EP4 as a Therapeutic Target for Aggressive Human Breast Cancer



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Breast Cancer: Needs to Identify Novel Therapeutic Targets
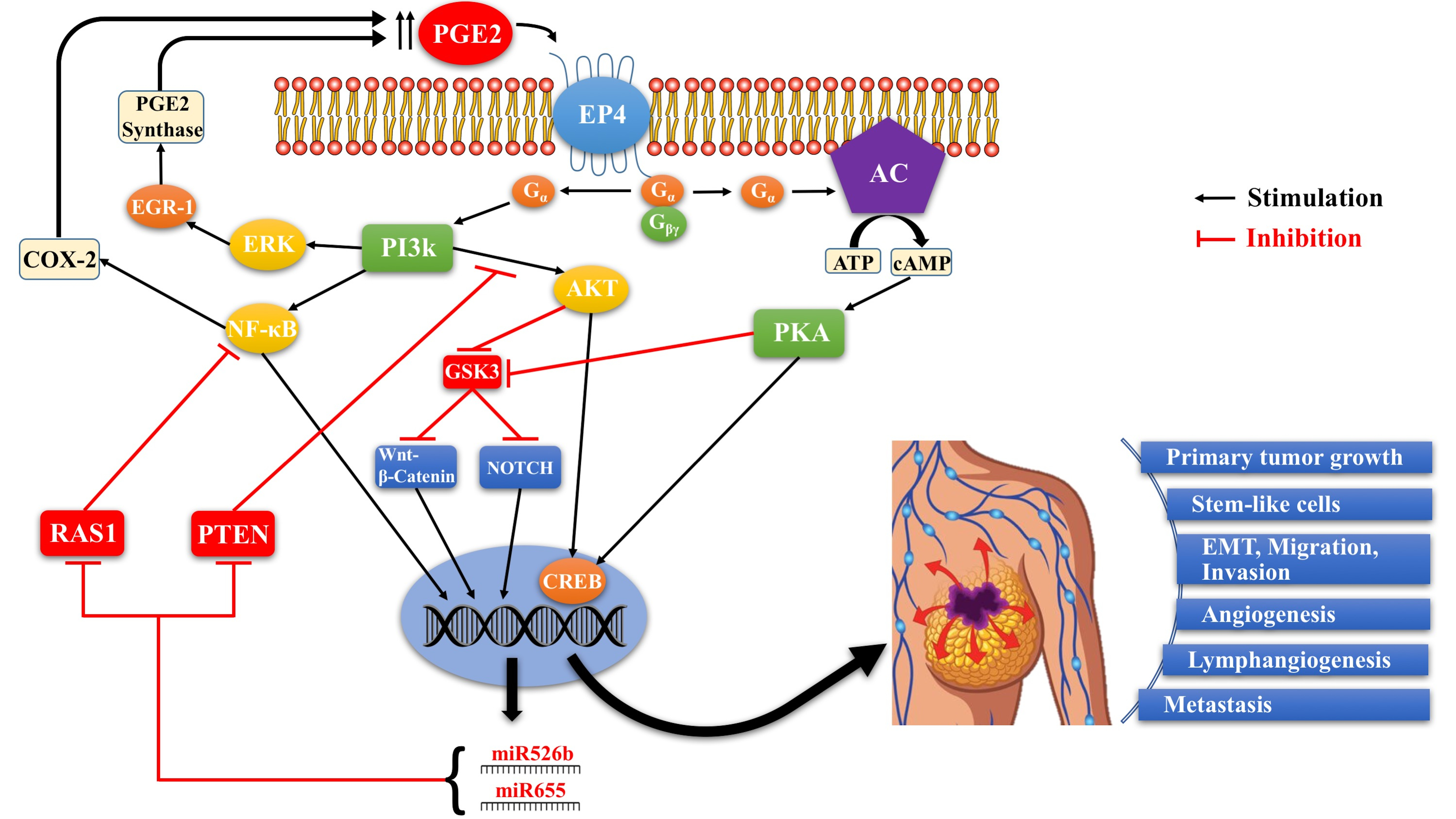
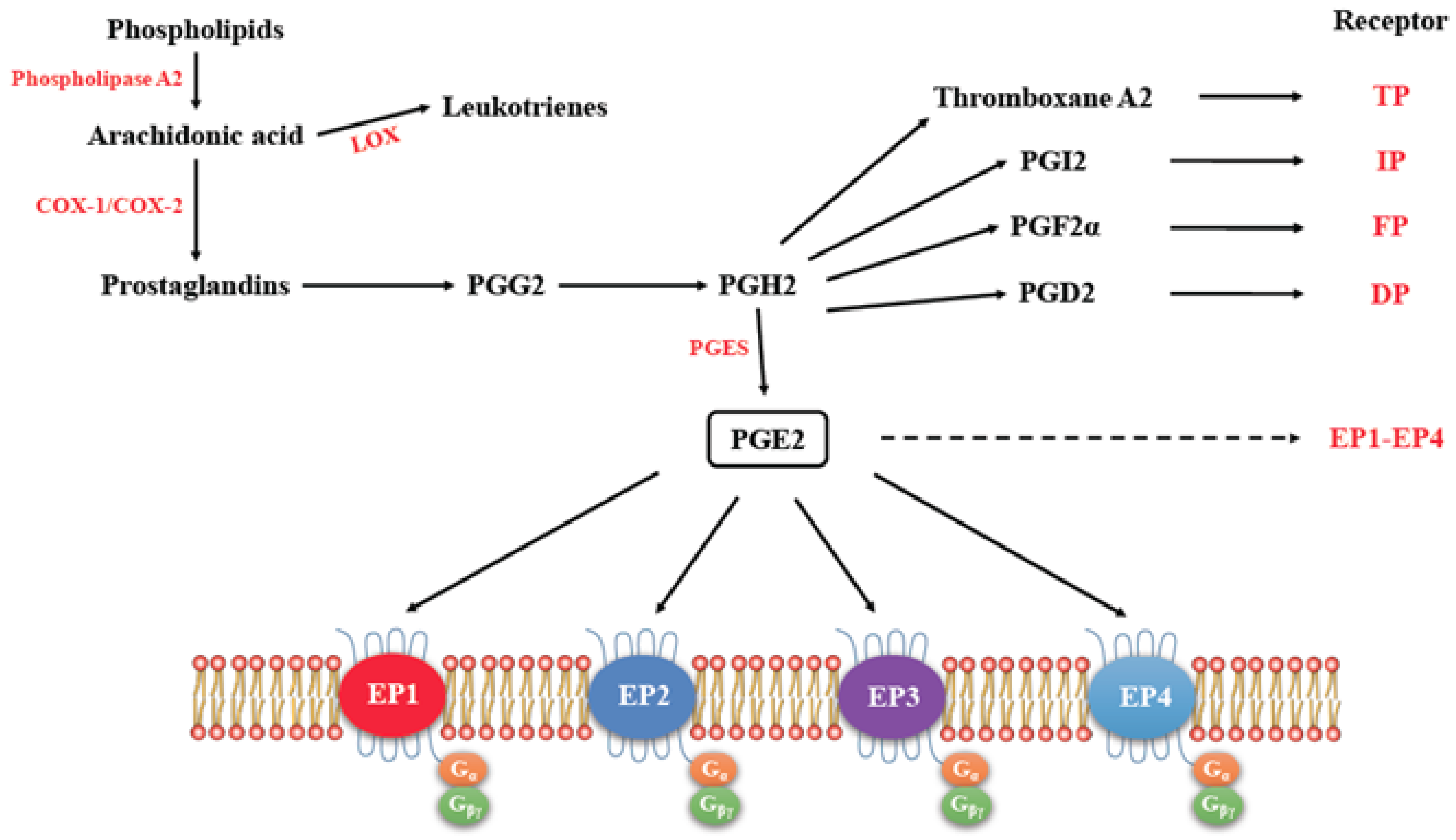
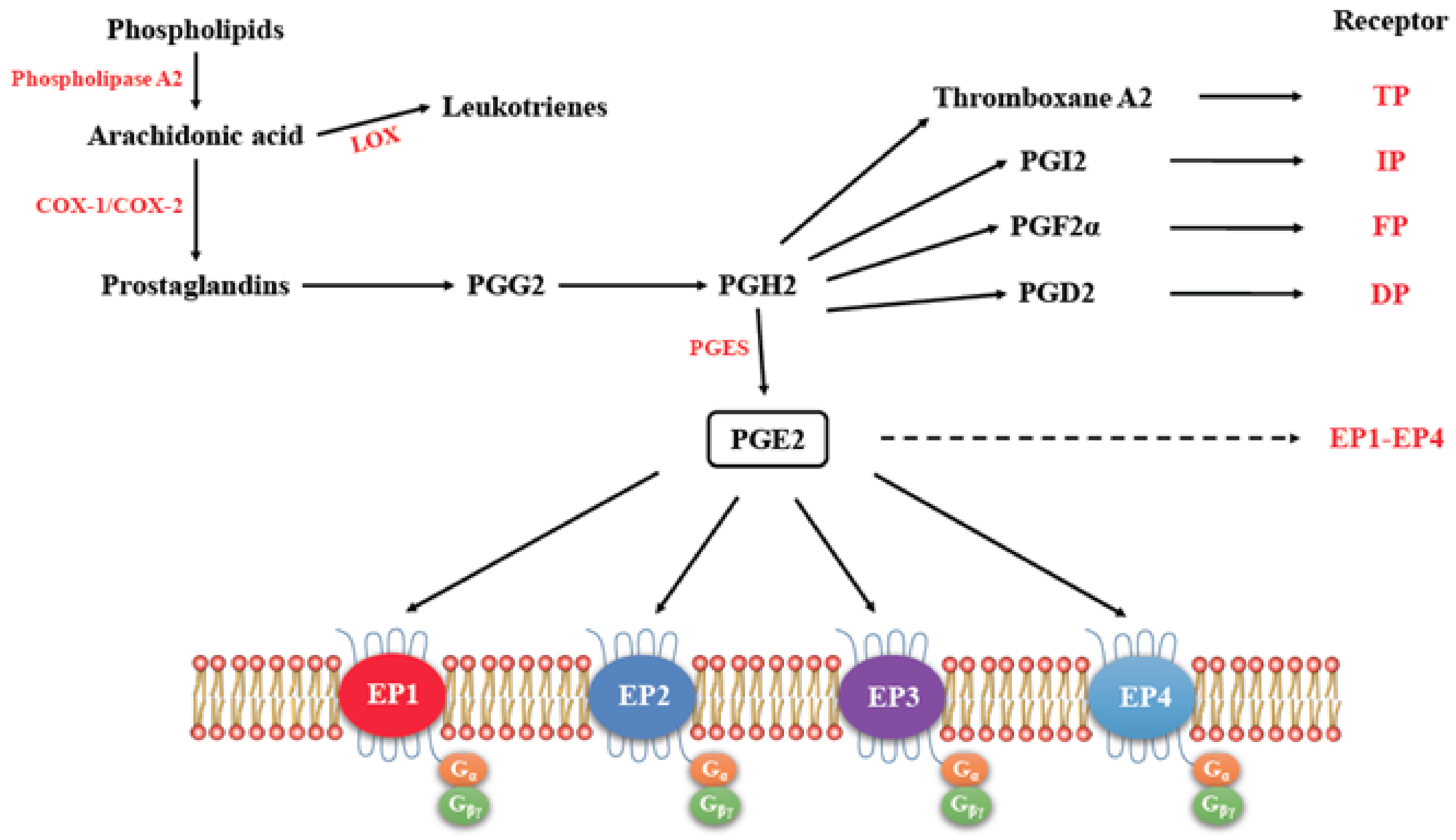
3. Cyclo-Oxygenase Pathway
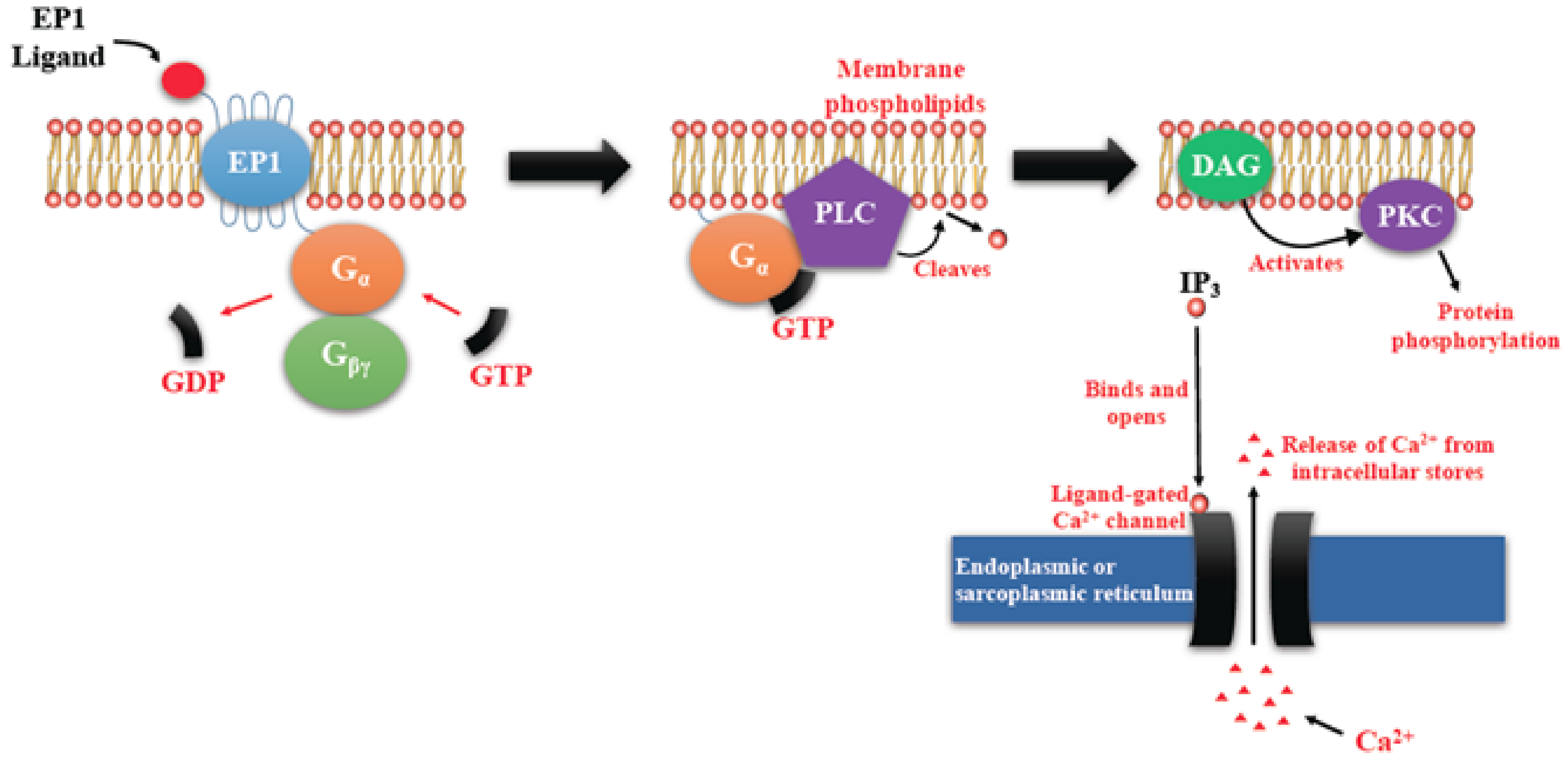
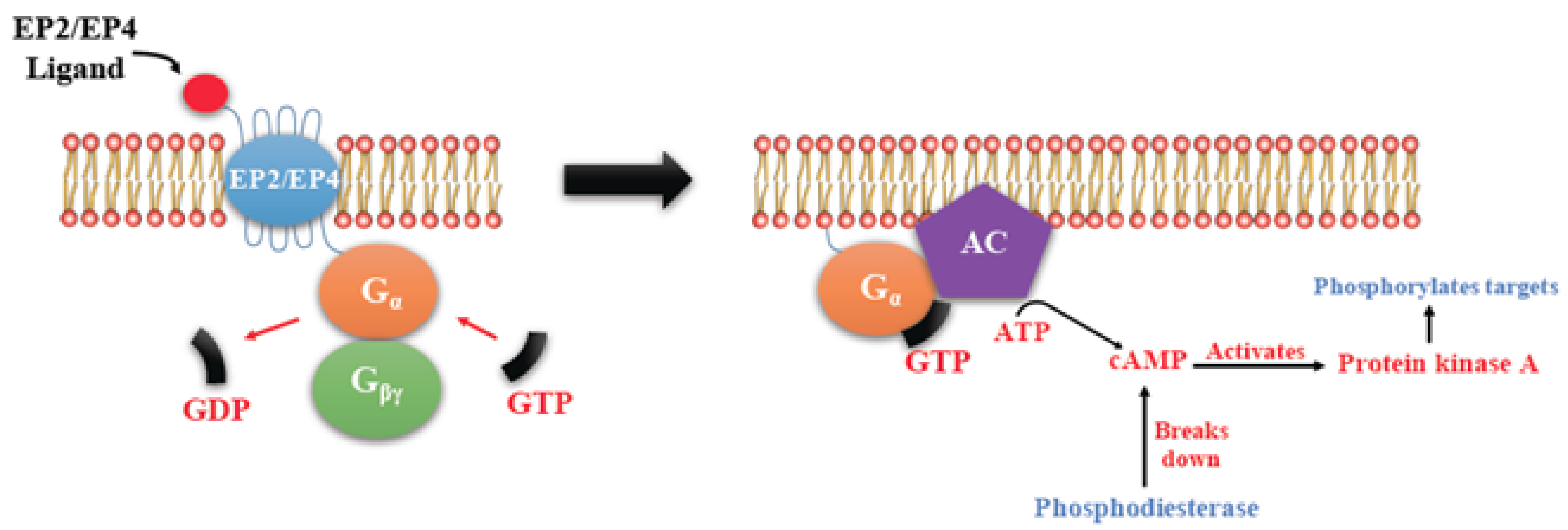
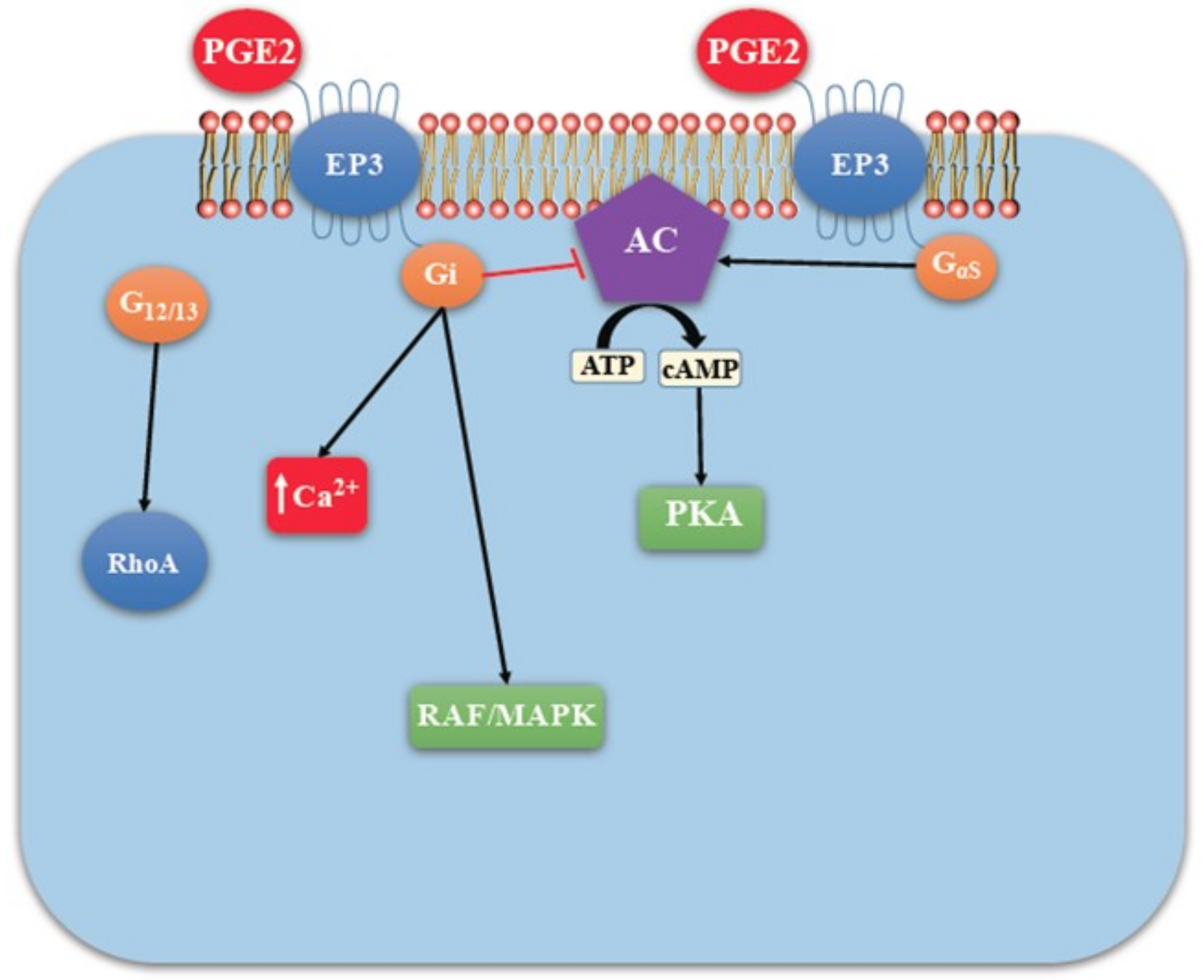
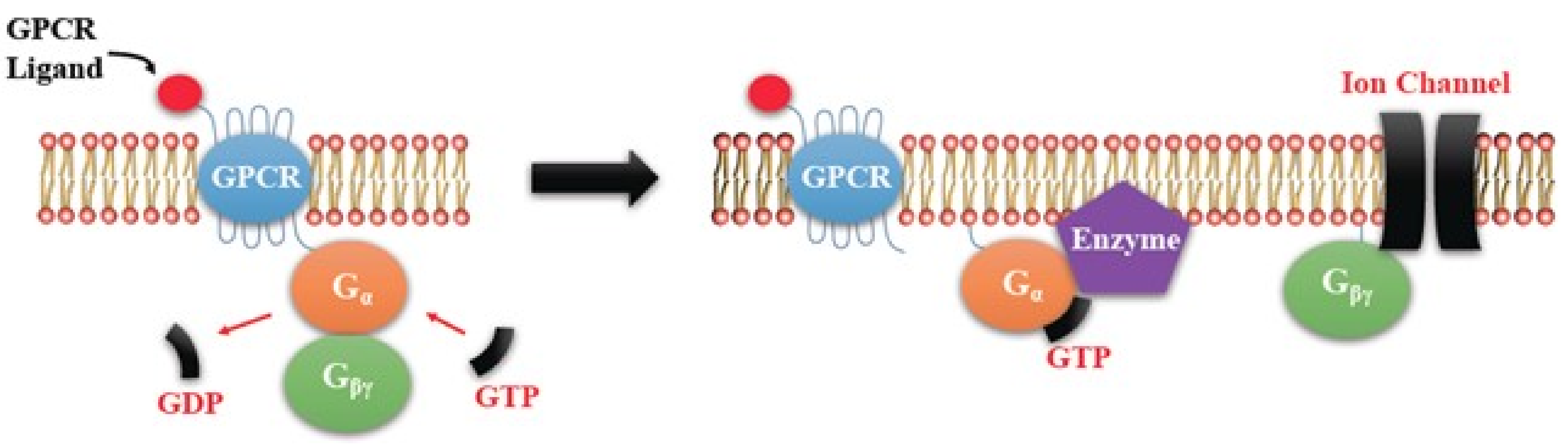
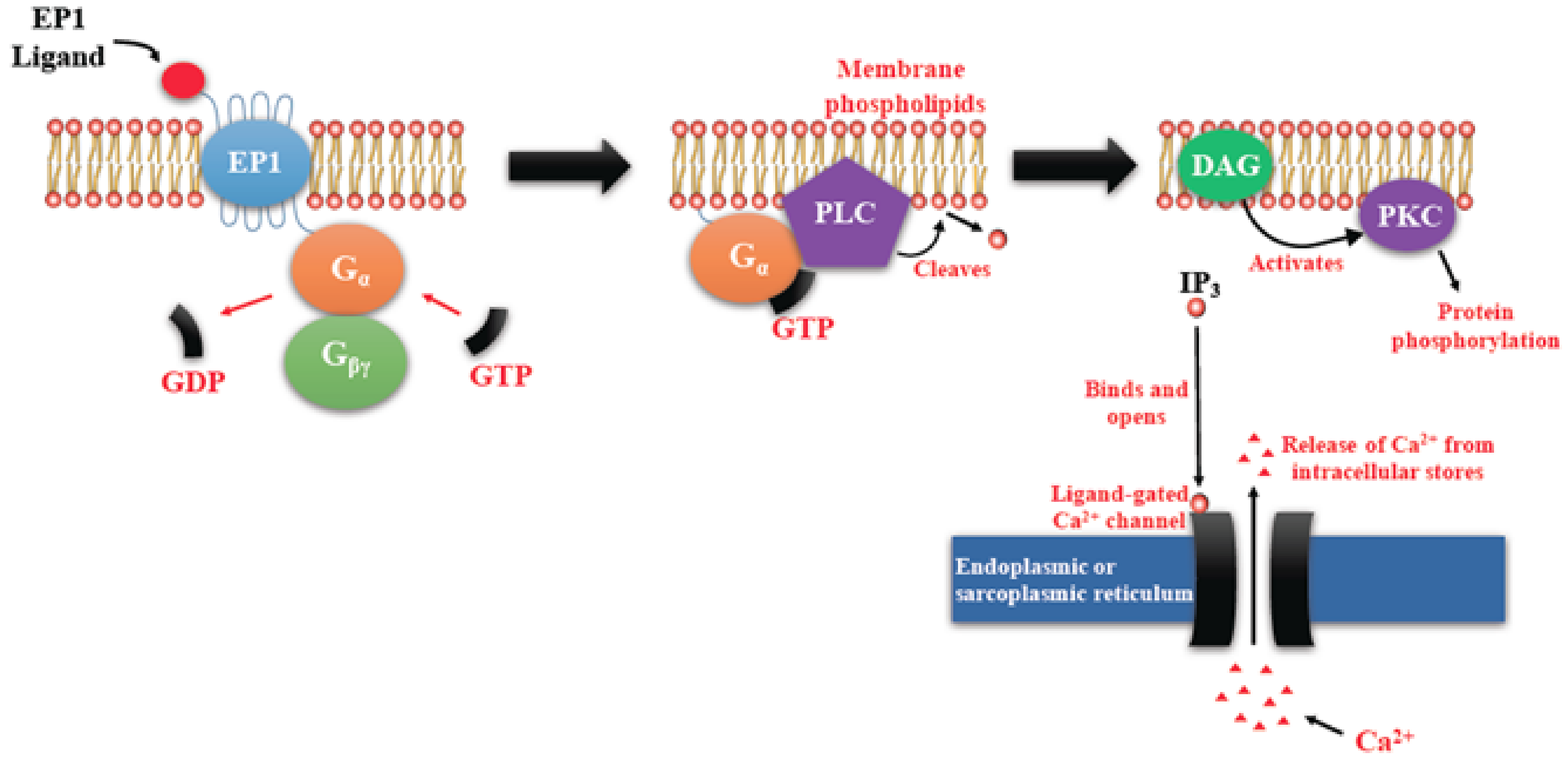
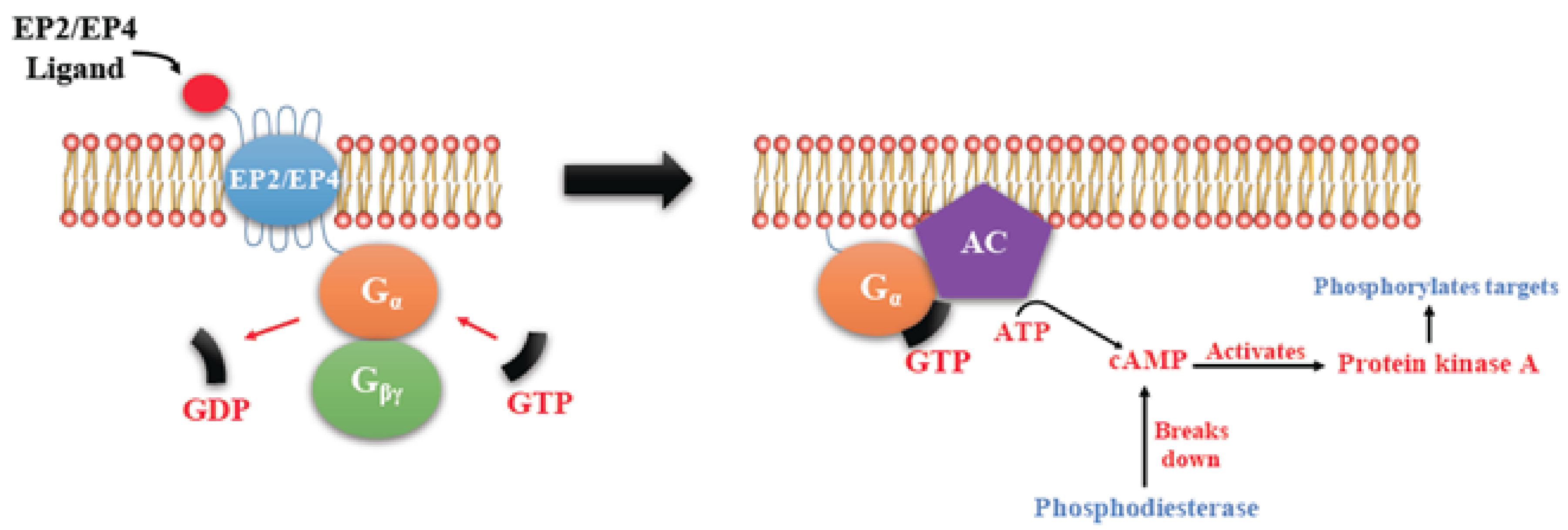
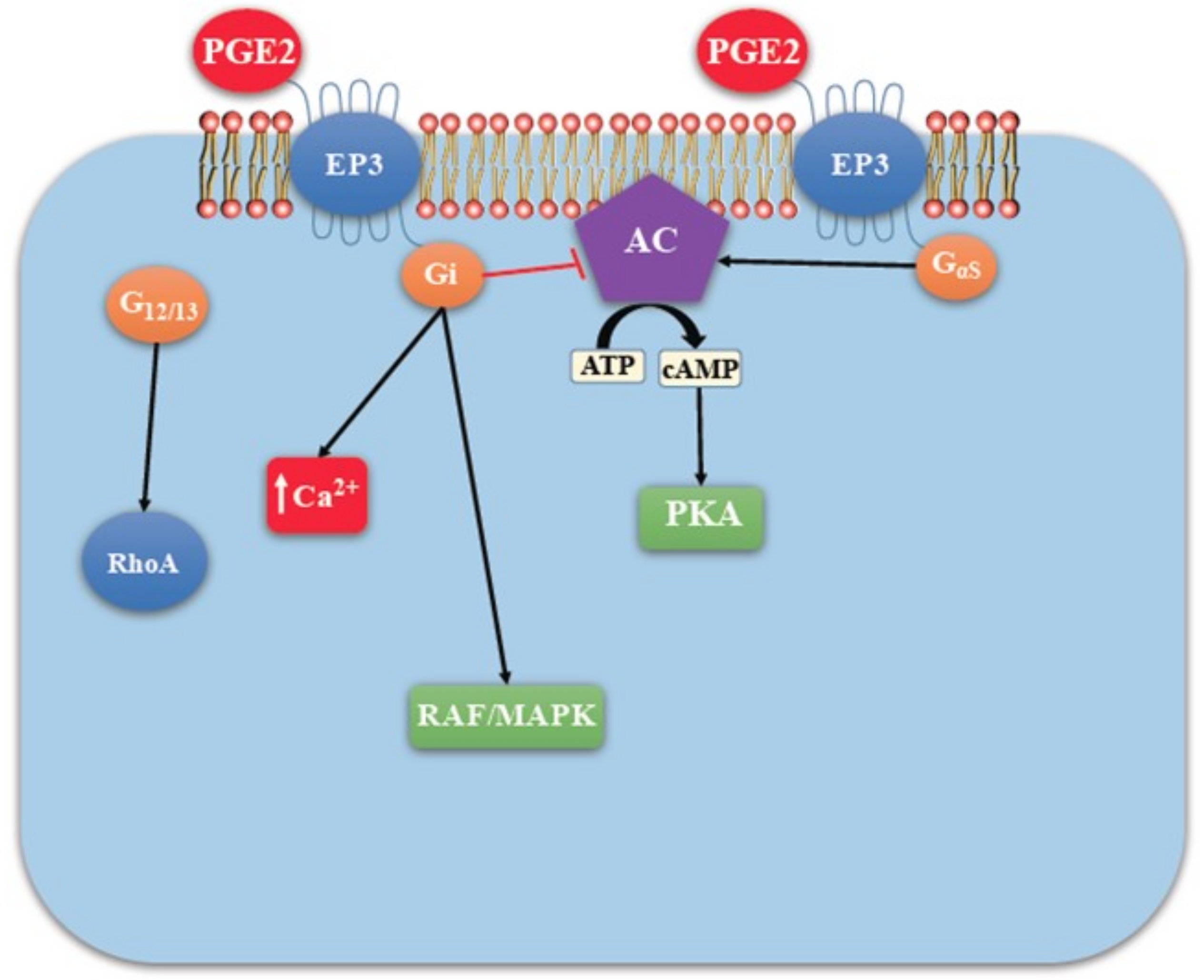
EP Receptors
4. COX2/PGE2 Mediated Cancer Progression
4.1. EP Receptor Functions in Health and Disease Including Tumorigenesis: A Brief Overview
4.1.1. Biological Functions of EP Receptors
4.1.2. EP Receptors in Tumorigenesis
4.1.3. EP Receptors in Colorectal Carcinogenesis
4.1.4. EP Receptors in Skin Carcinogenesis
4.1.5. EP Receptors in Carcinogenesis in the Mucosa of the Pharynx and the Esophagus
4.1.6. EP Receptors in Prostatic Carcinogenesis
4.1.7. EP Receptors in Urothelial Carcinogenesis
4.1.8. EP Receptors in Non-Small Cell Lung Cancer
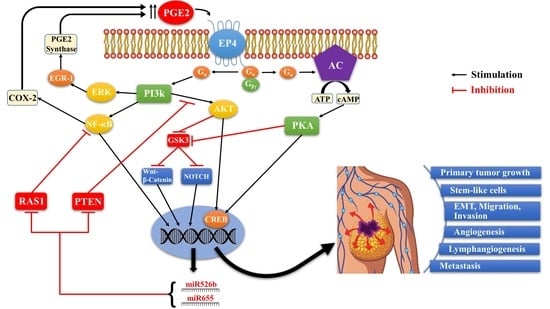
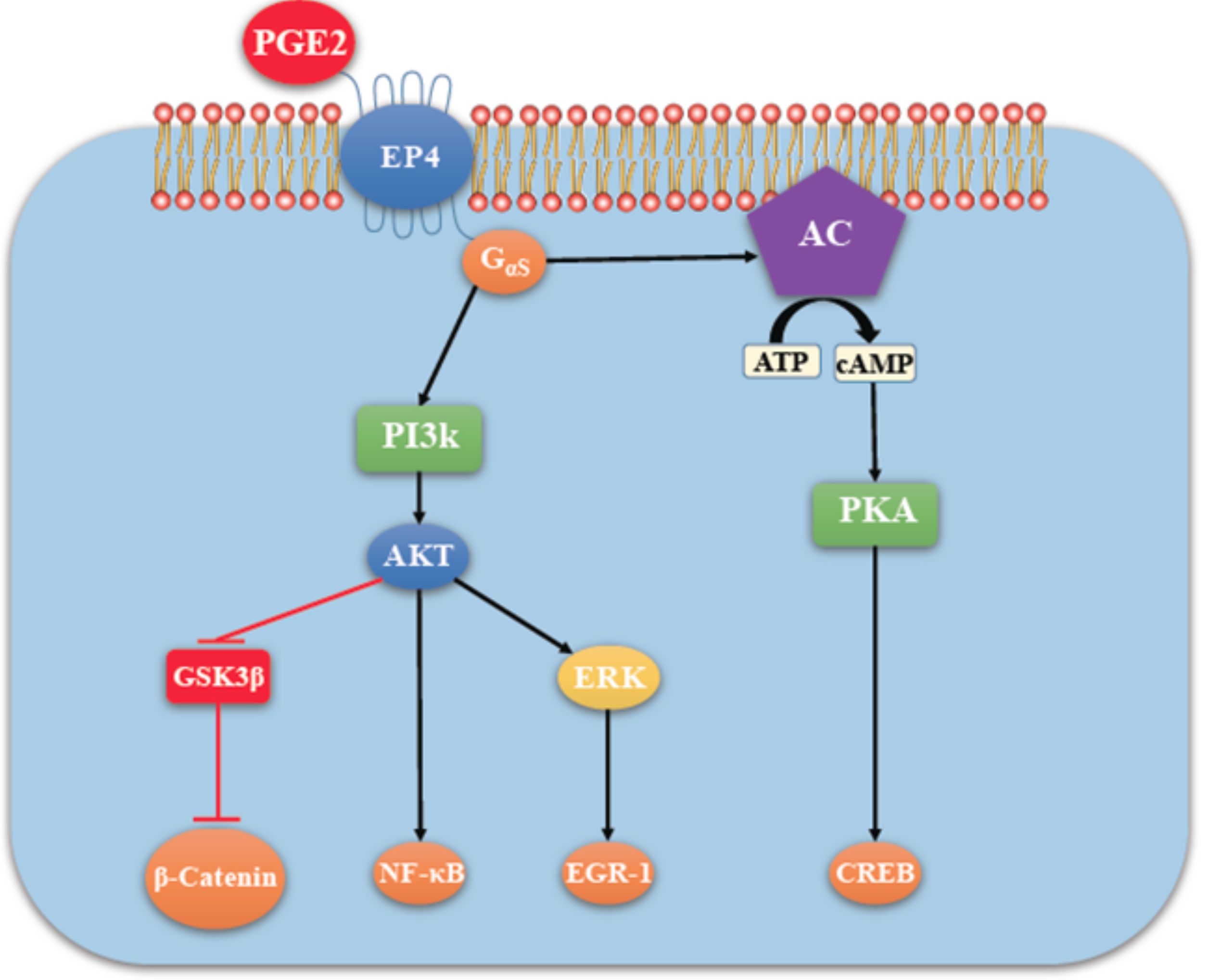
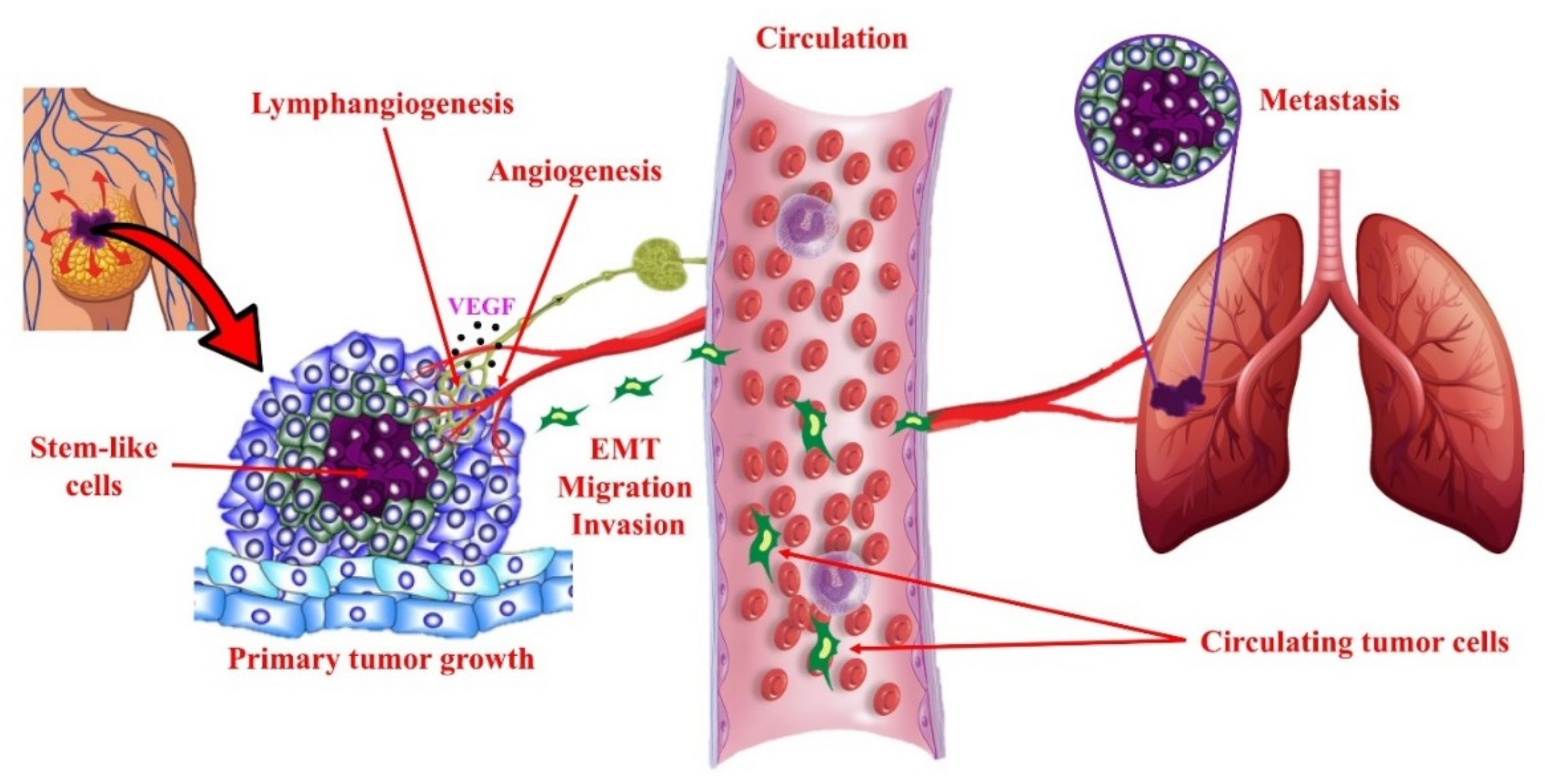
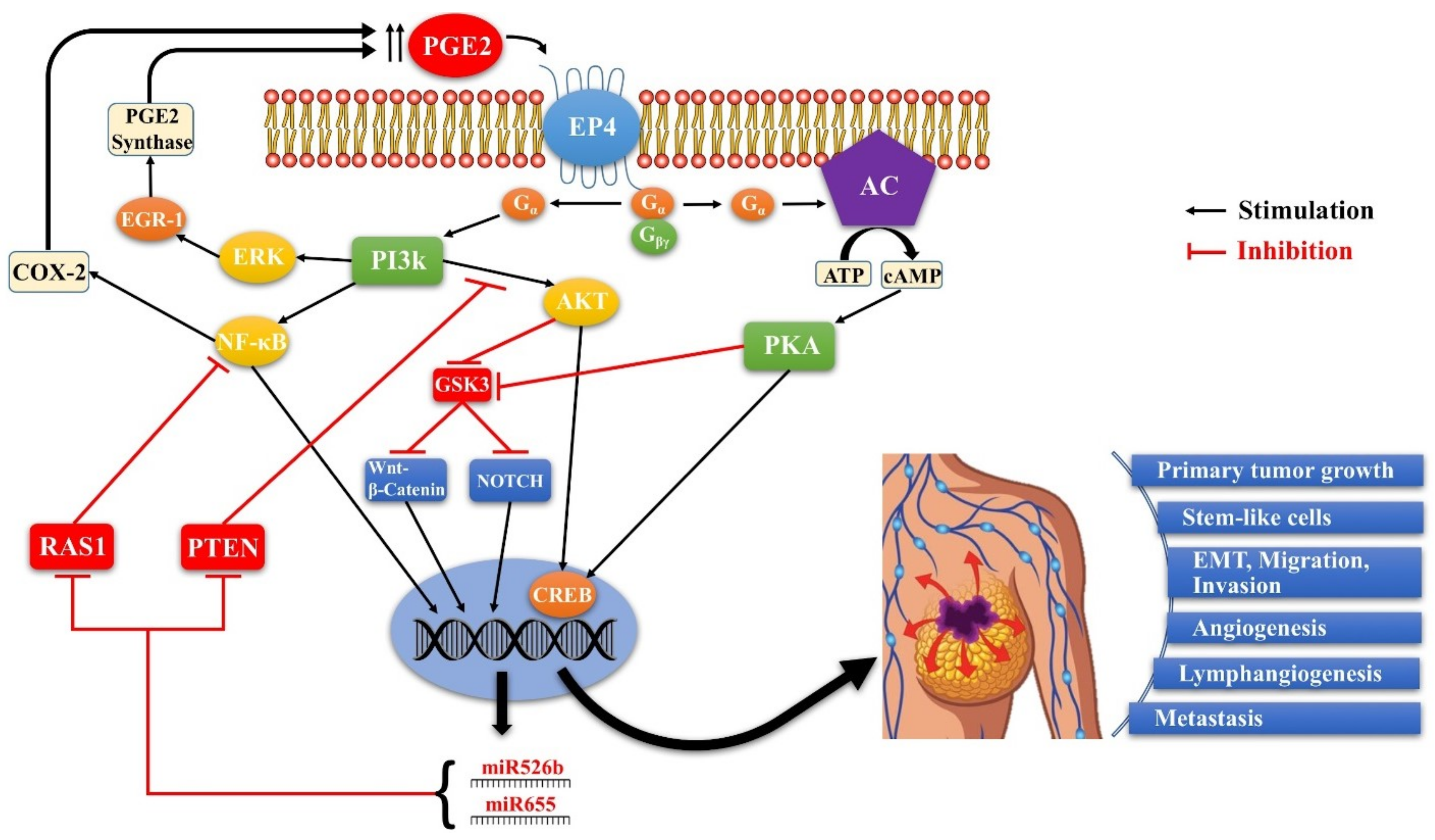
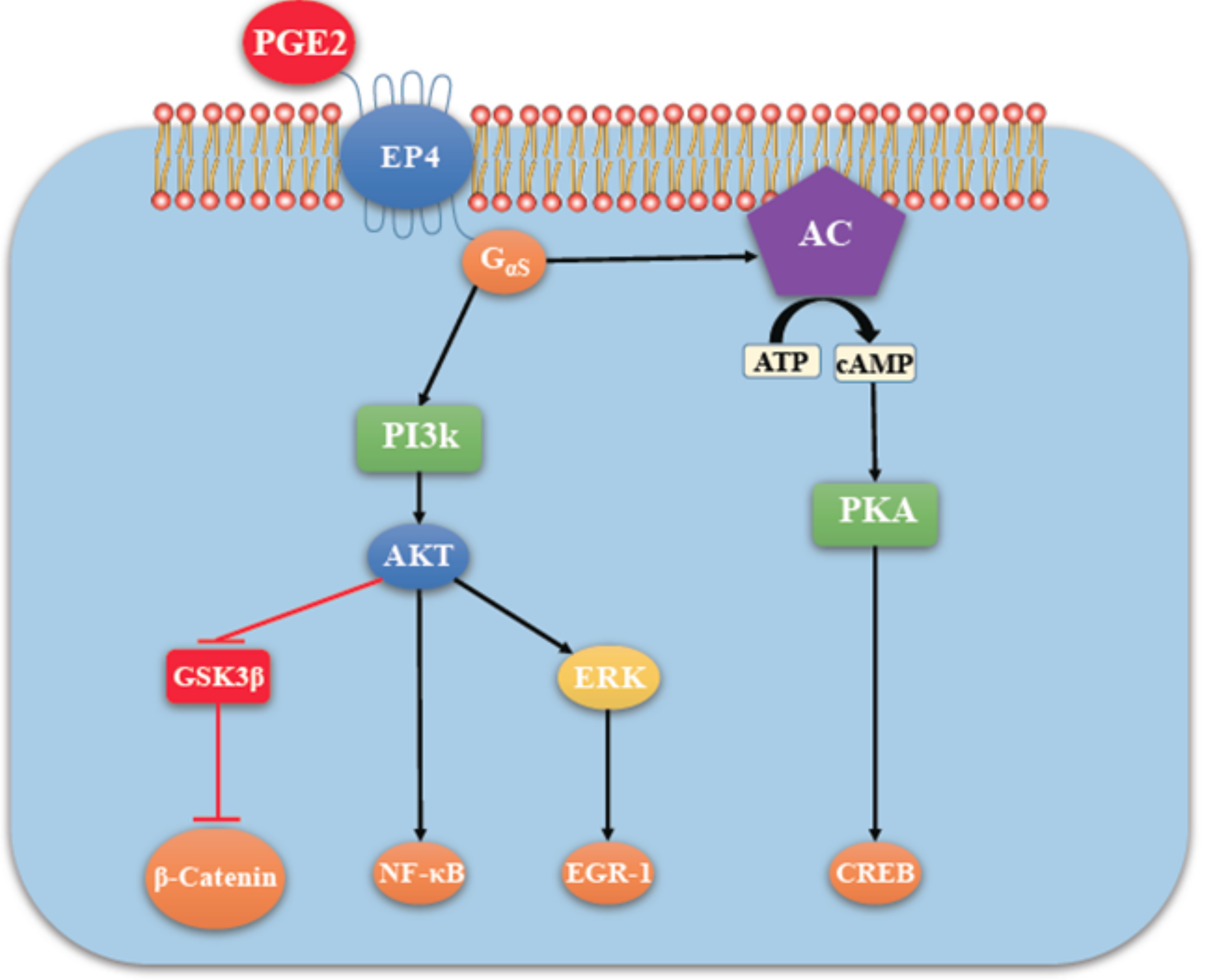
4.2. COX-2/PGE2 Mediated Breast Cancer Progression: Role of EP4 Receptor
4.3. Other Molecular Players Upstream or Downstream of EP4 that Promote Breast Cancer Progression
4.4. Rationale for the Choice of EP4 as a Potential Therapeutic Target in Breast Cancer
4.5. Functional Roles of COX-2 in the Absence or Presence of HER-2 in Breast Cancer
4.6. SLC-Linked MicroRNAs Induced by COX-2/EP4 Activity as Breast Cancer Biomarkers
4.7. Triple-Negative Breast Cancers (TNBC) Are Mostly COX2 Expressing
4.8. Proposed Combination of an EP4 Antagonist with an Immune Checkpoint Inhibitor for Treating TNBC
4.9. EP4 Antagonist in the Breast Cancer Clinic
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lappano, R.; Maggiolini, M. G-protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; An, S.; Ward, R.; Yang, Y.; Guo, X.; Li, W.; Xu, T. G-protein-coupled receptors as promising cancer targets. Cancer Lett. 2016, 376, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Jeon, J.; Kim, S.I. Personalized medicine in breast cancer: A systematic review. J. Breast