmol), p-dioxane (2 mL), glacial acetic acid (1 mL), and the corresponding 4-amino-N-(heteroaryl)benzenesulfonamide (22, III–VI) (1.45 mmol) was stirred at reflux for 26 h. After cooling to room temperature and standing overnight, the precipitate was collected by filtration, washed successively with p-dioxane (2 × 2 mL), water (4 × 2 mL), and ethanol (3 × 3 mL), and dried at temperatures gradually increasing to 110 °C.
N-(4-Chlorophenyl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (23). A mixture of 2,5-dimethoxytetrahydrofuran (198 mg, 1.5 mmol), p-dioxane (1 mL), glacial acetic acid (2 mL), and 4-amino-N-(4-chlorophenyl)benzenesulfonamide 12 (410 mg, 1.4 mmol) was stirred at reflux for 24 h. After cooling to room temperature and standing overnight, the small amount (8 mg) of insoluble side products was filtered out, and the filtrate was concentrated in vacuum. To the residue water, 5 mL was added and stirred at room temperature for 6 h. The precipitate was collected by filtration, washed successively with water (6 × 1 mL) and ethanol (3 × 1 mL), and dried at temperatures gradually increasing to 105 °C. Yield for 23 (434 mg, 90%): m.p. 141‒142 °C; IR (KBr) 3255 (NH), 1596 (NH def), 1507, 1489, 1436 (C=N, C=C), 1333 (SO2 asym), 1157 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.30 (t, 2H, H-3 and H-4, pyrrole), 7.11 (d, J = 8.8 Hz, 2H, H-3 and H-5, 4-ClPh), 7.30 (d, J = 8.8 Hz, 2H, H-2 and H-6, 4-ClPh), 7.47 (t, 2H, H-2 and H-5, pyrrole), 7.77 (s, 4H, PhSO2), 10.43 (s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6) δ 112.4, 119.7, 119.8, 122.4, 129.0, 129.2, 129.9, 135.6, 137.4, 143.6 ppm; anal. C 57.74, H 3.93, N 8.41% calcd. for C16H13ClN2O2S, C 57.80, H 4.02, N 8.43%.
N-(2-(Phenylthio)phenyl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (24). Method A. Starting from 4-amino-N-[2-(phenylthio)phenyl]benzenesulfonamide 13 (517 mg), the title compound 24 was obtained (413 mg, 70%): m.p. 121.8‒125.0 °C; IR (KBr) 3231 (NH), 1596 (NH def), 1509, 1475, 1440 (C=N, C=C), 1337 (SO2 asym), 1168 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.32 (t, 2H, H-3 and H-4, pyrrole), 7.02‒7.07 (m, 3H, arom.), 7.15‒7.18 (m, 2H, arom.), 7.21‒7.25 (m, 4H, arom.), 7.48 (t, 2H, H-2 and H-5, pyrrole), 7.73 (s, 4H, PhSO2), 9.86 (s, 1H, SO2NH) ppm; anal. C 65.00, H 4.46, N 6.89% calcd. for C22H18N2O2S2, C 64.69, H 4.58, N 6.88%.
N-(2-Phenoxyphenyl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (25). Method A. Starting from 4-amino-N-(2-phenoxyphenyl)benzenesulfonamide 14 (554 mg), the title compound 25 was obtained (396 mg, 70%): m.p. 146 °C; IR (KBr) 3309 (NH), 1598 (NH def), 1510, 1491, 1421 (C=N, C=C), 1338 (SO2 asym), 1168 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.31 (t, 2H, H-3 and H-4, pyrrole), 6.63 (d, J = 7.8 Hz, 2H, arom.), 6.70 (d, J = 7.3 Hz, 1H, arom.), 7.00 (t, 1H, arom.), 7.06‒7.13 (m, 2H, arom.), 7.19 (t, 2H, arom.), 7.40 (d, J = 7.3 Hz, 1H, arom.), 7.44 (t, 2H, H-2 and H-5, pyrrole), 7.66 (d, J = 8.8 Hz, 2H, PhSO2), 7.72 (d, J = 8.8 Hz, 2H, PhSO2), 9.96 (s, 1H, SO2NH); 13C NMR (125 MHz, DMSO-d6) δ 112.2, 119.0, 119.2, 119.5, 119.8, 124.0, 124.3, 126.7, 127.5, 128.4, 129.1, 130.3, 136.9, 143.4, 150.3, 156.7 ppm; anal. C 67.67, H 4.65, N 7.17% calcd. for C22H18N2O3S, C 67.66, H 4.54, N 7.27%.
