Differential Expression and Clinical Significance of Transforming Growth Factor-Beta Isoforms in GBM Tumors



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
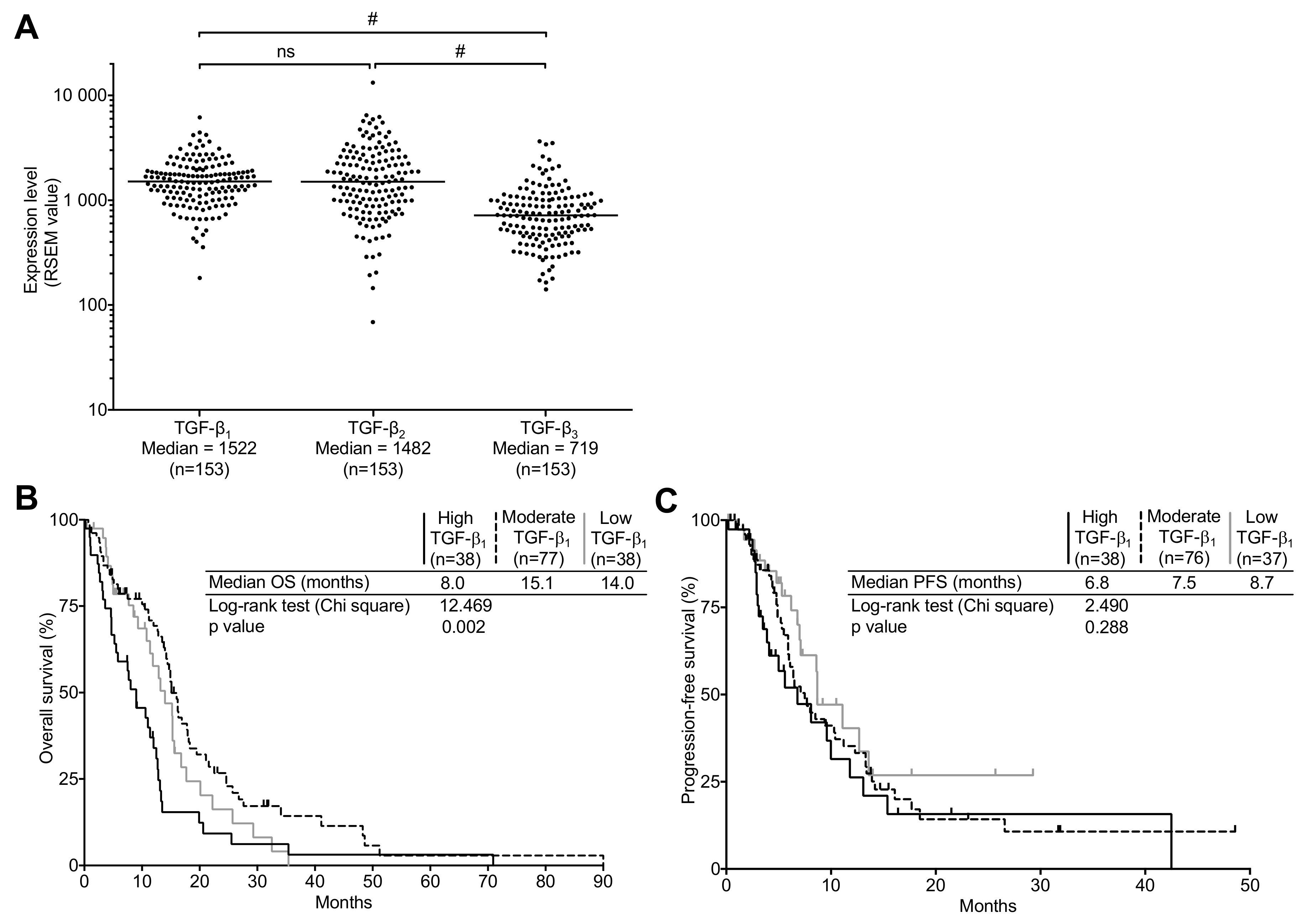
2.1. TGF-β Expression in the TCGA Cohort
2.2. Patient Characteristics
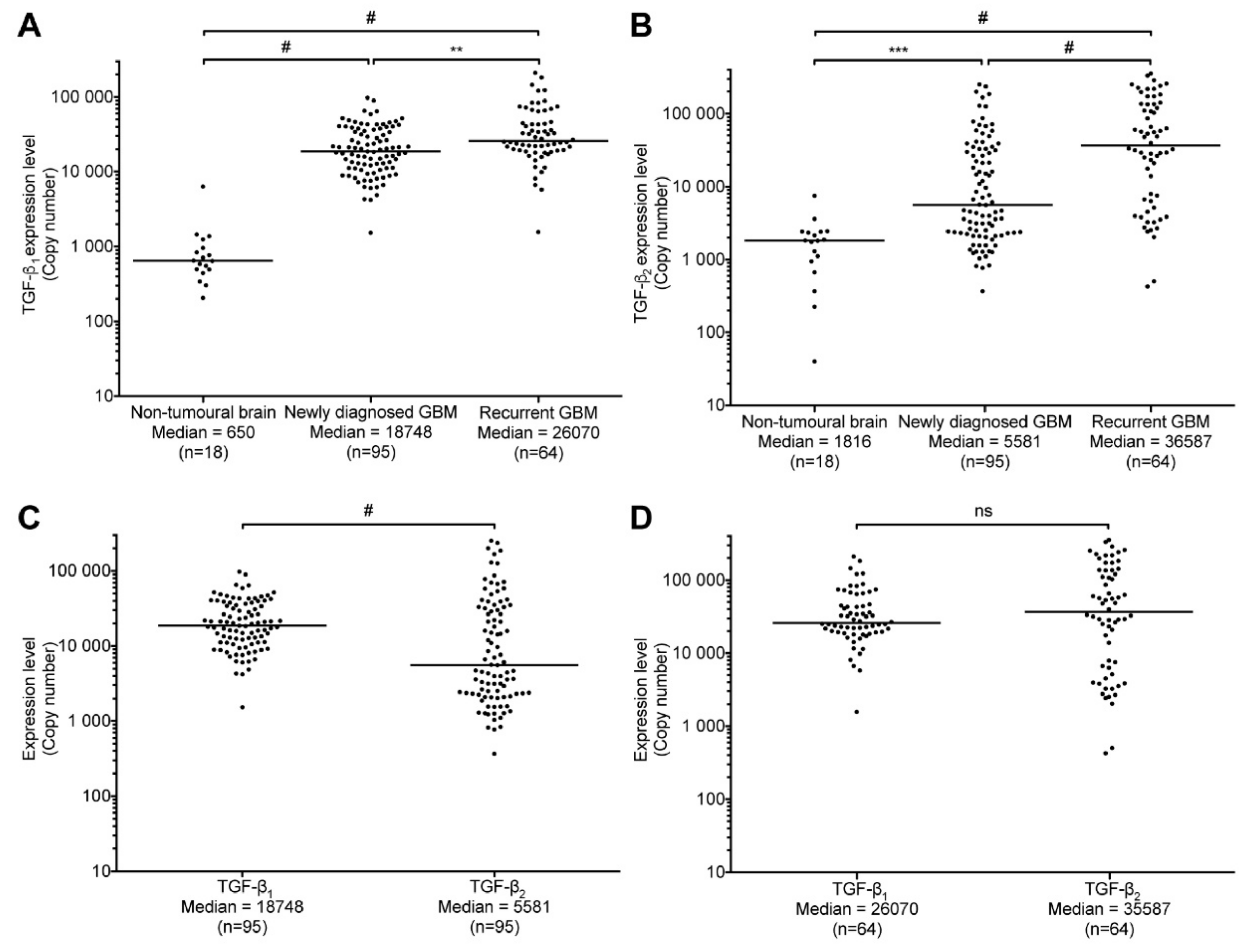
2.3. TGF-β1 and -β2 Expression in GBM Patients Compared to Non-Tumoral Brain Samples
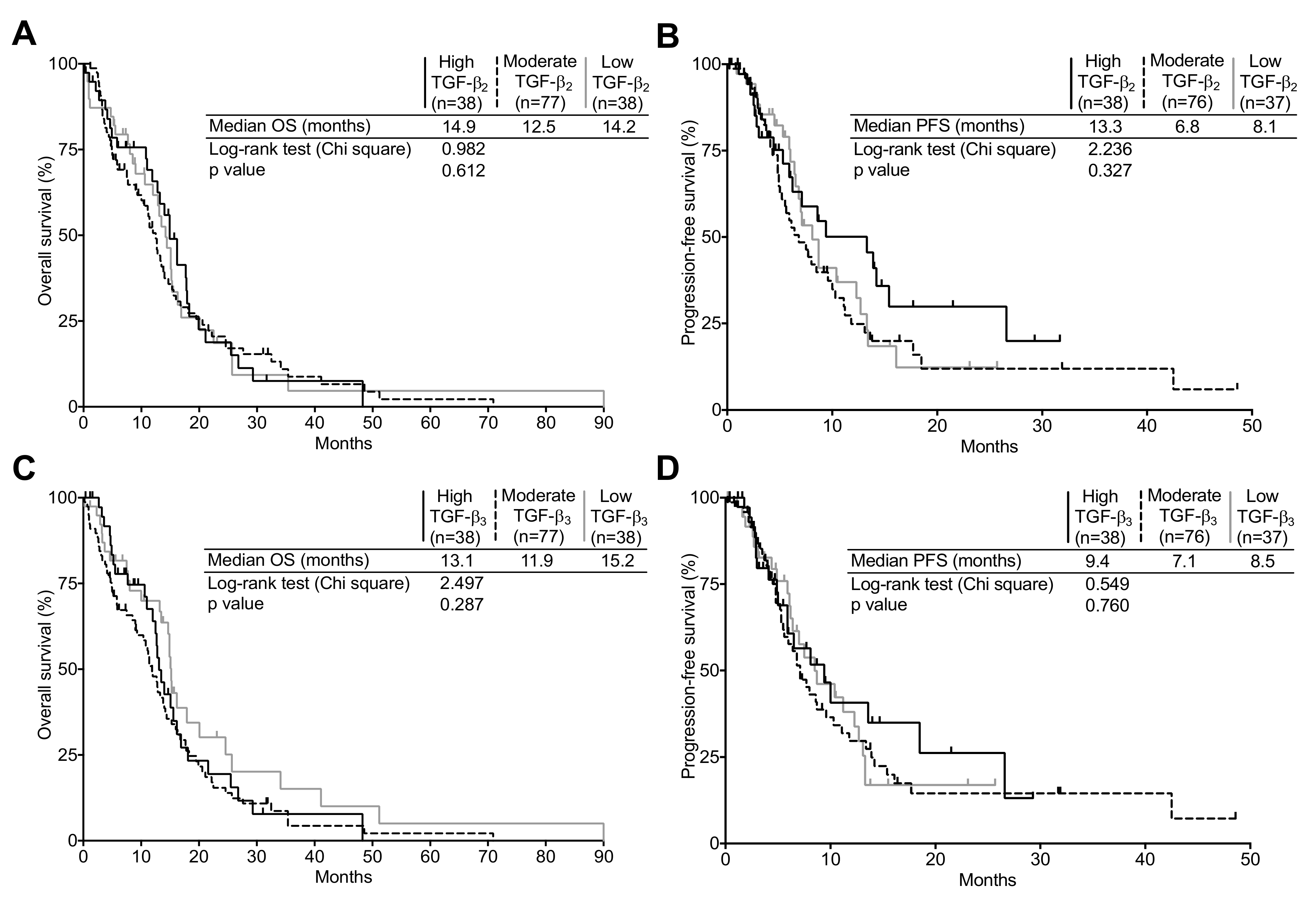
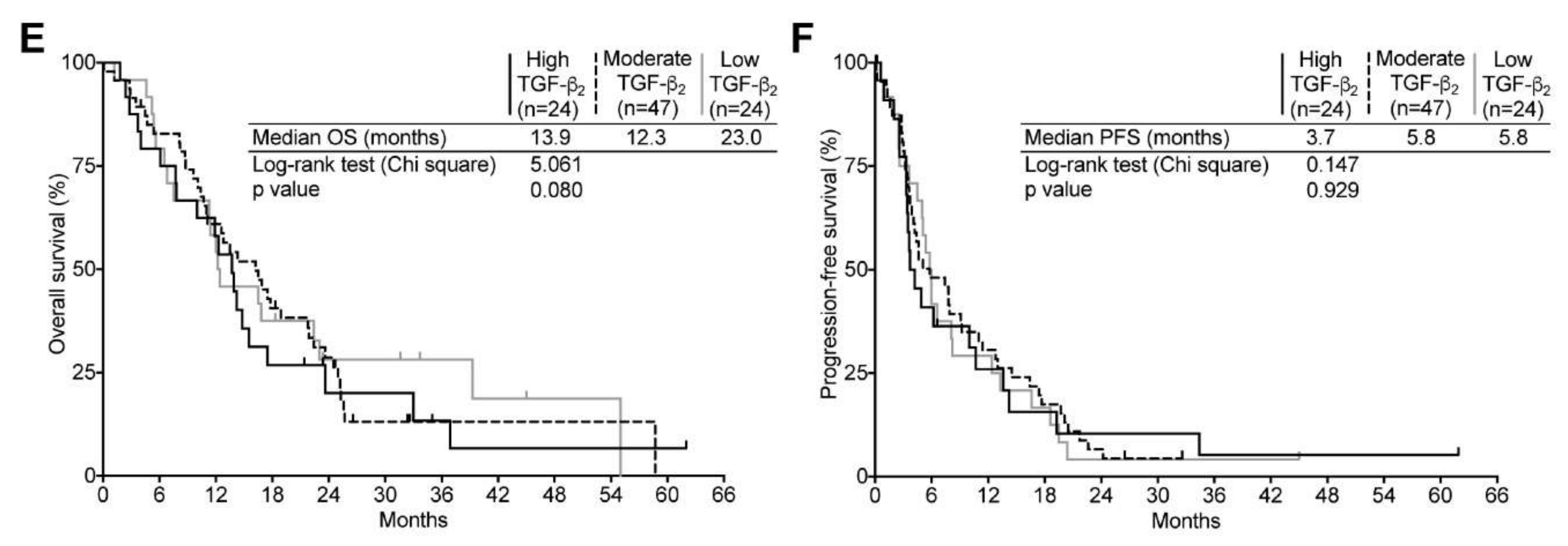
2.4. TGF-β Expression and Its Relation to Clinical Surrogates in Newly Diagnosed GBM
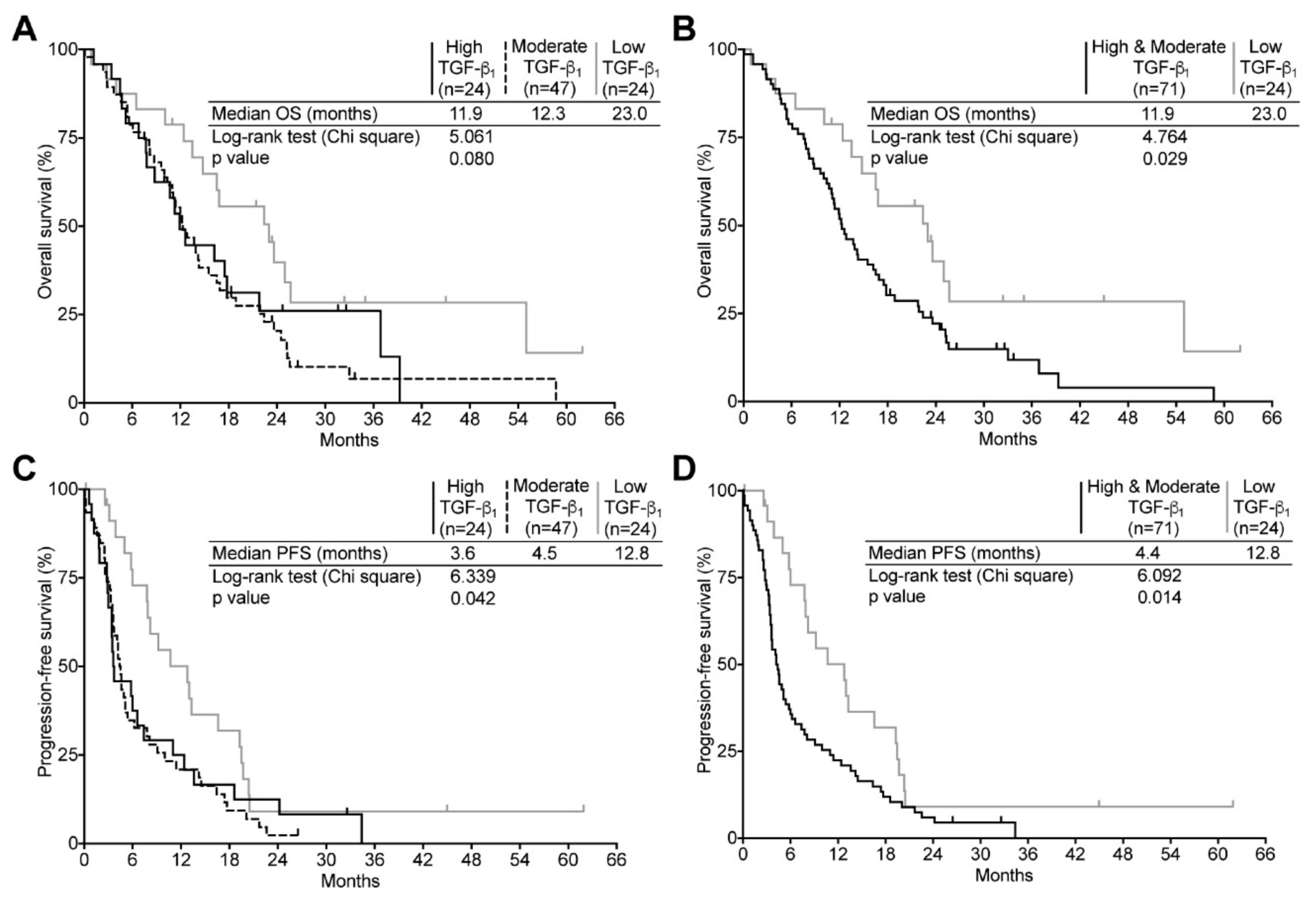
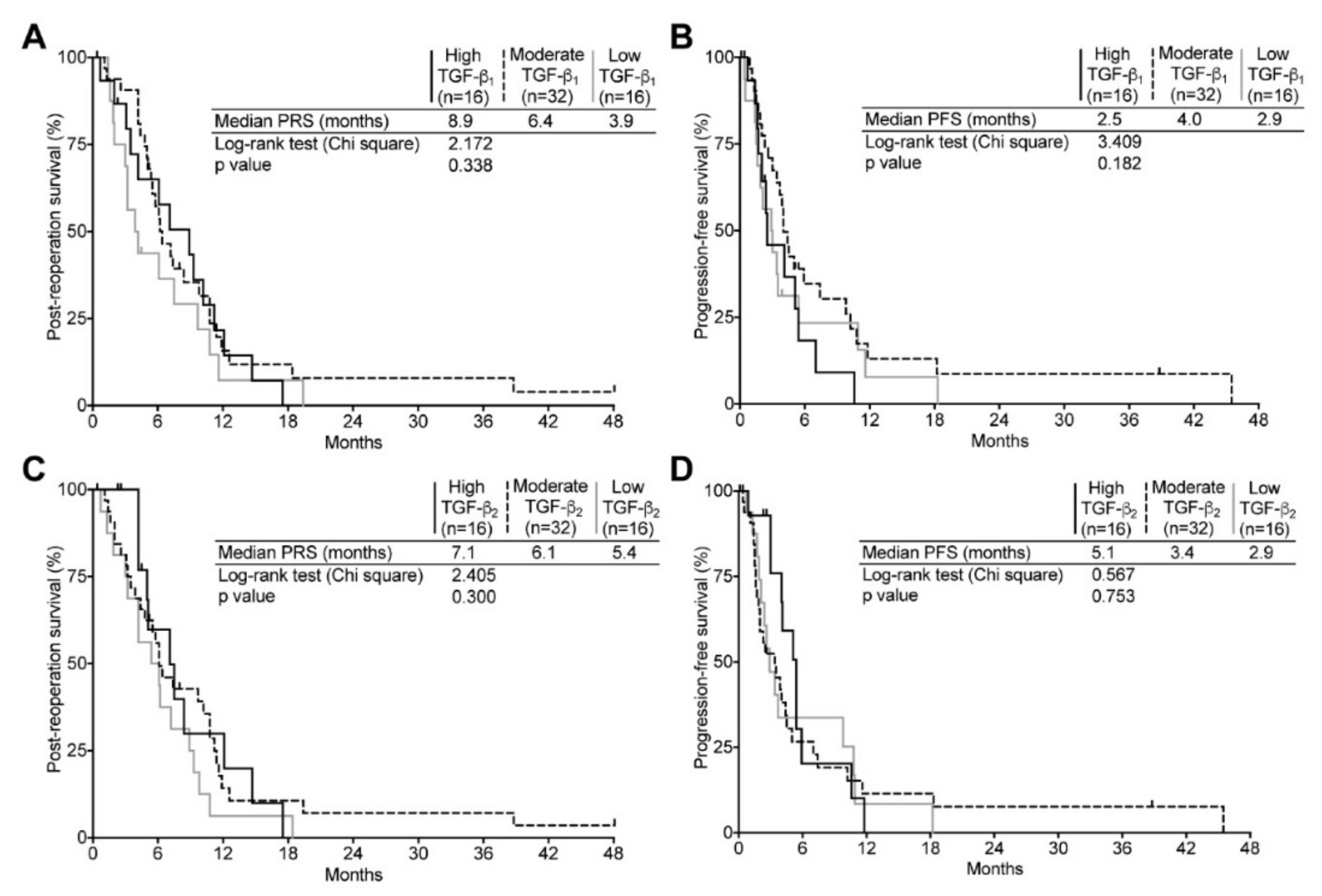
2.5. TGF-β Expression and Its Relation to Clinical Surrogates in Recurrent GBM
3. Discussion
4. Materials and Methods
4.1. TCGA Data Analysis
4.2. Tumor Specimens Acquisition and Diagnosis
4.3. Non-Tumoral Tissue
4.4. RNA Extraction and Quality Assessment
4.5. Reverse Transcription and Real-Time Polymerase Chain Reaction (qPCR)
4.6. Statistics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TGF-β | Transforming growth factor-beta |
| GBM | Glioblastoma |
| qPCR | Quantitative real-time polymerase chain reaction |
| OS | Overall survival |
| PRS | Post-reoperation survival |
| PFS | Progression-free survival |
References