Cancer 2012, 15, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Oh, D.S.; Wessels, L.; Weigelt, B.; Nuyten, D.S.; Nobel, A.B.; Van’t Veer, L.J.; Perou, C.M. Concordance among gene-expression—Based predictors for breast cancer. N. Engl. J. Med. 2006, 355, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R. Inflammation and breast cancer. Cyclooxygenase/prostaglandin signaling and breast cancer. Breast Cancer Res. 2007, 9, 210. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE 2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Markovič, T.; Jakopin, Ž.; Dolenc, M.S.; Mlinarič-Raščan, I. Structural features of subtype-selective EP receptor modulators. Drug Discov. Today 2017, 22, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Breyer, R.M.; Bagdassarian, C.K.; Myers, S.A.; Breyer, M.D. Prostanoid receptors: Subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 661–690. [Google Scholar] [CrossRef] [PubMed]
- Fujino, H.; Xu, W.; Regan, J.W. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J. Biol. Chem. 2003, 278, 12151–12156. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef] [PubMed]
- O’callaghan, G.; Houston, A. Prostaglandin E2 and the EP receptors in malignancy: Possible therapeutic targets? Br. J. Pharmacol. 2015, 172, 5239–5250. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, F.G.; Gorden, D.L.; Matta, P.; Shi, Q.; Matrisian, L.M.; DuBois, R.N. Role of β-arrestin 1 in the metastatic progression of colorectal cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Konya, V.; Marsche, G.; Schuligoi, R.; Heinemann, A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol. Ther. 2013, 138, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E. COX-2 Blockade in Cancer Prevention and Therapy; Springer Science & Business Media: Berlin, Germany, 2002. [Google Scholar]
- Liu, C.H.; Chang, S.; Narko, K.; Trifan, O.C.; Wu, M.; Smith, E.; Haudenschild, C.; Lane, T.F.; Hla, T. Over-expression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J. Biol. Chem. 2001, 276, 18563–18569. [Google Scholar] [CrossRef] [PubMed]
- Chulada, P.C.; Thompson, M.B.; Mahler, J.F.; Doyle, C.M.; Gaul, B.W.; Lee, C.; Tiano, H.F.; Morham, S.G.; Smithies, O.; Langenbach, R. Genetic disruption of Ptgs-1, as well as of Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000, 60, 4705–4708. [Google Scholar] [PubMed]