N-[2-(Phenylthio)pyridin-3-y]-4-(1H-pyrrol-1-yl)benzenesulfonamide (26). Method A. Starting from 4-amino-N-[2-(phenylthio)pyridin-3-y]benzenesulfonamide 15 (518 mg), the title compound 26 was obtained (236 mg, 40%): m.p. 113.4‒113.8 °C; IR (KBr) 3253 (NH), 1598 (NH def), 1510, 1475, 1439 (C=N, C=C), 1337 (SO2 asym), 1160 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.33 (t, 2H, H-3 and H-4, pyrrole), 7.15 (dd, J = 8.1, 4.7 Hz, 1H, arom.), 7.20 (d, J = 7.8 Hz, 2H, arom.), 7.27‒7.32 (m, 3H, arom.), 7.39 (d, J = 8.3 Hz, 1H, arom.), 7.51 (t, 2H, H-2 and H-5, pyrrole), 7.76 (d, J = 8.8 Hz, 2H, PhSO2), 7.81 (d, J = 8.8 Hz, 2H, PhSO2), 8.17 (d, J = 4.9 Hz, 1H, arom.), 10.19 (s, 1H, SO2NH) ppm; anal. C 61.89, H 4.20, N 10.31% calcd. for C21H17N3O2S2, C 61.71, H 4.13, N 10.23%.
N-(2-Phenoxypyridin-3-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (27). Method A. Starting from 4-amino-N-(2-phenoxypyridin-3-yl)benzenesulfonamide 16 (495 mg), the title compound 27 was obtained (369 mg, 65%): m.p. 163.7‒165.8 °C; IR (KBr) 3295 (NH), 1598 (NH def), 1512, 1489, 1451 (C=N, C=C), 1336 (SO2 asym), 1167 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.33 (t, 2H, H-3 and H-4, pyrrole), 6.72 (d, J = 7.8 Hz, 2H, arom.), 7.07‒7.13 (m, 2H, arom.), 7.23 (t, 2H, arom.), 7.46 (t, 2H, H-2 and H-5, pyrrole), 7.73‒7.80 (m, 5H, arom.), 7.86 (dd, J = 4.9, 1.5 Hz, 1H, arom.), 10.23 (s, 1H, SO2NH); 13C NMR (125 MHz, DMSO-d6) δ 112.1, 119.5, 119.6, 119.7, 121.6, 121.6, 124.9, 129.0, 129.7, 135.4, 136.4, 143.4, 144.2, 153.7, 156.7 ppm; anal. C 64.43, H 4.38, N 10.73% calcd. for C21H17N3O3S, C 64.09, H 4.41, N 10.66%.
4-(1H-Pyrrol-1-yl)-N-(quinolin-8-yl)benzenesulfonamide (28). Method A. Starting from 4-amino-N-(quinolin-8-yl)benzenesulfonamide 17 (434 mg), the title compound 28 was obtained (258 mg, 51%): m.p. 151.9‒153.0 °C dec.; IR (KBr) 3254 (NH), 1597 (NH def), 1505, 1471 (C=N, C=C), 1335 (SO2 asym), 1163 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.26 (t, 2H, H-3 and H-4, pyrrole), 7.41 (t, 2H, H-2 and H-5, pyrrole), 7.53 (t, 1H, arom.), 7.56 (dd, J = 8.3, 3.9 Hz, 1H, arom.), 7.66 (d, J = 8.3 Hz, 1H, arom.), 7.69 (d, J = 8.8 Hz, 2H, PhSO2), 7.72 (d, J = 7.9 Hz, 1H, arom.), 7.96 (d, J = 8.8 Hz, 2H, PhSO2), 8.33 (dd, J = 8.3, 2 Hz, 1H, arom.), 8.86 (dd, J = 4.4, 1.9 Hz, 1H, arom.), 10.06 (s, 1H, SO2NH); 13C NMR (125 MHz, DMSO-d6) δ 112.9, 117.3, 119.3, 119.5, 122.8, 123.6, 127.2, 128.6, 129.3, 134.1, 135.7, 136.0, 139.3, 143.4, 149.9 ppm; anal. C 65.31, H 4.33, N 12.03% calcd. for C19H15N3O2S, C 65.07, H 4.20, N 12.00%.
