Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
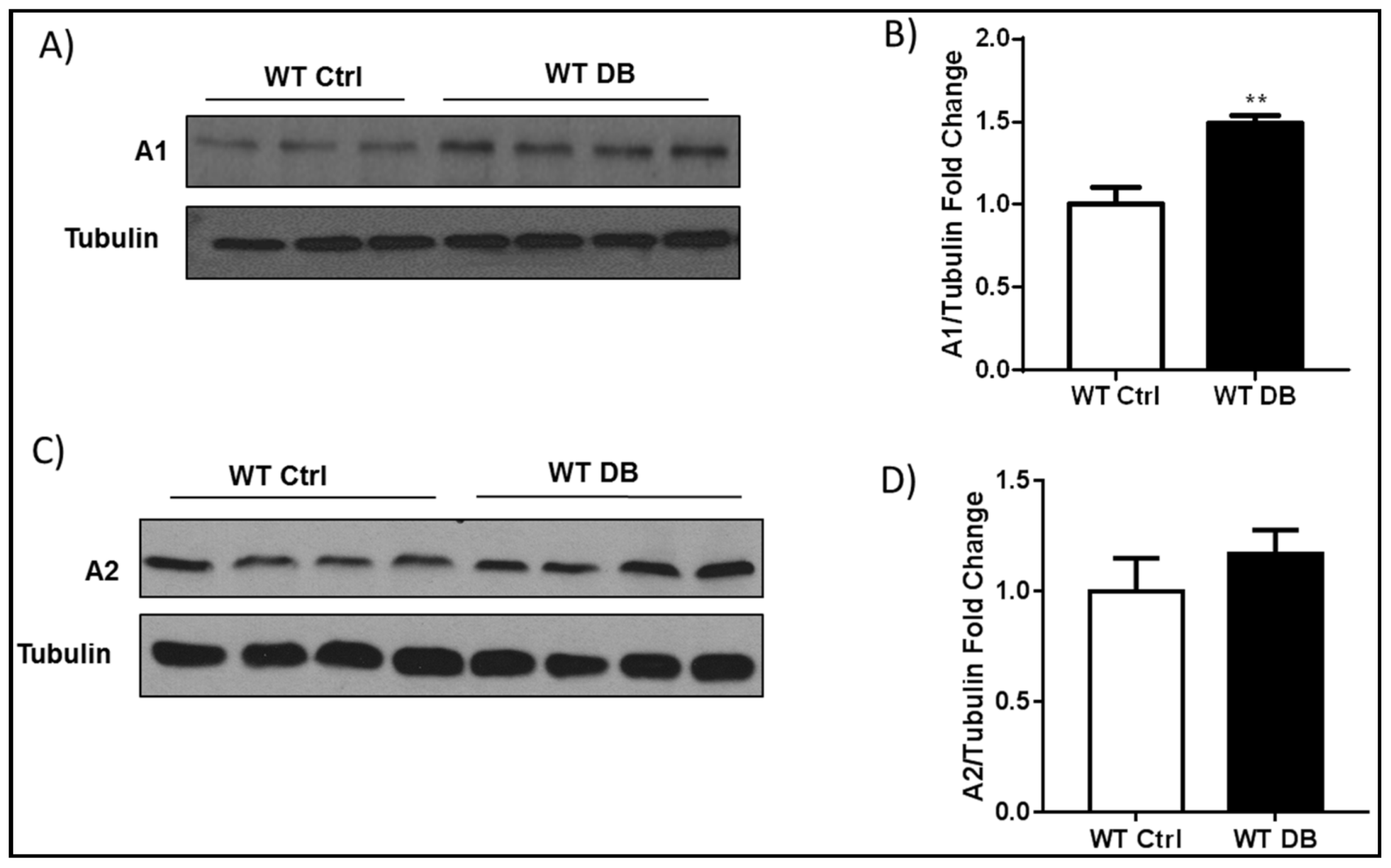
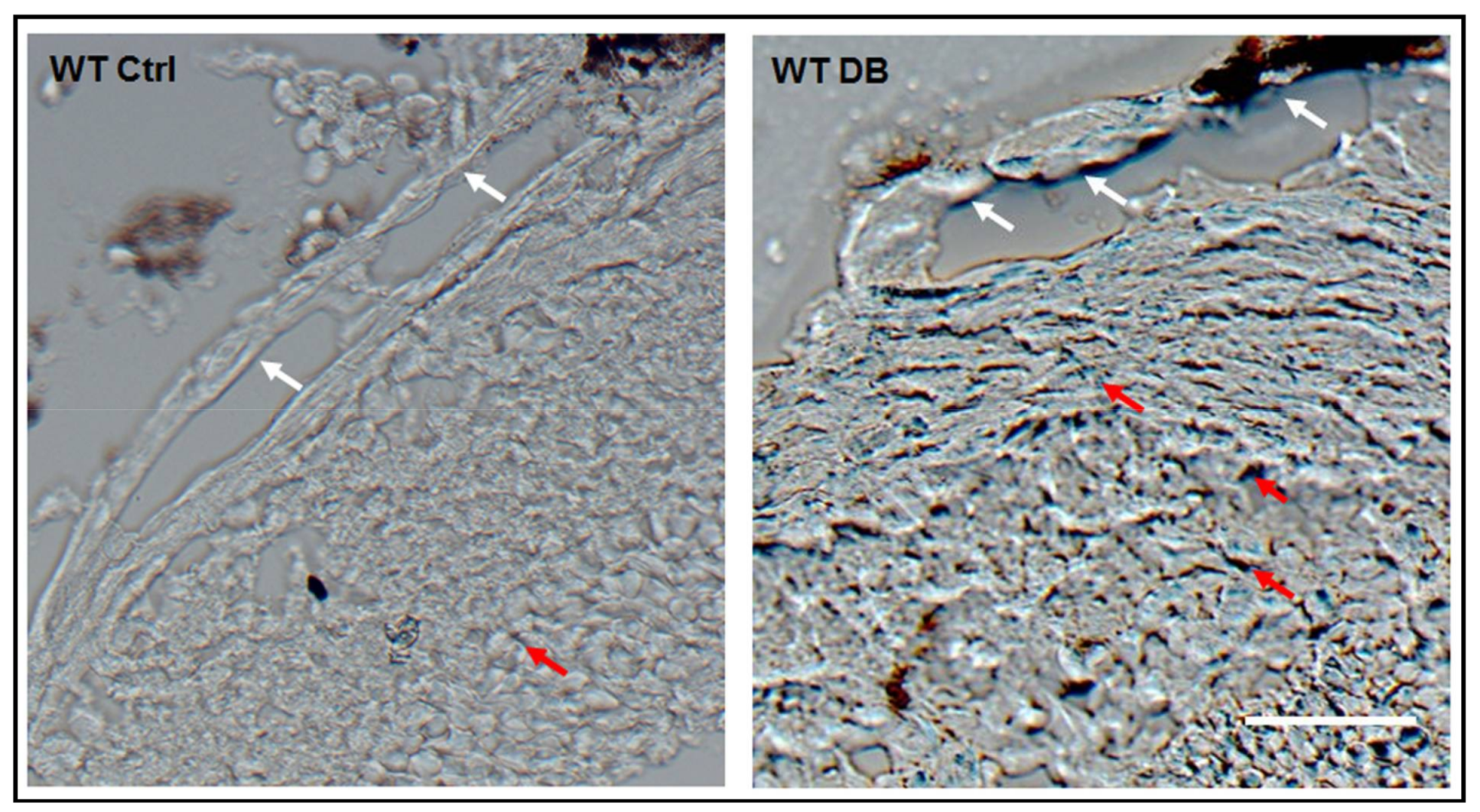
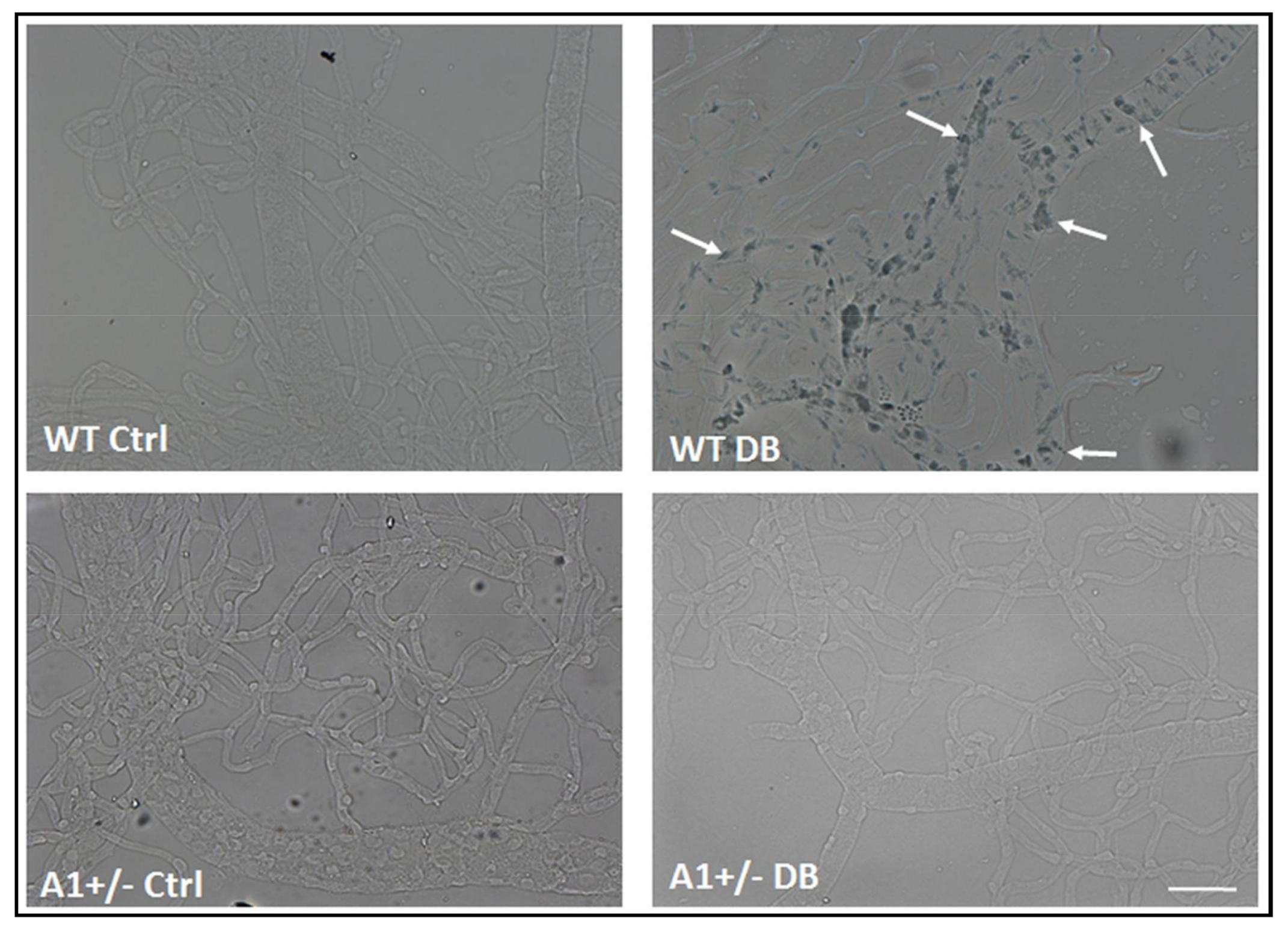
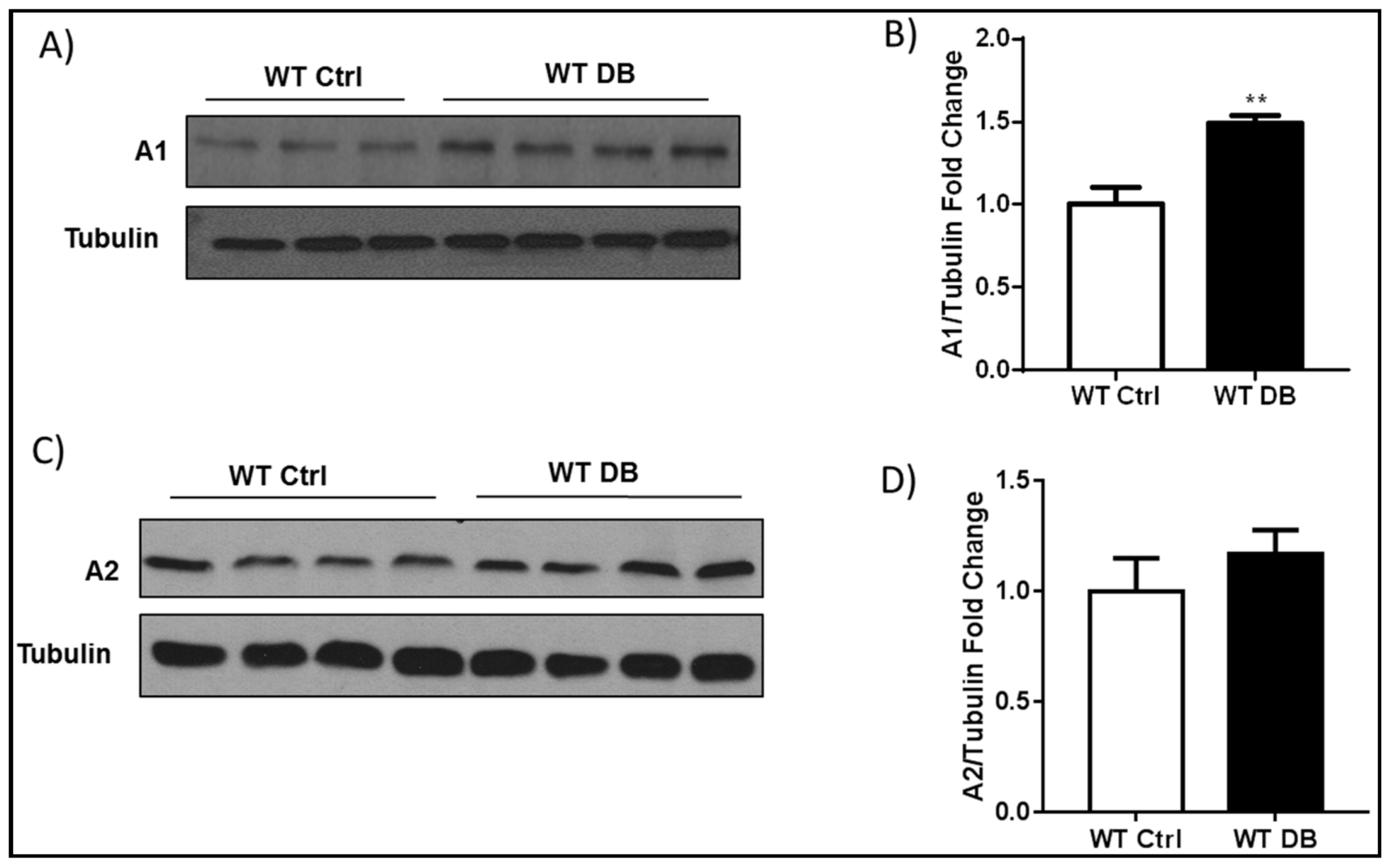
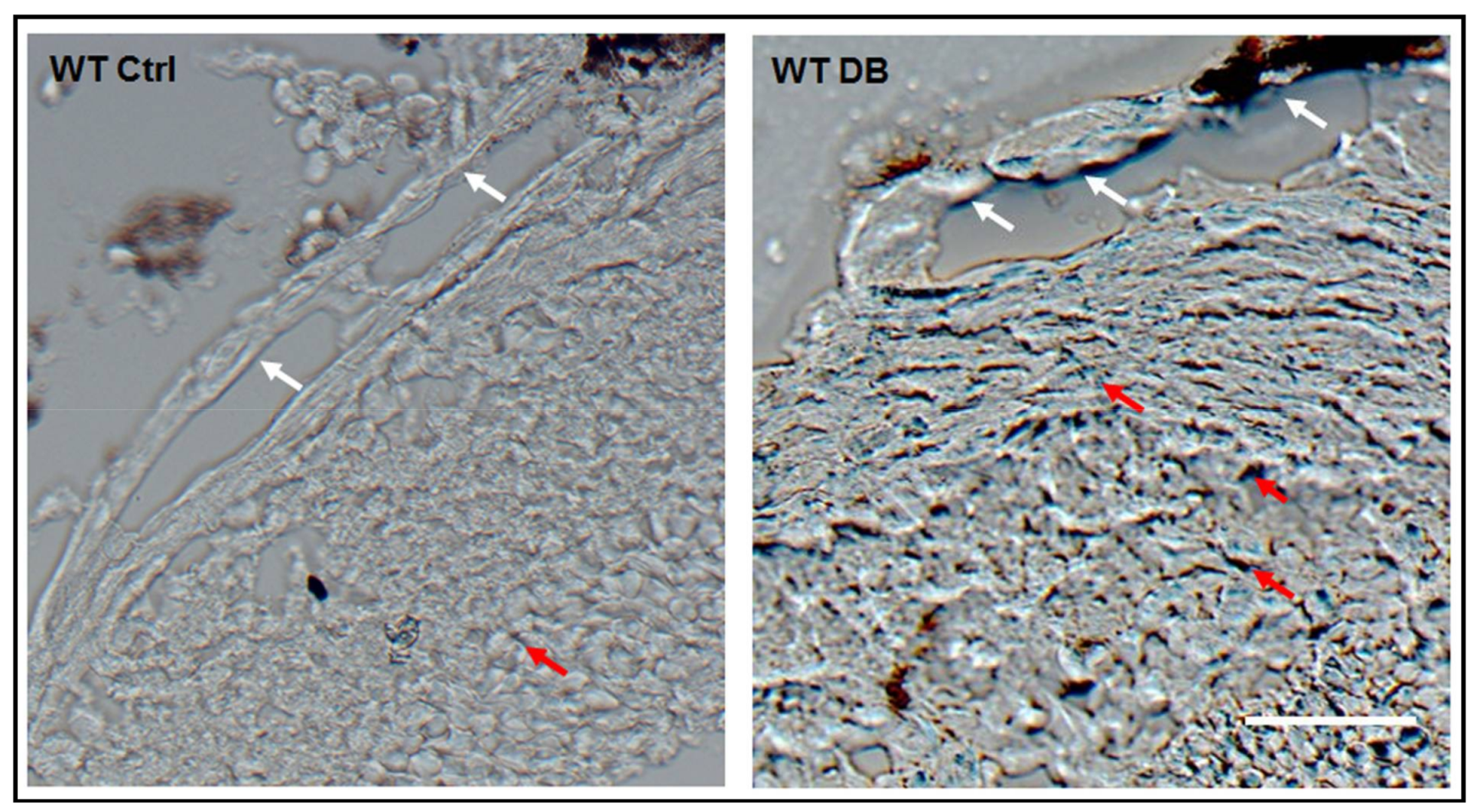
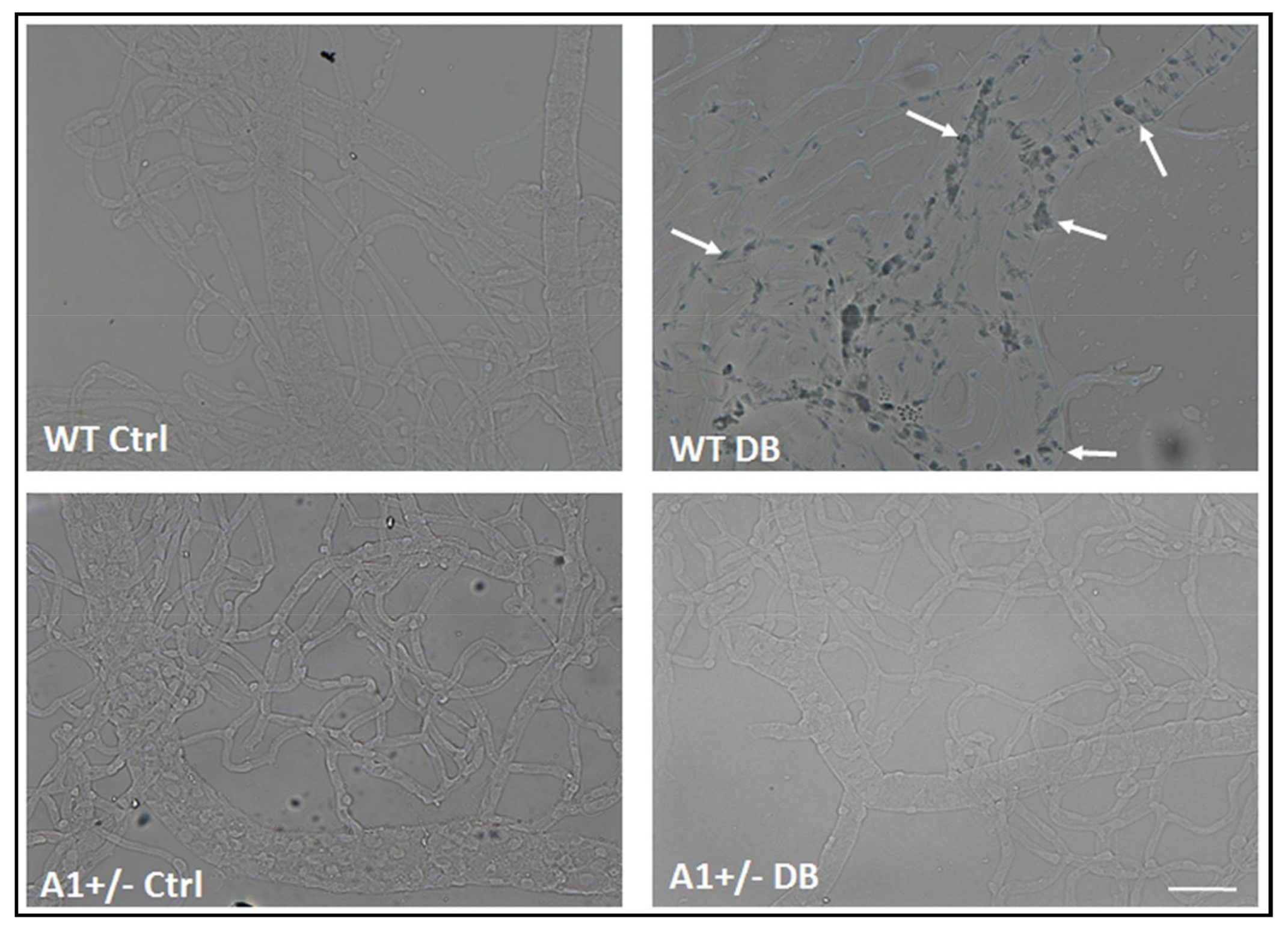
2.1. Diabetes-Induced Increases in Arginase 1 and Premature Cellular Senescence
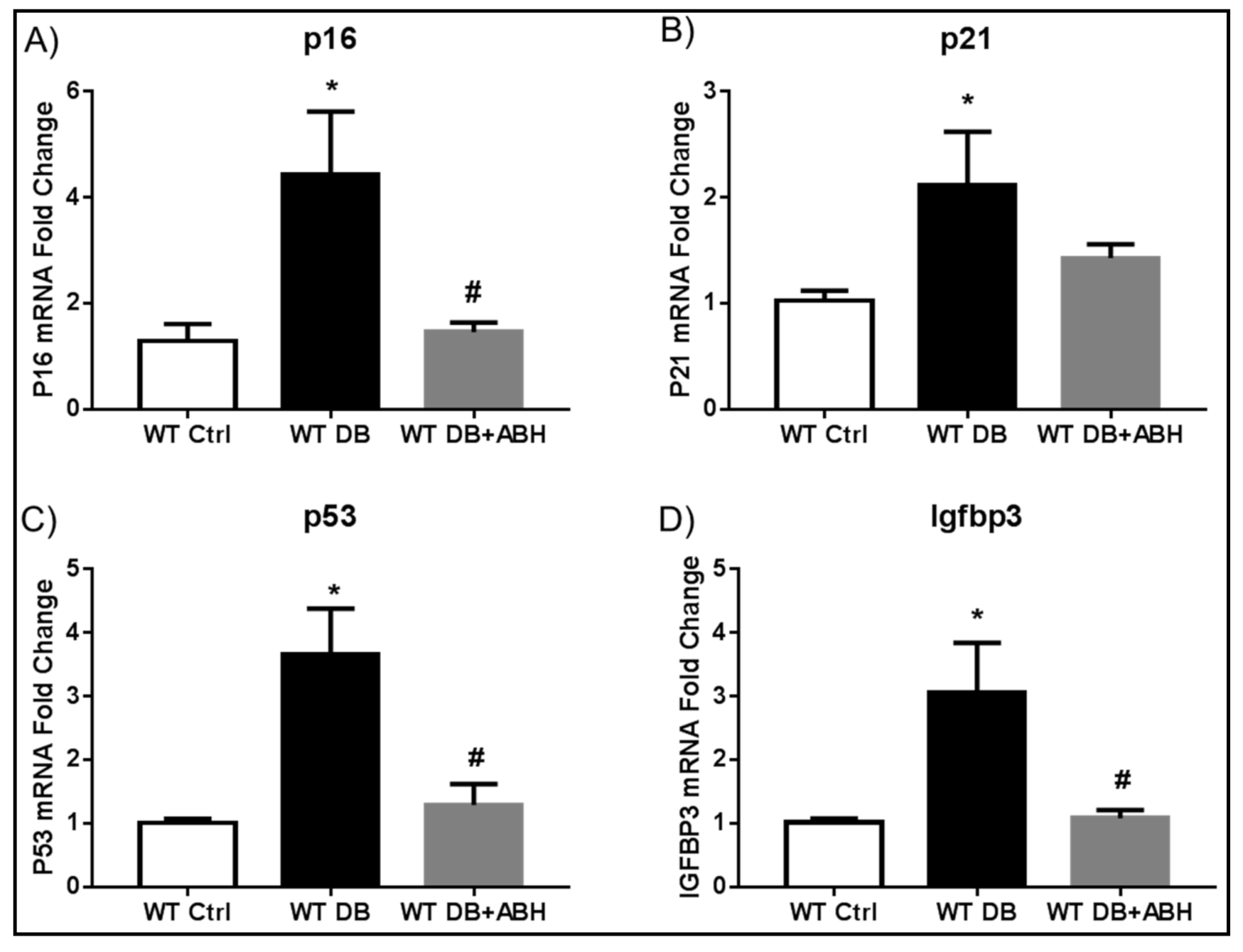
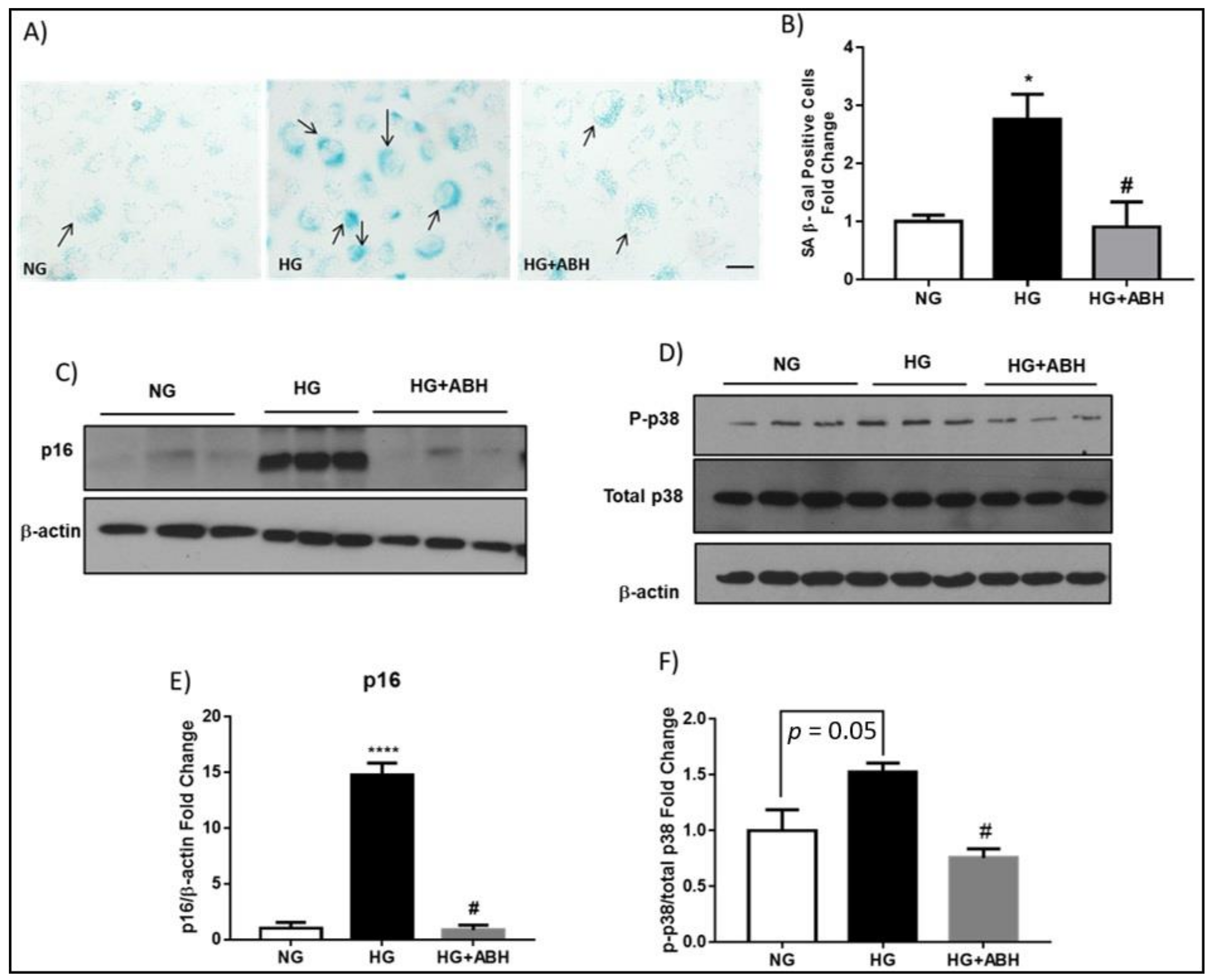
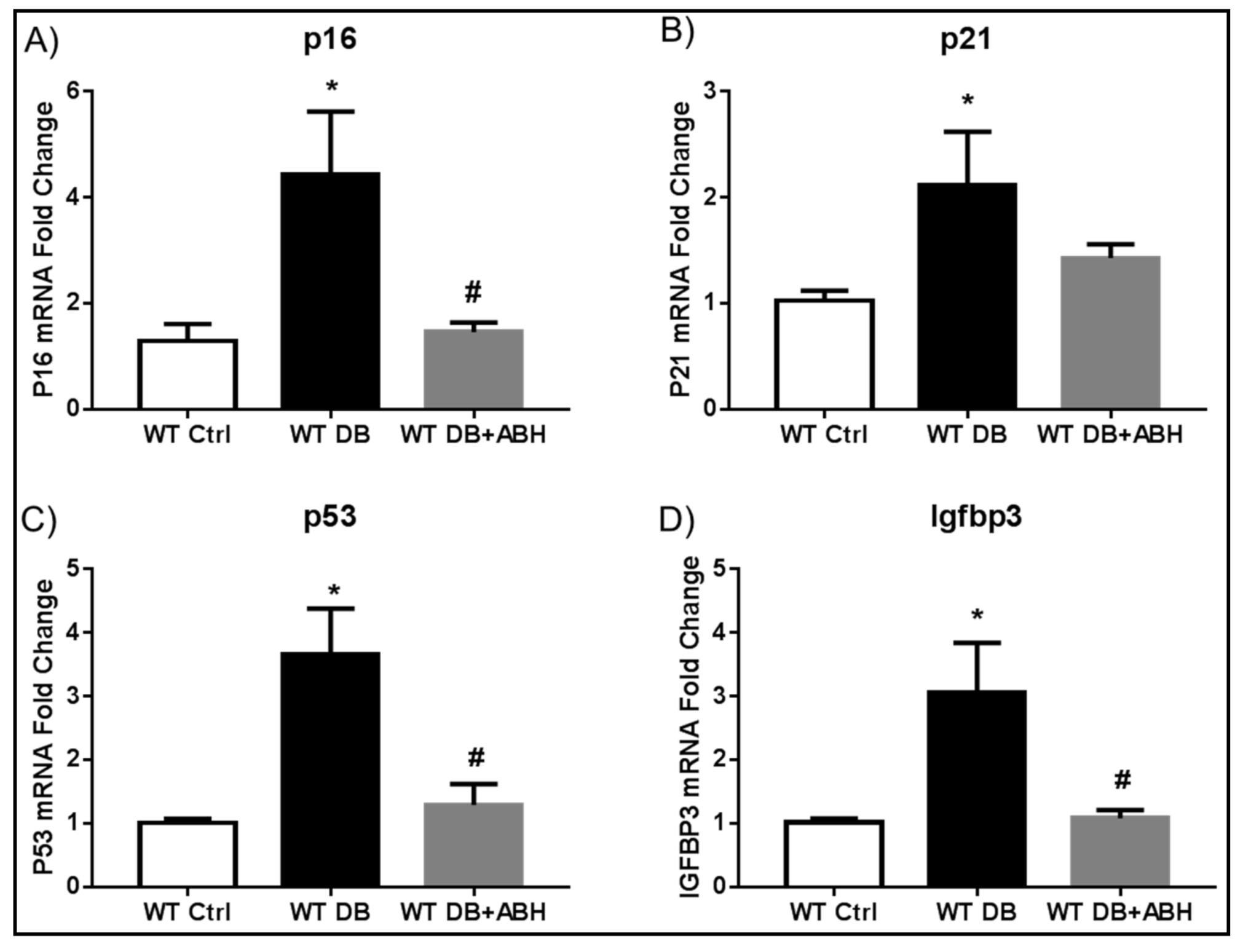
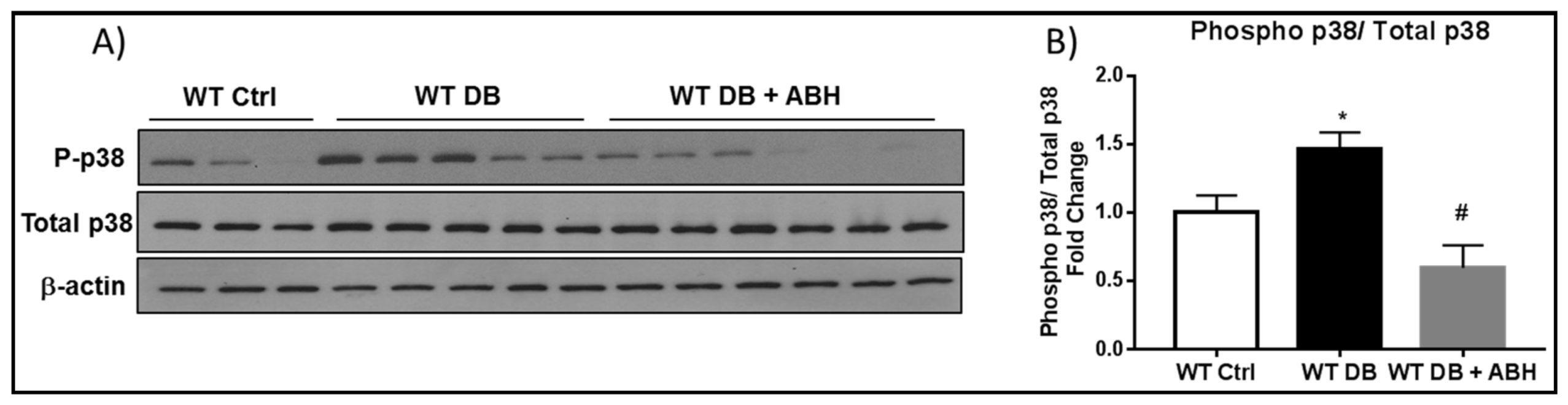
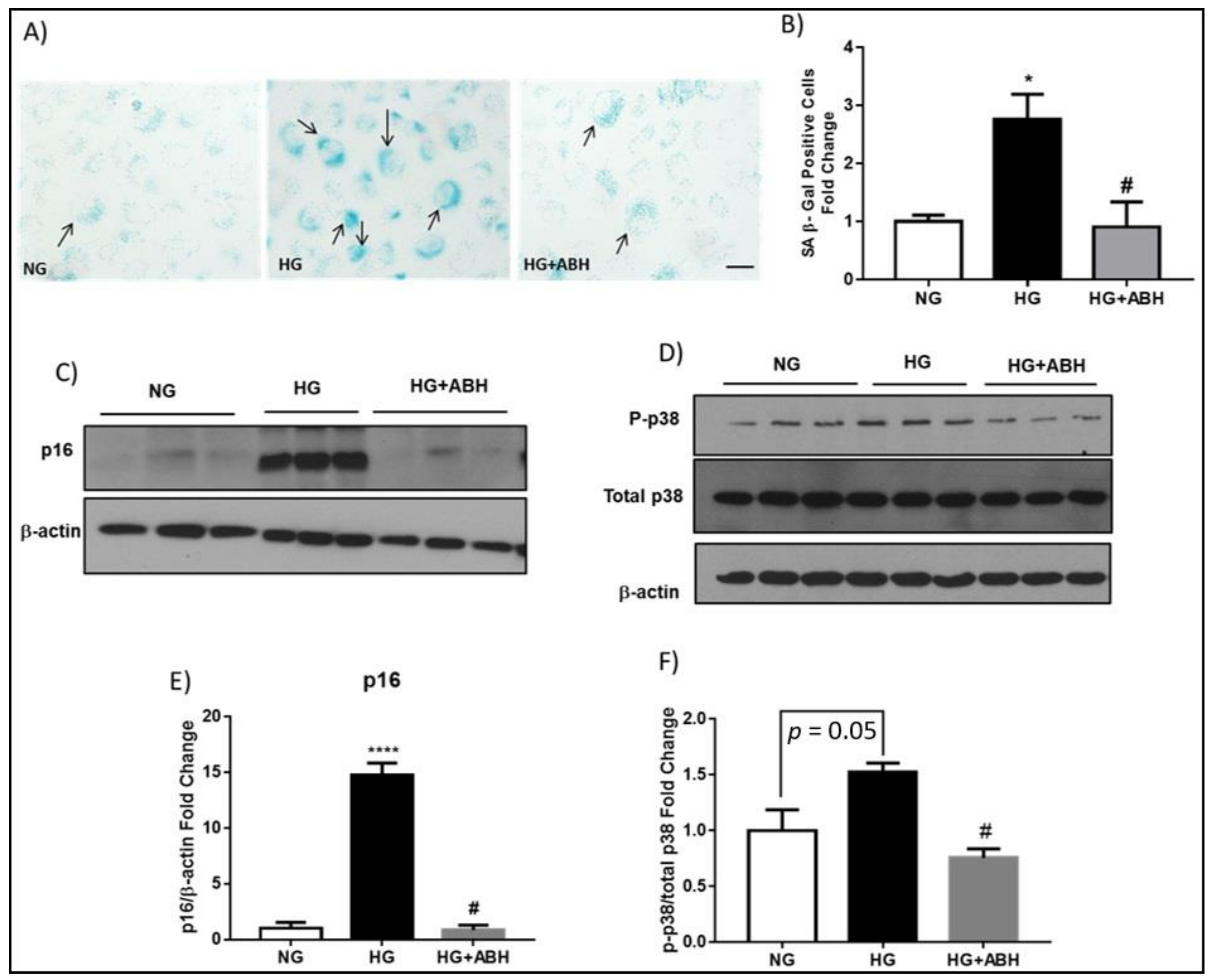
2.2. Suppression of Diabetes-Induced Increases in Senescence Markers by Inhibition of Arginase
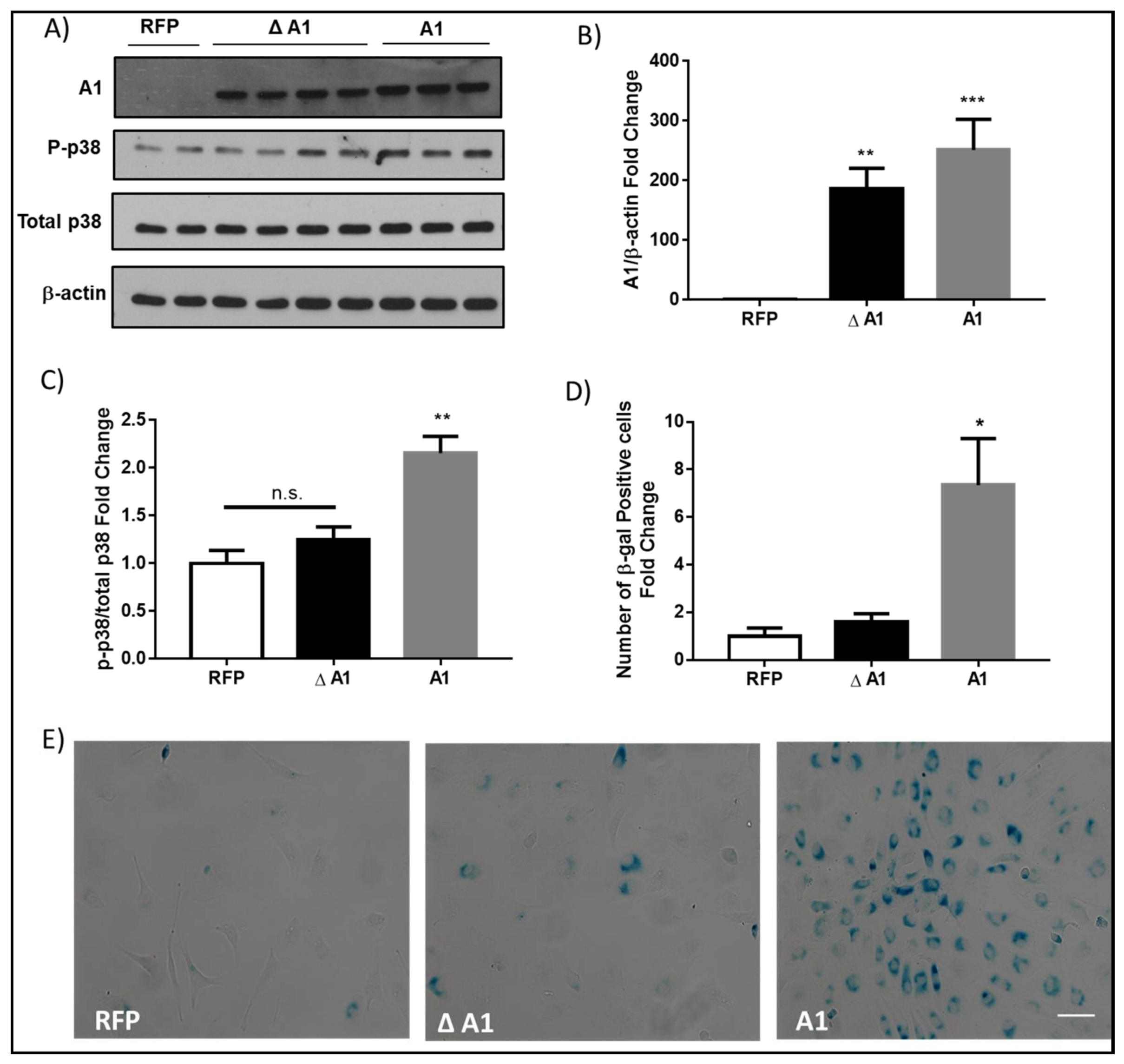
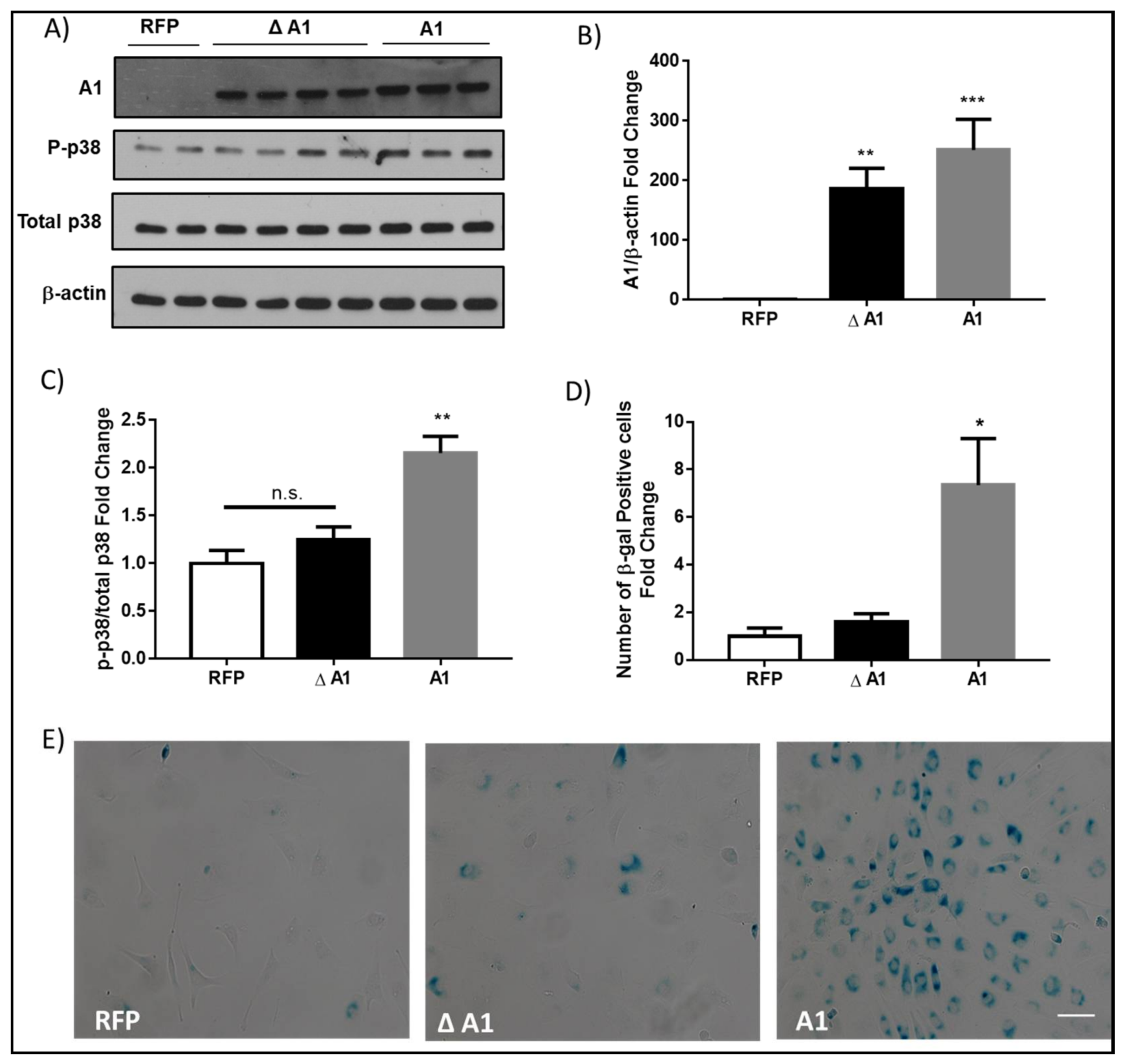
2.3. Induction of Endothelial Cell Senescence by High Glucose or Arginase 1 Overexpression and Blockade by Arginase Inhibition
3. Discussion
4. Materials and Methods
4.1. Animals and Diabetes Induction
4.2. Retinal Vessels Isolation
4.3. Cell Culture Studies
4.4. PCR Analyses
4.5. Western Blot
4.6. SA β-Galactosidase Activity
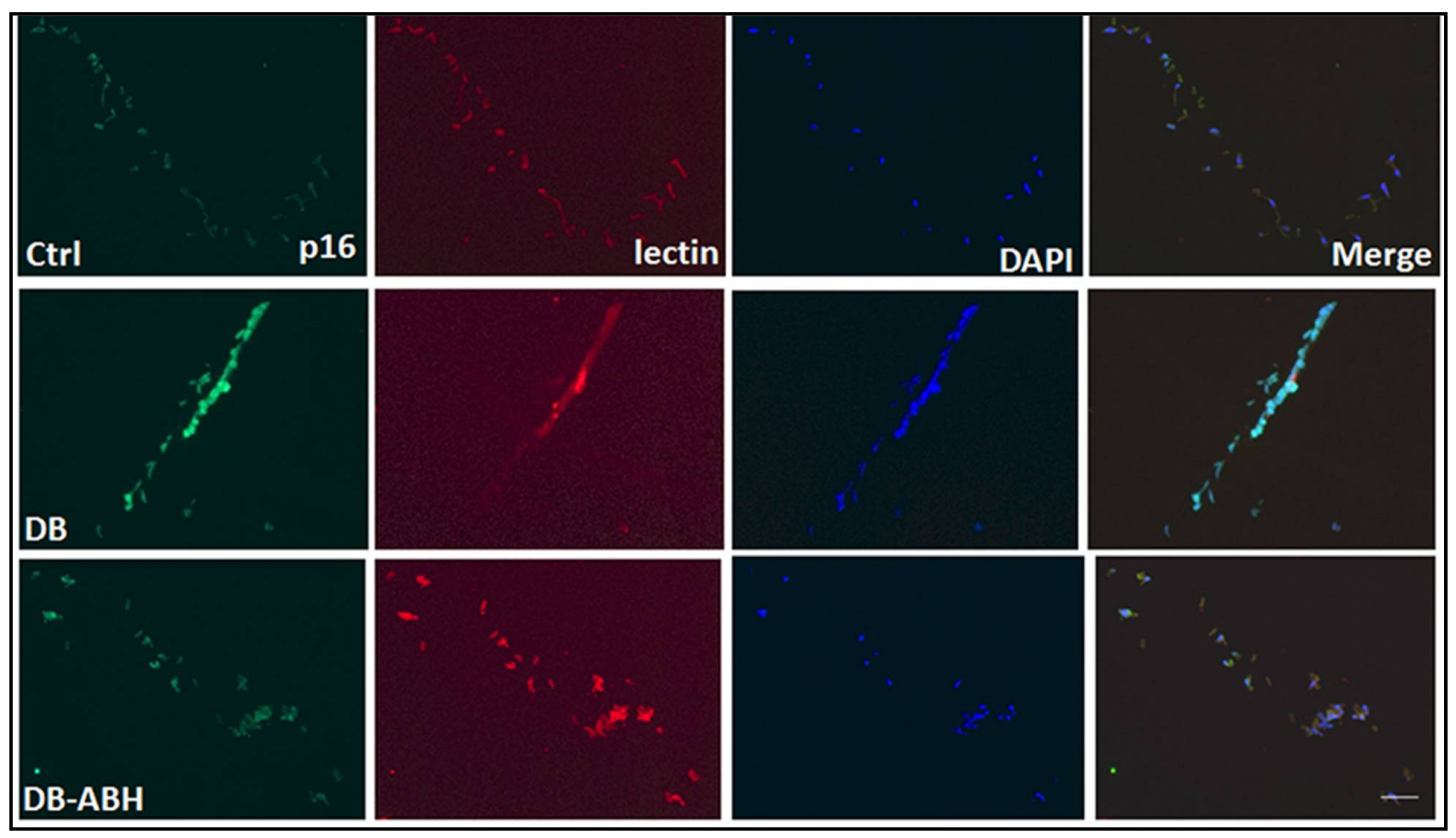
4.7. Immunofluorescence Analysis of Isolated Retinal Vessels
4.8. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| A1 | Arginase1 |
| A1+/− | A1 heterozygous knockout |
| A2 | Arginase 2 |
| ABH | [2(S)-Amino-6-boronohexanoic acid], arginase inhibitor |
| AGE | Advanced glycation end products |
| BRECs | Bovine retinal endothelial cells |
| BSA | Bovine serum albumin |
| DAPI | 4′,6′-diamino-2-phenylindole |
| ECs | Endothelial cells |
| FBS | Fetal bovine serum |
| HG | High glucose |
| HPRT | Hypoxanthine phosphoribosyl transferase |