- Gupta, A.K.; Schoen, R.E. Aberrant crypt foci: Are they intermediate endpoints of colon carcinogenesis in humans? Curr. Opin. Gastroenterol. 2009, 25, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Dannenberg, A.J. COX-2 inhibitors for the prevention of breast cancer. J. Mammary Gland Biol. Neoplasia 2003, 8, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, C.R.; Collet, J.P.; McNutt, M.; Belzile, E.; Boivin, J.F.; Hanley, J.A. Nested case–control study of the effects of non-steroidal anti-inflammatory drugs on breast cancer risk and stage. Br. J. Cancer 2000, 83, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Chlebowski, R.T.; Jackson, R.D.; Frid, D.J.; Ascenseo, J.L.; Anderson, G.; Loar, A.; Rodabough, R.J.; White, E.; McTiernan, A. Breast cancer and nonsteroidal anti-inflammatory drugs: Prospective results from the Women’s Health Initiative. Cancer Res. 2003, 63, 6096–6101. [Google Scholar] [PubMed]
- Oshima, M.; Dinchuk, J.E.; Kargman, S.L.; Oshima, H.; Hancock, B.; Kwong, E.; Trzaskos, J.M.; Evans, J.F.; Taketo, M.M. Suppression of intestinal polyposis in ApcΔ716 Knock-out mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996, 87, 803–809. [Google Scholar] [CrossRef]
- Tsujii, M.; Kawano, S.; DuBois, R.N. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc. Natl. Acad. Sci. USA 1997, 94, 3336–3340. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Yatabe, Y.; Achiwa, H.; Muramatsu, H.; Kozaki, K.; Nakamura, S.; Ogawa, M.; Mitsudomi, T.; Sugiura, T.; Takahashi, T. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res. 1998, 58, 3761–3764. [Google Scholar] [PubMed]
- Chan, G.; Boyle, J.O.; Yang, E.K.; Zhang, F.; Sacks, P.G.; Shah, J.P.; Edelstein, D.; Soslow, R.A.; Koki, A.T.; Woerner, B.M. Cyclooxygenase-2 expression is up-regulated in squamous cell carcinoma of the head and neck. Cancer Res. 1999, 59, 991–994. [Google Scholar] [PubMed]
- Tucker, O.N.; Dannenberg, A.J.; Yang, E.K.; Zhang, F.; Teng, L.; Daly, J.M.; Soslow, R.A.; Masferrer, J.L.; Woerner, B.M.; Koki, A.T. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999, 59, 987–990. [Google Scholar] [PubMed]
- Parrett, M.; Harris, R.; Joarder, F.; Ross, M.; Clausen, K.; Robertson, F. Cyclooxygenase-2 gene expression in human breast cancer. Int. J. Oncol. 1997, 10, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Ristimäki, A.; Sivula, A.; Lundin, J.; Lundin, M.; Salminen, T.; Haglund, C.; Joensuu, H.; Isola, J. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002, 62, 632–635. [Google Scholar] [PubMed]