N-(2-Methylquinolin-8-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (29). Method A. Starting from 4-amino-N-(2-methylquinolin-8-yl)benzenesulfonamide 18 (454 mg), the title compound 29 was obtained (316 mg, 60%): m.p. 167.6‒168.7 °C; IR (KBr) 3225 (NH), 1596 (NH def), 1506, 1472 (C=N, C=C), 1327 (SO2 asym), 1166 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.66 (s, 3H, CH3), 6.27 (t, 2H, H-3 and H-4, pyrrole), 7.40‒7.46 (m, 4H, arom. and H-2 and H-5, pyrrole), 7.60 (d, J = 7.8 Hz, 1H, arom.), 7.64 (d, J = 7.3 Hz, 1H, arom.), 7.69 (d, J = 8.8 Hz, 2H, PhSO2), 7.94 (d, J = 8.8 Hz, 2H, PhSO2), 8.20 (d, J = 8.8 Hz, 1H, arom.), 9.83 (s, 1H, SO2NH) ppm; anal. C 66.10, H 4.71, N 11.56% calcd. for C20H17N3O2S, C 66.02, H 4.69, N 11.56%.
N-(7-Methylquinolin-8-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (30). Method A. Starting from 4-amino-N-(7-methylquinolin-8-yl)benzenesulfonamide 19 (454 mg), the title compound 30 was obtained (343 mg, 65%): m.p. 142‒145 °C; IR (KBr) 3235 (NH), 1599 (NH def), 1516, 1503, 1466 (C=N, C=C), 1335 (SO2 asym), 1167 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.59 (s, 3H, CH3), 6.29 (t, 2H, H-3 and H-4, pyrrole), 7.29 (dd, J = 8.3, 3.9 Hz, 1H, arom.), 7.41 (t, 2H, H-2 and H-5, pyrrole), 7.53 (d, J = 8.3 Hz, 1H, arom.), 7.56 (br. s, 4H, arom.), 7.78 (d, J = 8.3 Hz, 1H, arom.), 8.21 (d, J = 8.3 Hz, 1H, arom.), 8.39 (d, J = 3.9 Hz, 1H, arom.), 9.74 (s, 1H, SO2NH); 13C NMR (125 MHz, DMSO-d6) δ 19.9, 111.9, 118.5, 119.5, 121.1, 127.0, 127.1, 129.3, 130.3, 131.5, 136.2, 137.3, 138.9, 142.5, 144.5, 149.7 ppm; anal. C 66.10, H 4.71, N 11.56% calcd. for C20H17N3O2S, C 65.82, H 4.68, N 11.51%.
N-(2-Methylquinolin-6-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (31). Method B. Starting from 4-amino-N-(2-methylquinolin-6-yl)benzenesulfonamide 20 (439 mg), the title compound 31 was obtained (280 mg, 55%): m.p. 286‒288 °C; IR (KBr) 3265, 3140 (SO2NH), 1596 (NH def), 1510, 1475 (C=N, C=C), 1340 (SO2 asym), 1160 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.57 (s, 3H, CH3), 6.26 (br. s, 2H, H-3 and H-4, pyrrole), 7.33 (d, J = 6.8 Hz, 1H, H-3, quinoline), 7.42-7.46 (m, 3H, arom.), 7.59 (s, 1H, H-5, quinoline), 7.73-7.96 (m, 5H, arom.), 8.14 (d, J = 6.4 Hz, 1H, H-8, quinoline), 10.58 (s, 1H, SO2NH) ppm; 13C NMR (125 MHz, DMSO-d6) δ 24.7, 111,9, 116.3, 119.3, 122.9, 124.0, 126.7, 128.8, 129.6, 135.4, 143.1, 144.7, 158.0 ppm; anal. C 66.09, H 4.71, N 11.56% calcd. for C20H17N3O2S, C 66.12, H 4.81, N 11.55%.