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2005–2009. Neuro Oncol. 2012, 14 (Suppl 5), v1–v49. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells-much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Hegi, M.E.; Neyns, B.; Goldbrunner, R.; Schlegel, U.; Clement, P.M.J.; Grabenbauer, G.G.; Ochsenbein, A.F.; Simon, M.; Dietrich, P.-Y.; et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 2712–2718. [Google Scholar] [CrossRef] [PubMed]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.E.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.J.; Buter, J.; Honkoop, A.H.; Boerman, D.; de Vos, F.Y.F.; et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Field, K.M.; Jordan, J.T.; Wen, P.Y.; Rosenthal, M.A.; Reardon, D.A. Bevacizumab and glioblastoma: Scientific review, newly reported updates, and ongoing controversies. Cancer 2015, 121, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Peereboom, D.M.; Ahluwalia, M.S.; Ye, X.; Supko, J.G.; Hilderbrand, S.L.; Phuphanich, S.; Nabors, L.B.; Rosenfeld, M.R.; Mikkelsen, T.; Grossman, S.A. NABTT 0502: A phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. 2013, 15, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Nabors, L.B.; Gorlia, T.; Leske, H.; Rushing, E.; Bady, P.; Hicking, C.; Perry, J.; Hong, Y.-K.; Roth, P.; et al. Cilengitide in newly diagnosed glioblastoma: Biomarker expression and outcome. Oncotarget 2016, 7, 15018–15032. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.-O.; Poirier, M.-B.; Fortin, D. Transforming growth factor-beta and its implication in the malignancy of gliomas. Target. Oncol. 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Peñuelas, S.; Anido, J.; Prieto-Sanchez, R.M.; Folch, G.; Barba, I.; Cuartas, I.; GarcIa-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. TGF-beta Increases Glioma-Initiating Cell Self-Renewal through the Induction of LIF in Human Glioblastoma. Cancer Cell 2009, 15, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-beta. J. Neurooncol. 2001, 53, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Pepper, M.S. Transforming growth factor-beta: Vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997, 8, 21–43. [Google Scholar] [CrossRef]
- Ahuja, S.S.; Paliogianni, F.; Yamada, H.; Balow, J.E.; Boumpas, D.T. Effect of transforming growth factor-beta on early and late activation events in human T cells. J. Immunol. 1993, 150, 3109–3118. [Google Scholar] [PubMed]
- Hardee, M.E.; Marciscano, A.E.; Medina-Ramirez, C.M.; Zagzag, D.; Narayana, A.; Lonning, S.M.; Barcellos-Hoff, M.H. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-β. Cancer Res. 2012, 72, 4119–4129. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Kato, M.; Yamashita, H.; Nistér, M.; Miyazono, K.; Heldin, C.