- Parhar, R.S.; Lala, P.K. Changes in the host natural killer cell population in mice during tumor development: 2. The mechanism of suppression of NK activity. Cell. Immunol. 1985, 93, 265–279. [Google Scholar] [CrossRef]
- Lala, P.K.; Parhar, R.S.; Singh, P. Indomethacin therapy abrogates the prostaglandin-mediated suppression of natural killer activity in tumor-bearing mice and prevents tumor metastasis. Cell. Immunol. 1986, 99, 108–118. [Google Scholar] [CrossRef]
- Lala, P.K.; Al-Mutter, N.; Orucevic, A. Effects of chronic indomethacin therapy on the development and progression of spontaneous mammary tumors in C3H/HEJ mice. Int. J. Cancer 1997, 73, 371–380. [Google Scholar] [CrossRef]
- Kundu, N.; Fulton, A.M. Selective cyclooxygenase (COX)-1 or COX-2 inhibitors control metastatic disease in a murine model of breast cancer. Cancer Res. 2002, 62, 2343–2346. [Google Scholar] [PubMed]
- Reader, J.; Holt, D.; Fulton, A. Prostaglandin E2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011, 30, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, A.J.; Lippman, S.M.; Mann, J.R.; Subbaramaiah, K.; DuBois, R.N. Cyclooxygenase-2 and epidermal growth factor receptor: Pharmacologic targets for chemoprevention. J. Clin. Oncol. 2005, 23, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Hull, M.A.; Ko, S.C.; Hawcroft, G. Prostaglandin EP receptors: Targets for treatment and prevention of colorectal cancer? Mol. Cancer Ther. 2004, 3, 1031–1039. [Google Scholar] [PubMed]
- Fujino, H. The roles of EP4 prostanoid receptors in cancer malignancy signaling. Biol. Pharm. Bull. 2016, 39, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Chell, S.D.; Witherden, I.R.; Dobson, R.R.; Moorghen, M.; Herman, A.A.; Qualtrough, D.; Williams, A.C.; Paraskeva, C. Increased EP4 receptor expression in colorectal cancer progression promotes cell growth and anchorage independence. Cancer Res. 2006, 66, 3106–3113. [Google Scholar] [CrossRef] [PubMed]
- Shoji, Y.; Takahashi, M.; Takasuka, N.; Niho, N.; Kitamura, T.; Sato, H.; Maruyama, T.; Sugimoto, Y.; Narumiya, S.; Sugimura, T. Prostaglandin E receptor EP 3 deficiency modifies tumor outcome in mouse two-stage skin carcinogenesis. Carcinogenesis 2005, 26, 2116–2122. [Google Scholar] [CrossRef] [PubMed]