| HUVECs | Human umbilical vein endothelial cells |
| Igfbp3 | Insulin-like growth factor binding protein-3 |
| MOI | Multiple of infections |
| NG | Normal glucose |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NOX2 | NADPH oxidase 2 |
| OCT | Optimal cutting temperature |
| P16INK4a | cyclin-dependent kinase inhibitor 2A |
| P21 | cyclin-dependent kinase inhibitor 1 |
| P53 | Tumor suppressor p53 |
| PBS | Phosphate-buffered Saline |
| RFP | Red fluorescence protein |
| SA β-Gal | Senescence associated β-galactosidase |
| STZ | Streptozotocin |
| WT | Wild type |
| WT Ctrl | Wild type control |
| WT DB | Wild type diabetic |
| Δ A1 | Inactive mutant arginase 1 |
References
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Lemtalsi, T.; Toque, H.A.; Xu, Z.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. NOX2-Induced Activation of Arginase and Diabetes-Induced Retinal Endothelial Cell Senescence. Antioxidants 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Lamoke, F.; Shaw, S.; Yuan, J.; Ananth, S.; Duncan, M.; Martin, P.; Bartoli, M. Increased Oxidative and Nitrative Stress Accelerates Aging of the Retinal Vasculature in the Diabetic Retina. PLoS ONE 2015, 10, e0139664. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Rojas, M.; Narayanan, S.P.; Zhang, W.; Xu, Z.; Lemtalsi, T.; Jittiporn, K.; Caldwell, R.W.; Caldwell, R.B. Arginase as a mediator of diabetic retinopathy. Front. Immunol. 2013, 4, 173. [Google Scholar] [CrossRef] [PubMed]