-H.; Funa, K. Enhanced expression of transforming growth factor-β and its type-I and type-II receptors in human glioblastoma. Int. J. Cancer 1995, 62, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Kjellman, C.; Olofsson, S.P.; Hansson, O.; Von Schantz, T.; Lindvall, M.; Nilsson, I.; Salford, L.G.; Sjögren, H.O.; Widegren, B. Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. Int. J. Cancer 2000, 89, 251–258. [Google Scholar] [CrossRef]
- Bayin, N.S.; Ma, L.; Thomas, C.; Baitalmal, R.; Sure, A.; Fansiwala, K.; Bustoros, M.; Golfinos, J.G.; Pacione, D.; Snuderl, M.; et al. Patient-Specific Screening Using High-Grade Glioma Explants to Determine Potential Radiosensitization by a TGF-β Small Molecule Inhibitor. Neoplasia 2016, 18, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Rodón, L.; Gonzàlez-Juncà, A.; del Mar Inda, M.; Sala-Hojman, A.; Martínez-Sáez, E.; Seoane, J. Active CREB1 Promotes a Malignant TGFβ2 Autocrine Loop in Glioblastoma. Cancer Discov 2014, 4, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocaña, A.; Peñuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFβ-Smad Activity Confers Poor Prognosis in Glioma Patients and Promotes Cell Proliferation Depending on the Methylation of the PDGF-B Gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Frei, K.; Gramatzki, D.; Tritschler, I.; Schroeder, J.J.; Espinoza, L.; Rushing, E.J.; Weller, M. Transforming