4-(1H-pyrrol-1-yl)-N-(4H-1,2,4-triazol-4-yl)benzenesulfonamide (32). Method B. Starting from 4-amino-N-(4H-1,2,4-triazol-4-yl)benzenesulfonamide 21 (335 mg), the title compound 32 was obtained (219 mg, 54%): m.p. 251‒252 °C dec.; IR (KBr) 3150 (NH), 1595 (NH def), 1515, 1475, 1425 (C=N, C=C), 1340 (SO2 asym), 1165 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.35 (br. s, 2H, H-3 and H-4, pyrrole), 7.57 (br. s, 2H, H-2 and H-5, pyrrole), 7.70 (d, J = 8.3 Hz, 2H, H-3 and H-5, PhSO2), 7.88 (d, J = 8.3 Hz, 2H, H-2 and H-6, PhSO2), 8.37 (s, 2H, H-3 and H-5, 1,2,4-triazole), 11.90 (br. s, 1H, SO2NH) ppm; 13C NMR (DMSO-d6) δ 112.7, 119.9, 120.0, 130.4, 143.5, 144.7 ppm; anal. C 49.82, H 3.83, N 24.20% calcd. for C12H11N5O2S, C 49.80, H 3.93, N 24.19%.
N-(Benzo-2,1,3-thiadiazol-4-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (33). Method C. Starting from 4-amino-N-(benzo-2,1,3-thiadiazol-4-yl)benzenesulfonamide 22 (444 mg), the title compound 33 was obtained (336 mg, 65%): m.p. 325‒327 °C dec.; IR (KBr) 3320, 3225 (NH), 1595 (NH def), 1540, 1510, 1495 (C=N, C=C), 1340 (SO2 asym), 1170 (SO2 sym) cm−1; 1H NMR (220 MHz, DMSO-d6) δ 6.30 (br. s, 2H, H-3 and H-4, pyrrole), 7.46 (br. s, 2H, H-2 and H-5, pyrrole), 7.64 (t, J = 7.8 Hz, 1H, H-6, benzothiadizole), 7.68-7.80 (m, 4H, H-3 and H-5, PhSO2 and H-5 and H-7 benzothiadiazole), 7.90 (d, J = 8.0 Hz, 2H, H-2 and H-6 PhSO2), 11.0 (br. s, 1H, SO2NH) ppm; 13C NMR (DMSO-d6) δ 111.9, 117.2, 118.1, 119.2, 119.4, 129.0, 129.7, 130.7, 136.0, 143.2, 149.0, 155.1 ppm; anal. C 53.91, H 3.39, N 15.72% calcd. for C16H12 N4O2S2, C 54.02, H 3.48, N 15.81%.
N-(5-Methylisoxazol-3-yl)-4-(1H-pyrrol-1-yl)-benzenesulfonamide (34). Method A. Starting from 4-amino-N-(5-methylisoxazol-3-yl)benzenesulfonamide I (367 mg), the title compound 34 was obtained (387 mg, 88%): m.p. 203‒204.8 °C; IR (KBr) 3235 (NH), 1599 (NH def), 1516, 1503, 1466 (C=N, C=C), 1337 (SO2 asym), 1168 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 2.29 (s, 3H, CH3), 6.17 (s, 1H, arom.), 6.32 (s, 2H, H-3 and H-4, pyrrole), 7.49 (s, 2H, H-2 and H-5, pyrrole), 7.82 (d, J = 8.8 Hz, 2H, H-2 and H-6 PhSO2), 7.89 (d, J = 8.8 Hz, 2H, H-2 and H-6 PhSO2.), 11.46 (s, 1H, SO2NH); 13C NMR (125 MHz, DMSO-d6) δ 12.7, 96.1, 112.4, 119.8, 119.9, 129.3, 135.9, 143.9, 158.2, 171.2 ppm; anal. C 55.43, H 4.32, N 13.85% calcd. for C14H13N3O3S, C 54.96, H 4.24, N 13.75%.