- Elms, S.C.; Toque, H.A.; Rojas, M.; Xu, Z.; Caldwell, R.W.; Caldwell, R.B. The role of arginase I in diabetes-induced retinal vascular dysfunction in mouse and rat models of diabetes. Diabetologia 2013, 56, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.P.; Suwanpradid, J.; Saul, A.; Xu, Z.; Still, A.; Caldwell, R.W.; Caldwell, R.B. Arginase 2 deletion reduces neuro-glial injury and improves retinal function in a model of retinopathy of prematurity. PLoS ONE 2011, 6, e22460. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.P.; Xu, Z.; Putluri, N.; Sreekumar, A.; Lemtalsi, T.; Caldwell, R.W.; Caldwell, R.B. Arginase 2 deficiency reduces hyperoxia-mediated retinal neurodegeneration through the regulation of polyamine metabolism. Cell Death Dis. 2014, 5, e1075. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.J.; Platt, D.H.; Tawfik, H.E.; Labazi, M.; El-Remessy, A.B.; Bartoli, M.; Caldwell, R.B.; Caldwell, R.W. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ. Res. 2008, 102, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Shosha, E.; Xu, Z.; Yokota, H.; Saul, A.; Rojas, M.; Caldwell, R.W.; Caldwell, R.B.; Narayanan, S.P. Arginase 2 promotes neurovascular degeneration during ischemia/reperfusion injury. Cell Death Dis. 2016, 7, e2483. [Google Scholar] [CrossRef] [PubMed]
- Suwanpradid, J.; Rojas, M.; Behzadian, M.A.; Caldwell, R.W.; Caldwell, R.B. Arginase 2 deficiency prevents oxidative stress and limits hyperoxia-induced retinal vascular degeneration. PLoS ONE 2014, 9, e110604. [Google Scholar] [CrossRef] [PubMed]
- Toque, H.A.; Nunes, K.P.; Yao, L.; Xu, Z.; Kondrikov, D.; Su, Y.; Webb, R.C.; Caldwell, R.B.; Caldwell, R.W. Akita spontaneously type 1 diabetic mice exhibit elevated vascular arginase and impaired vascular endothelial and nitrergic function. PLoS ONE 2013, 8, e72277. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Romero, M.J.; Shatanawi, A.; Alkilany, A.M.; Caldwell, R.B.; Caldwell, R.W. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br. J. Pharmacol. 2012, 165, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Kim, M.S.; Seu, Y.B.; Chung, H.Y.; Kim, J.H.; Kim, J.R. Regulation of replicative senescence by insulin-like growth factor-binding protein 3 in human umbilical vein endothelial cells. Aging Cell 2007, 6, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Benatar, T.; Yang, W.; Amemiya, Y.; Evdokimova, V.; Kahn, H.; Holloway, C.; Seth, A. IGFBP7 reduces breast tumor growth by induction of senescence and apoptosis pathways. Breast Cancer Res. Treat. 2012, 133, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Shuang, E.; Kijima, R.; Honma, T.; Yamamoto, K.; Hatakeyama, Y.; Kitano, Y.; Kimura, T.; Nakagawa, K.; Miyazawa, T.; Tsuduki, T. 1-Deoxynojirimycin attenuates high glucose-accelerated senescence in human umbilical vein endothelial cells. Exp. Gerontol. 2014, 55, 63–69. [Google Scholar]
- Senthil, K.K.J.; Gokila, V.M.; Wang, S.Y. Activation of Nrf2-mediated anti-oxidant genes by antrodin C prevents hyperglycemia-induced senescence and apoptosis in human endothelial cells. Oncotarget 2017, 8, 96568–96587. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.G.; Tabor, D.E.; Kern, R.M.; Goodman, B.K.; Wissmann, P.B.; Kang, D.S.; Grody, W.W.; Cederbaum, S.D. Identification of mutations (D128G, H141L) in the liver arginase gene of patients with hyperargininemia. Hum. Mutat. 