growth factor-β pathway activity in glioblastoma. Oncotarget 2015, 6, 5963–5977. [Google Scholar] [CrossRef] [PubMed]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the tgf-β2 inhibitor trabedersen: Results of a randomized and controlled phase iib study. Neuro Oncol. 2011, 13, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Sanchez, J.; Ramos, A.; Hilario, A.; De Velasco, G.; Castellano, D.; Garcia De La Torre, M.; Rodón, J.; Lahn, M.F. Brain perfusion and permeability in patients with advanced, refractory glioblastoma treated with lomustine and the transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate. Oncol. Lett. 2015, 9, 2442–2448. [Google Scholar] [CrossRef] [PubMed]
- Gadji, M.; Fortin, D.; Tsanaclis, A.M.; Garini, Y.; Katzir, N.; Wienburg, Y.; Yan, J.; Klewes, L.; Klonisch, T.; Drouin, R.; Mai, S. Three-dimensional nuclear telomere architecture is associated with differential time to progression and overall survival in glioblastoma patients. Neoplasia 2010, 12, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Desmarais, G.; Fortin, D.; Bujold, R.; Wagner, R.; Mathieu, D.; Paquette, B. Infiltration of glioma cells in brain parenchyma stimulated by radiation in the F98/Fischer rat model. Int. J. Radiat. Biol. 2012, 88, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.