- Rundhaug, J.E.; Simper, M.S.; Surh, I.; Fischer, S.M. The role of the EP receptors for prostaglandin E 2 in skin and skin cancer. Cancer Metastasis Rev. 2011, 30, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Abrahao, A.C.; Castilho, R.M.; Squarize, C.H.; Molinolo, A.A.; Santos-Pinto, D. d.; Gutkind, J.S. A role for COX2-derived PGE2 and PGE2-receptor subtypes in head and neck squamous carcinoma cell proliferation. Oral Oncol. 2010, 46, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Lowry, M.C.; Reynolds, J.V.; Cathcart, M. The Role of PGE2 and its Corresponding Receptors (Ep1-4) in Oesophageal Carcinogenesis: Novel Therapeutics for Chemoprevention and/or Intervention. Carcinog. Mutagen. 2014. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Campbell, W.B. Roles of eicosanoids in prostate cancer. Future Lipidol. 2008, 3, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, Z.; Ogawa, O.; Yoshikawa, T.; Sakamoto, H.; Shibasaki, N.; Goto, T.; Wang, L.; Terada, N. An EP4 Antagonist ONO-AE3-208 Suppresses Cell Invasion, Migration, and Metastasis of Prostate Cancer. Cell Biochem. Biophys. 2014, 70, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, E.; Inoue, S.; Mizushima, T.; Chen, J.; Ide, H.; Kawahara, T.; Reis, L.O.; Baras, A.S.; Netto, G.J.; Miyamoto, H. Prostaglandin receptors induce urothelial tumourigenesis as well as bladder cancer progression and cisplatin resistance presumably via modulating PTEN expression. Br. J. Cancer 2018, 118, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.G.; Al-Sarraf, N.; Baird, A.; Cathcart, M.; McGovern, E.; O’Byrne, K.J. Regulation of EP receptors in non-small cell lung cancer by epigenetic modifications. Eur. J. Cancer 2009, 45, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Rozic, J.G.; Chakraborty, C.; Lala, P.K. Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor cell migration, invasiveness and angiogenesis. Int. J. Cancer 2001, 93, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Timoshenko, A.V.; Xu, G.; Chakrabarti, S.; Lala, P.K.; Chakraborty, C. Role of prostaglandin E 2 receptors in migration of murine and human breast cancer cells. Exp. Cell Res. 2003, 289, 265–274. [Google Scholar] [CrossRef]