1994, 4, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Elms, S.; Chen, F.; Wang, Y.; Qian, J.; Askari, B.; Yu, Y.; Pandey, D.; Iddings, J.; Caldwell, R.B.; Fulton, D.J. Insights into the arginine paradox: Evidence against the importance of subcellular location of arginase and eNOS. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H651–H666. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.P.; Rojas, M.; Suwanpradid, J.; Toque, H.A.; Caldwell, R.W.; Caldwell, R.B. Arginase in retinopathy. Prog. Retin. Eye Res. 2013, 36, 260–280. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.J.; Iddings, J.A.; Platt, D.H.; Ali, M.I.; Cederbaum, S.D.; Stepp, D.W.; Caldwell, R.B.; Caldwell, R.W. Diabetes-induced vascular dysfunction involves arginase I. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H159–H166. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Chandra, S.; Toque, H.A.; Bhatta, A.; Rojas, M.; Caldwell, R.B.; Caldwell, R.W. Prevention of diabetes-induced arginase activation and vascular dysfunction by Rho kinase (ROCK) knockout. Cardiovasc. Res. 2013, 97, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, A.; Yao, L.; Xu, Z.; Toque, H.A.; Chen, J.; Atawia, R.T.; Fouda, A.Y.; Bagi, Z.; Lucas, R.; Caldwell, R.B.; et al. Obesity-induced vascular dysfunction and arterial stiffening requires endothelial cell arginase 1. Cardiovasc. Res. 2017, 113, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Beleznai, T.; Feher, A.; Spielvogel, D.; Lansman, S.L.; Bagi, Z. Arginase 1 contributes to diminished coronary arteriolar dilation in patients with diabetes. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H777–H783. [Google Scholar] [CrossRef] [PubMed]