-O.; Poirier, M.-B.; Fortin, D. Chloroquine inhibits the malignant phenotype of glioblastoma partially by suppressing TGF-beta. Investig. New Drugs 2015, 33, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Jobling, M.F.; Mott, J.D.; Finnegan, M.T.; Jurukovski, V.; Erickson, A.C.; Walian, P.J.; Taylor, S.E.; Ledbetter, S.; Lawrence, C.M.; Rifkin, D.B.; et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat. Res. 2006, 166, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Gatherer, D.; Ten Dijke, P.; Baird, D.T.; Akhurst, R.J. Expression of TGF-beta isoforms during first trimester human embryogenesis. Development 1990, 110, 445–460. [Google Scholar] [PubMed]
- Saharinen, J.; Hyytiäinen, M.; Taipale, J.; Keski-Oja, J. Latent transforming growth factor-beta binding proteins (LTBPs)—Structural extracellular matrix proteins for targeting TGF-beta action. Cytokine Growth Factor Rev. 1999, 10, 99–117. [Google Scholar] [CrossRef]
- Seystahl, K.; Papachristodoulou, A.; Burghardt, I.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P.; Weller, M. Biological Role and Therapeutic Targeting of TGF-β3 in Glioblastoma. Mol. Cancer Ther. 2017, 16, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, M.C.; Mitchell, H.; Penheiter, S.G.; Doré, J.J.; Suzuki, K.; Edens, M.; Sharma, D.K.; Pagano, R.E.; Leof, E.B. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 2005, 65, 10431–10440. [Google Scholar] [CrossRef] [PubMed]