- Timoshenko, A.V.; Lala, P.K.; Chakraborty, C. PGE2-mediated upregulation of iNOS in murine breast cancer cells through the activation of EP4 receptors. Int. J. Cancer 2004, 108, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Timoshenko, A.V.; Chakraborty, C.; Wagner, G.F.; Lala, P.K. COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. Br. J. Cancer 2006, 94, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Majumder, M.; Girish, G.V.; Mohindra, V.; Maruyama, T.; Lala, P.K. Targeting COX-2 and EP4 to control tumor growth, angiogenesis, lymphangiogenesis and metastasis to the lungs and lymph nodes in a breast cancer model. Lab. Investig. 2012, 92, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Xin, X.; Liu, L.; Girish, G.V.; Lala, P.K. Prostaglandin E2 receptor EP4 as the common target on cancer cells and macrophages to abolish angiogenesis, lymphangiogenesis, metastasis, and stem-like cell functions. Cancer Sci. 2014, 105, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Nandi, P.; Girish, G.V.; Majumder, M.; Xin, X.; Tutunea-Fatan, E.; Lala, P.K. PGE2 promotes breast cancer-associated lymphangiogenesis by activation of EP4 receptor on lymphatic endothelial cells. BMC Cancer 2017, 17, 11. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.; Ma, X.; Kundu, N.; Fulton, A. Prostaglandin E2 (PGE2) suppresses natural killer cell function primarily through the PGE2 receptor EP4. Cancer Immunol. Immunother. 2011, 60, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Holt, D.; Kundu, N.; Reader, J.; Goloubeva, O.; Take, Y.; Fulton, A.M. A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2-mediated immunosuppression and inhibits breast cancer metastasis. OncoImmunology 2013, 2, e22647. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Sugata, Y.; Fujiwara, T.; Matsumoto, R.; Nishibori, M.; Shimizu, K.; Maeda, M.; Kimura, Y.; Kariya, S.; Hattori, H.; et al. E prostanoid 2 (EP2)/EP4-mediated suppression of antigen-specific human T-cell responses by prostaglandin E2. Immunology 2006, 118, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Albu, D.I.; Wang, Z.; Wu, J.; Huang, K.; Li, W.; Liu, D.; Kuznetsov, G.; Chen, Q.; Bao, X.; Woodall-Jappe, M. Abstract 275: ER-886046, an antagonist of PGE2 receptor type-4, induces an effective antitumor immune response in mice by attenuating intratumoral MDSCs and TAMs. Cancer Res. 2015, 75. [Google Scholar] [CrossRef]
- Harizi, H.; Grosset, C.; Gualde, N. Prostaglandin E2 modulates dendritic cell function via EP2 and EP4 receptor subtypes. J. Leukoc. Biol. 2003, 73, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Xin, X.; Liu, L.; Tutunea-Fatan, E.; Rodriguez-Torres, M.; Vincent, K.; Postovit, L.; Hess, D.; Lala, P.K. COX-2 Induces Breast Cancer Stem Cells via EP4/PI3K/AKT/NOTCH/WNT Axis. Stem Cells 2016, 34, 2290–2305. [Google Scholar] [CrossRef] [PubMed]