N-(Pyridin-2-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (35). Method A. Starting from 4-amino-N-(pyridin-2-yl)benzenesulfonamide II (361 mg), the title compound 35 was obtained (391 mg, 90%): m.p. 244‒246.4 °C; IR (KBr) 3138 (NH), 1634 (NH def), 1535, 1512, 1466 (C=N, C=C), 1359 (SO2 asym), 1142 (SO2 sym) cm−1; 1H NMR (500 MHz, DMSO-d6) δ 6.29 (s, 2H, H-3 and H-4, pyrrole), 6.86 (t, 1H, arom.), 7.17 (d, 1H, arom.), 7.45 (s, 2H, H-2 and H-5, pyrrole), 7.71 (d, 1H, arom.), 7.74 (d, J = 8.8 Hz, 2H, H-2 and H-6 PhSO2), 7.90 (d, J = 8.8 Hz, 2H, H-2 and H-6 PhSO2.), 8.00 (d, 1H, arom.), 12.06 (s, 1H, SO2NH); 13C NMR (125 MHz, DMSO-d6) δ 112.2, 114.6, 116.1, 119.6, 119.8, 129.0, 138.8, 141.4, 143.0, 153.9 ppm; anal. C 60.18, H 4.38, N 14.04% calcd. for C15H13N3O2S, C 60.00, H 4.19, N 13.88%.
N-(Pyrimidin-2-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (36). Method C. Starting from 4-amino-N-(pyrimidin-2-yl)benzenesulfonamide III (363 mg), the title compound 36 was obtained (379 mg, 87%): m.p. 260‒261°C dec.; IR (KBr) 3150 (NH), 1600 (NH def), 1585, 1510, 1445 (C=N, C=C), 1345 (SO2 asym), 1160 (SO2 sym) cm−1; 1H NMR (200 MHz, DMSO-d6) δ 6.30 (br. s, 2H, H-3 and H-4, pyrrole), 7.04 (t, J = 4.9 Hz, 1H, H-5, pyrimidine), 7.48 (br. s, 2H, H-2 and H-5, pyrrole), 7.85 (d, J = 8.8 Hz, 2H, H-3 and H-5, PhSO2), 8.01 (d, J = 8.8 Hz, 2H, H-2 and H-6, PhSO2), 8.50 (d, J = 4.9 Hz, 2H, H-4 and H-6, (pyrimidine), 11.88 (br. s, 1H, SO2NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ 112.3, 119.4, 119.8, 130.1, 136.9, 143.5, 157.5, 159.0 ppm; anal. C 55.99, H 4.02, N 18.65% calcd. for C14H12N4O2S, C 56.20, H 4.22, N 18.72%.
N-(4-Methylpyrimidin-2-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (37). Method C. Starting from 4-amino-N-(4-methylpyrimidin-2-yl)benzenesulfonamide IV (383 mg) the title compound 37 was obtained (392 mg, 86%): m.p. 256‒257 °C dec.; IR (KBr) 3235 (NH), 1600 (NH def), 1565, 1510, 1405 (C=N, C=C), 1345 (SO2 asym), 1150 (SO2 sym) cm−1; 1H NMR (200 MHz, DMSO-d6) δ 2.31 (s, 3H CH3), 6.30 (br. s, 2H, H-3 and H-4, pyrrole), 6.90 (d, J = 4.9 Hz, 1H, H-5, pyrimidine), 7.47 (br. s, 2H, H-2 and H-5, pyrrole), 7.77 (d, J = 8.3 Hz, 2H, H-3 and H-5, PhSO2), 8.01 (d, J = 8.3 Hz, 2H, H-2 and H-6, PhSO2), 8.32 (d, J = 4.9 Hz, 1H, H-6, pyrimidine), 11.82 (br. s, 1H, SO2NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ 23.9, 112.3, 119.2, 119.8, 130.3, 137.2, 143.4, 157.2 ppm; anal. C 57.31, H 4.49, N 17.82% calcd. for C15H14N4O2S, C 57.45, H 4.66, N 17.90%.