- El-Bassossy, H.M.; El-Fawal, R.; Fahmy, A. Arginase inhibition alleviates hypertension associated with diabetes: Effect on endothelial dependent relaxation and NO production. Vasc. Pharmacol. 2012, 57, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Gronros, J.; Jung, C.; Lundberg, J.O.; Cerrato, R.; Ostenson, C.G.; Pernow, J. Arginase inhibition restores in vivo coronary microvascular function in type 2 diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1174–H1181. [Google Scholar] [CrossRef] [PubMed]
- Kovamees, O.; Shemyakin, A.; Checa, A.; Wheelock, C.E.; Lundberg, J.O.; Ostenson, C.G.; Pernow, J. Arginase Inhibition Improves Microvascular Endothelial Function in Patients With Type 2 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2016, 101, 3952–3958. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Gao, T.; Cooper, T.K.; Morris, S.M., Jr.; Awad, A.S. Arginase inhibition mediates renal tissue protection in diabetic nephropathy by a nitric oxide synthase 3-dependent mechanism. Kidney Int. 2013, 84, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Toque, H.A.; Nunes, K.P.; Yao, L.; Liao, J.K.; Webb, R.C.; Caldwell, R.B.; Caldwell, R.W. Activated Rho kinase mediates diabetes-induced elevation of vascular arginase activation and contributes to impaired corpora cavernosa relaxation: Possible involvement of p38 MAPK activation. J. Sex. Med. 2013, 10, 1502–1515. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Gao, T.; Cooper, T.K.; Morris, S.M., Jr.; Awad, A.S. Arginase inhibition: A new treatment for preventing progression of established diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2015, 309, F447–F455. [Google Scholar] [CrossRef] [PubMed]