- Kundu, N.; Ma, X.; Kochel, T.; Goloubeva, O.; Staats, P.; Thompson, K.; Martin, S.; Reader, J.; Take, Y.; Collin, P.; et al. Prostaglandin E receptor EP4 is a therapeutic target in breast cancer cells with stem-like properties. Breast Cancer Res Treat 2014, 143, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer Stem Cells: An Old Idea—A Paradigm Shift. Cancer Res. 2006, 66, 6. [Google Scholar] [CrossRef] [PubMed]
- Tysnes, B.B. Tumor-Initiating and-Propagating Cells: Cells That We Would to Identify and Control. Neoplasia 2010, 12, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Filipenko, I.; Schwalm, S.; Reali, L.; Pfeilschifter, J.; Fabbro, D.; Huwiler, A.; Zangemeister-Wittke, U. Upregulation of the S1P3 receptor in metastatic breast cancer cells increases migration and invasion by induction of PGE2 and EP2/EP4 activation. Biochim. Biophys. Acta 2016, 11, 1840–1851. [Google Scholar] [CrossRef] [PubMed]
- Hiken, J.F.; McDonald, J.I.; Decker, K.F.; Sanchez, C.; Hoog, J.; VanderKraats, N.D.; Jung, K.L.; Akinhanmi, M.; Ellis, M.J.; Edwards, J.R. Epigenetic activation of the prostaglandin receptor EP4 promotes resistance to endocrine therapy for breast cancer. Oncogene 2017, 36, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Tönisen, F.; Perrin, L.; Bayarmagnai, B.; van den Dries, K.; Cambi, A.; Gligorijevic, B. EP4 receptor promotes invadopodia and invasion in human breast cancer. Eur. J. Cell Biol. 2017, 2, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-R.; Hou, M.-F.; Chang, H.-C.; Hung, W.-C. Cyclooxygenase-2 Up-regulates CCR7 via EP2/EP4 Receptor Signaling Pathways to Enhance Lymphatic Invasion of Breast Cancer Cells. J. Biol. Chem. 2008, 283, 11155–11163. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.W.; Pan, M.-R.; Hou, M.-F.; Hung, W.-C. Cyclooxygenase-2 up-regulates CCR7 expression via AKT-mediated phosphorylation and activation of Sp1 in breast cancer cells. Cell Physiol. 2013, 228, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, D.; Ang, R.; Waxman, J. COX inhibitors and breast cancer. Br. J. Cancer 2006, 94, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Beebe-Donk, J.; Alshafie, G.A. Reduction in the risk of human breast cancer by selective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer 2006, 6. [Google Scholar] [CrossRef] [PubMed]