N-(4,6-Dimethylpyrimidin-2-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (38). Method C. Starting from 4-amino-N-(4,6-dimethylpyrimidin-2-yl)benzenesulfonamide V (404 mg), the title compound 38 was obtained (376 mg, 79%): m.p. 224‒227 °C dec.; IR (KBr) 3245 (NH), 1600 (NH def), 1585, 1510, 1475 (C=N, C=C), 1335 (SO2 asym), 1160 (SO2 sym) cm−1; 1H NMR (200 MHz, DMSO-d6) δ 2.24 (s, 6H, 2 × CH3), 6.29 (br. s, 2H, H-3 and H-4, pyrrole), 6.74 (s, 1H, H-5, pyrimidine), 7.46 (br. s, 2H, H-2 and H-5, pyrrole), 7.76 (d, J = 8.3 Hz, 2H, H-3 and H-5, PhSO2), 8.02 (d, J = 8.3 Hz, 2H, H-2 and H-6, PhSO2), 11.86 (br. s, 1H, SO2NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ 23.5, 112.2, 119.0, 119.8, 130.5, 143.2, 156.8 ppm; anal. C 58.51, H 4.91, N 17.06% calcd. for C16H16N4O2S, C 58.85, H 5.30, N 17.52%.
N-(2,6-Dimethoxypirimidin-4-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (39). Method C. Starting from 4-amino-N-(2,6-dimethoxypirimidin-4-yl)benzenesulfonamide VI (450 mg) the title compound 39 was obtained (423 mg, 81%): m.p. 162‒163 °C dec.; IR (KBr) 3240, 3150 (NH), 1595 (NH def), 1510, 1490, 1455 (C=N, C=C), 1335 (SO2 asym), 1160 (SO2 sym) cm−1; 1H NMR (200 MHz, DMSO-d6) δ 3.78 (s, 3H CH3O, pyrimidine), 3.81 (s, 3H, CH3O-2, pyrimidine), 5.99 (s, 1H, H-5, pyrimidine), 6.34 (br. s, 2H, H-3 and H-4, pyrrole), 7.51 (br. s, 2H, H-2 and H-5, pyrrole), 7.84 (d, J = 8.4 Hz, 2H, H-3 and H-5, PhSO2), 8.00 (d, J = 8.4 Hz, 2H, H-2 and H-6, PhSO2), 11.86 (br. s, 1H, SO2NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ 54.1, 54.8, 85.0, 112.0, 119.3, 119.5, 129.3, 136.0, 143.4, 160.1, 164.2, 171.9 ppm; anal. C 53.32, H 4.47, N 15.54% calcd. for C16H16N4O4S, C 53.48, H 4.60, N 15.70%.
N-(6-Methoxypyridazin-3-yl)-4-(1H-pyrrol-1-yl)benzenesulfonamide (40). Method B. Starting from 4-amino-N-(6-methoxypyridazin-3-yl)benzenesulfonamide VII (392 mg), the title compound 40 was obtained (241 mg, 52%): 191‒192 °C; IR (KBr) 3215 (NH), 1645 (NH def), 1525, 1510, 1470 (C=N, C=C), 1335 (SO2 asym), 1145 (SO2 sym) cm−1; 1H NMR (200 MHz, DMSO-d6) δ 3.83 (s, 3H, CH3O), 6.30 (br. s, 2H, H-3 and H-4, pyrrole), 7.37 (d, J = 7.9 Hz, 1H, H-5, pyridazine), 7.48 (br. s, 2H, H-2 and H-5, pyrrole), 7.72‒7.81 (m, 3H, H-3 and H-5, PhSO2, and H-4, pyridazine), 7.87 (d, J = 8.6 Hz, 2H, H-2 and H-6, PhSO2), 13.00 (br. s, 1H, SO2NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ 53.54, 110.9, 117.8, 118.5, 119.2, 125.0, 128.2, 140.5, 144.3, 158.7, 159.9 ppm; anal. C 54.53, H 4.27, N 16.96% calcd. for C15H14N4O3S, C 54.52, H 4.30, N 16.95%.