- Edlund, S.; Landström, M.; Heldin, C.-H.; Aspenström, P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Hakami, N.Y.; Wong, H.; Shah, M.H.; Dusting, G.J.; Jiang, F.; Peshavariya, H.M. Smad-independent pathway involved in transforming growth factor β1-induced Nox4 expression and proliferation of endothelial cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 388, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Weller, M. Trabedersen to target transforming growth factor-β: When the journey is not the reward, in reference to Bogdahn et al. (Neuro-Oncology 2011; 13: 132–142). Neuro. Oncol. 2011, 13, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Basque, J.; Martel, M.; Leduc, R.; Cantin, A.M. Lysosomotropic drugs inhibit maturation of transforming growth factor-β. Can. J. Physiol. Pharmacol. 2008, 86, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, I.; Tritschler, F.; Opitz, C.A.; Frank, B.; Weller, M.; Wick, W. Pirfenidone inhibits TGF-beta expression in malignant glioma cells. Biochem. Biophys. Res. Commun. 2007, 354, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Skuli, N.; Karpel-Massler, G.; Frosina, G.; Ryken, T.; Halatsch, M.-E. Blocking epithelial-to-mesenchymal transition in glioblastoma with a sextet of repurposed drugs: The EIS regimen. Oncotarget 2017, 8, 60727–60749. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed]