- Shatanawi, A.; Romero, M.J.; Iddings, J.A.; Chandra, S.; Umapathy, N.S.; Verin, A.D.; Caldwell, R.B.; Caldwell, R.W. Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am. J. Physiol. Cell Physiol. 2011, 300, C1181–C1192. [Google Scholar] [CrossRef] [PubMed]
- Shatanawi, A.; Lemtalsi, T.; Yao, L.; Patel, C.; Caldwell, R.B.; Caldwell, R.W. Angiotensin II limits NO production by upregulating arginase through a p38 MAPK-ATF-2 pathway. Eur. J. Pharmacol. 2015, 746, 106–114. [Google Scholar] [CrossRef] [PubMed]
- De Magalhaes, J.P.; Passos, J.F. Stress, cell senescence and organismal ageing. Mech. Ageing Dev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, T.; Fukuo, K.; Yasuda, O.; Hotta, M.; Miyazaki, J.; Takemura, Y.; Kawamoto, H.; Ichijo, H.; Ogihara, T. Apoptosis signal-regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes 2006, 55, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Ma, X.; Liu, J.; Fan, Y.; Deng, X. High glucose-induced endothelial progenitor cell dysfunction. Diabetes Vasc. Dis. Res. 2017, 14, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yu, Y.; Montani, J.P.; Ming, X.F.; Yang, Z. Arginase-I enhances vascular endothelial inflammation and senescence through eNOS-uncoupling. BMC Res. Notes 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Zhang, W.; Romero, M.J.; Caldwell, R.W. Vascular dysfunction in retinopathy-an emerging role for arginase. Brain Res. Bull. 2010, 81, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.V.; Gealekman, O.; Chen, J.; Zhang, F.; Togashi, N.; Crabtree, M.; Gross, S.S.; Nasjletti, A.; Goligorsky, M.S. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ. Res. 2004, 94, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yu, Y.; Montani, J.P.; Yang, Z.; Ming, X.F. Arginase-II induces vascular smooth muscle cell senescence and apoptosis through p66Shc and p53 independently of its l-arginine ureahydrolase activity: Implications for atherosclerotic plaque vulnerability. J. Am. Heart Assoc. 2013, 2, e000096. [Google Scholar] [CrossRef] [PubMed]
- Yepuri, G.; Velagapudi, S.; Xiong, Y.; Rajapakse, A.G.; Montani, J.P.; Ming, X.F.; Yang, Z. Positive crosstalk between arginase-II and S6K1 in vascular endothelial inflammation and aging. Aging Cell 2012, 11, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Behzadian, M.A.; Wang, X.L.; Jiang, B.; Caldwell, R.B. Angiostatic role of astrocytes: Suppression of vascular endothelial cell growth by TGF-beta and other inhibitory factor(s). Glia 1995, 15, 480–490. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shosha, E.; Xu, Z.; Narayanan, S.P.; Lemtalsi, T.; Fouda, A.Y.; Rojas, M.; Xing, J.; Fulton, D.; Caldwell, R.W.; Caldwell, R.B. Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1. Int. J. Mol. Sci. 2018, 19, 1215. https://doi.org/10.3390/ijms19041215
Shosha E, Xu Z, Narayanan SP, Lemtalsi T, Fouda AY, Rojas M, Xing J, Fulton D, Caldwell RW, Caldwell RB. Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1. International Journal of Molecular Sciences. 2018; 19(4):1215. https://doi.org/10.3390/ijms19041215
Chicago/Turabian StyleShosha, Esraa, Zhimin Xu, S. Priya Narayanan, Tahira Lemtalsi, Abdelrahman Y. Fouda, Modesto Rojas, Ji Xing, David Fulton, R. William Caldwell, and Ruth B. Caldwell. 2018. "Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1" International Journal of Molecular Sciences 19, no. 4: 1215. https://doi.org/10.3390/ijms19041215