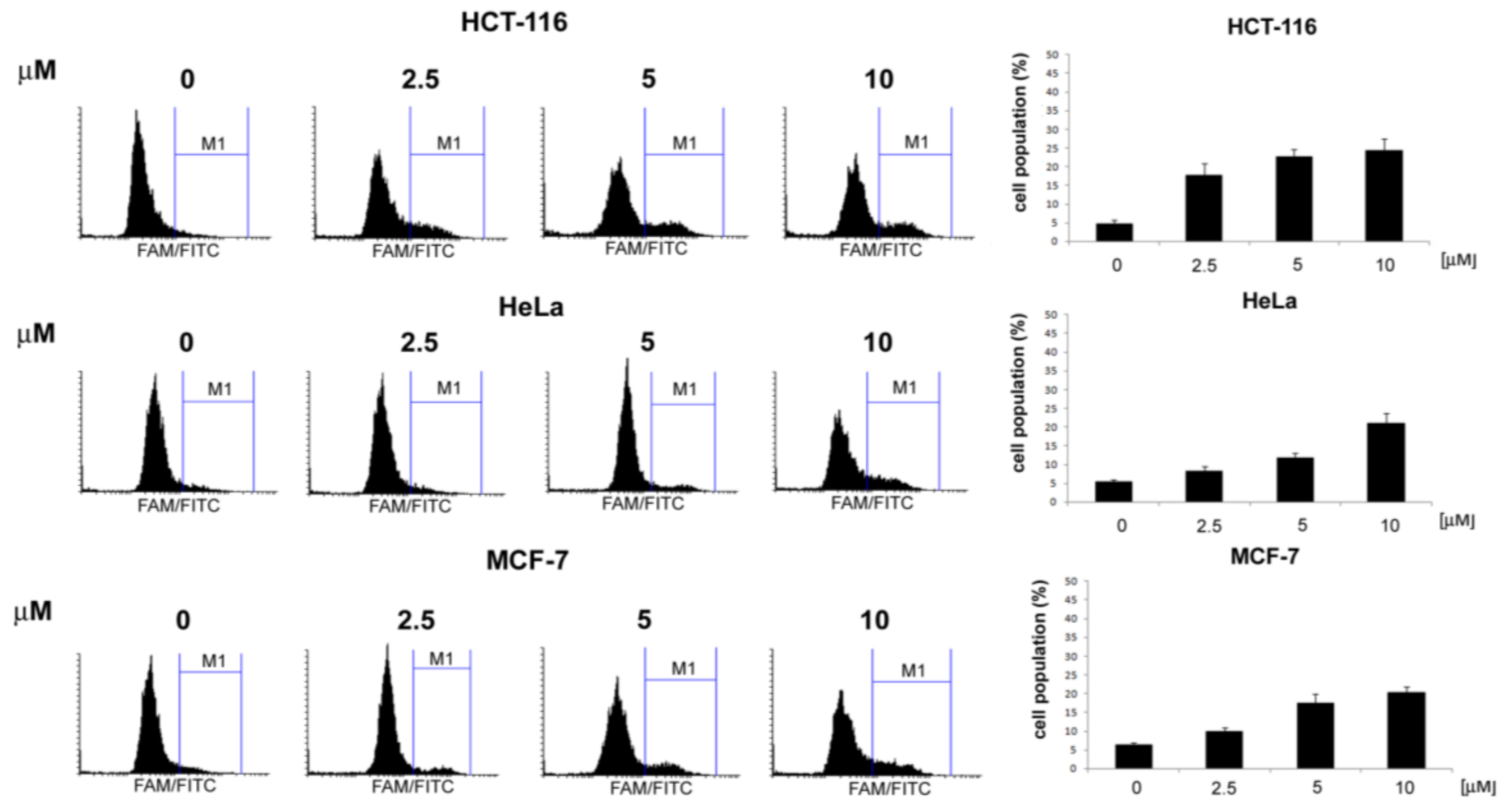
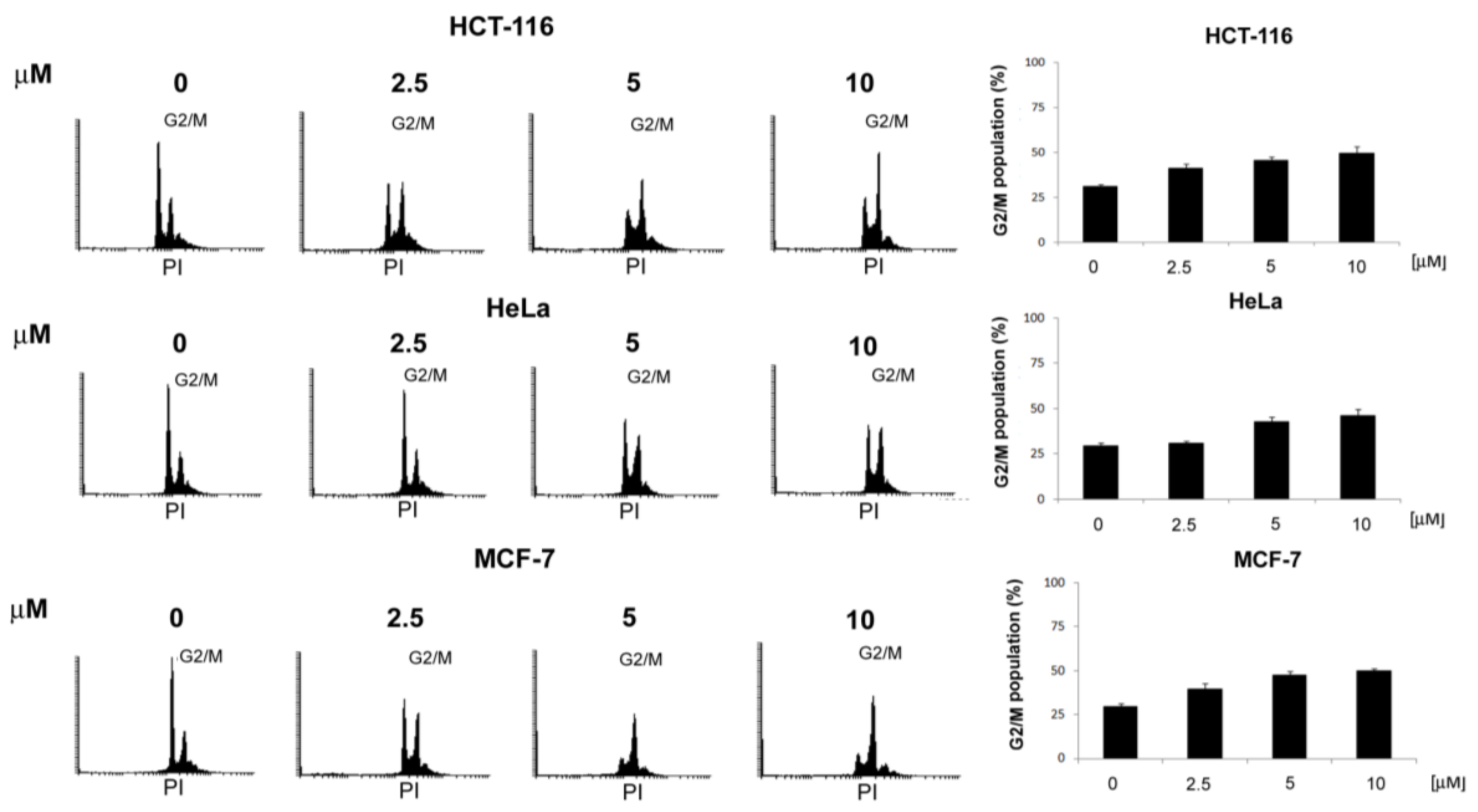
 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}