- Bender, R.; Lange, S. Adjusting for multiple testing—When and how? J. Clin. Epidemiol. 2001, 54, 343–349. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, L.-O.; Poirier, M.-B.; Fortin, D. Differential Expression and Clinical Significance of Transforming Growth Factor-Beta Isoforms in GBM Tumors. Int. J. Mol. Sci. 2018, 19, 1113. https://doi.org/10.3390/ijms19041113
Roy L-O, Poirier M-B, Fortin D. Differential Expression and Clinical Significance of Transforming Growth Factor-Beta Isoforms in GBM Tumors. International Journal of Molecular Sciences. 2018; 19(4):1113. https://doi.org/10.3390/ijms19041113
Chicago/Turabian StyleRoy, Laurent-Olivier, Marie-Belle Poirier, and David Fortin. 2018. "Differential Expression and Clinical Significance of Transforming Growth Factor-Beta Isoforms in GBM Tumors" International Journal of Molecular Sciences 19, no. 4: 1113. https://doi.org/10.3390/ijms19041113
APA StyleRoy, L.-O., Poirier, M.-B., & Fortin, D. (2018). Differential Expression and Clinical Significance of Transforming Growth Factor-Beta Isoforms in GBM Tumors. International Journal of Molecular Sciences, 19(4), 1113. https://doi.org/10.3390/ijms19041113