- Ashok, V.; Dash, C.; Rohan, T.E.; Sprafka, J.M.; Terry, P.D. Selective cyclooxygenase-2 (COX-2) inhibitors and breast cancer risk. Breast 2011, 20, 66–70. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, G.A. Coxibs and Cardiovascular Disease. N. Engl. J. Med. 2004, 351, 1709–1711. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.J. COX-2 Inhibitors, Other NSAIDs, and Cardiovascular Risk: The Seduction of Common Sense. JAMA 2006, 296, 1653–1656. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, M.; Tamosiuniene, R.; Chen, G.; Neilan, T.G.; Bradford, A.; O’Byrne, K.J.; Fitzgerald, D.J.; Pidgeon, G.P. Cyclooxygenase-2-linked attenuation of hypoxia-induced pulmonary hypertension and intravascular thrombosis. J. Pharmacol. Exp. Ther. 2008, 326, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Hara, A.; Yuhki, K.; Fujino, T.; Ma, H.; Okada, Y.; Takahata, O.; Yamada, T.; Murata, T.; Narumiya, S.; et al. Roles of Prostaglandin I2 and Thromboxane A2 in Cardiac Ischemia-Reperfusion Injury: A Study Using Mice Lacking Their Respective Receptors. Circulation 2001, 104, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Meyer-Kirchrath, J.; Kaber, G.; Jacoby, C.; Flogel, U.; Schrader, J.; Ruther, U.; Schror, K.; Hohlfeld, T. Cardiospecific Over-expression of the Prostaglandin EP3 Receptor Attenuates Ischemia-Induced Myocardial Injury. Circulation 2005, 112, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Thiemermann, C.; Zacharowski, K. Selective activation of E-type prostanoid3-receptors reduces myocardial infarct size. Pharmacol. Ther. 2000, 87, 61–67. [Google Scholar] [CrossRef]
- Hishikari, K.; Suzuki, J.; Ogawa, M.; Isobe, K.; Takahashi, T.; Onishi, M.; Takayama, K.; Isobe, M. Pharmacological activation of the prostaglandin E2 receptor EP4 improves cardiac function after myocardial ischaemia/reperfusion injury. Cardiovas. Res. 2009, 81, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.N.; Timoshenko, A.V.; Cai, J.; Lala, P.K. Relationship between cyclooxygenase-2 and human epidermal growth factor receptor 2 in vascular endothelial growth factor C up-regulation and lymphangiogenesis in human breast cancer. Cancer Sci. 2010, 101, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Subbaramaiah, K.; Howe, L.R.; Port, E.R.; Brogi, E.; Fishman, J.; Liu, C.H.; Hla, T.; Hudis, C.; Dannenberg, A.J. HER-2/neu Status Is a Determinant of Mammary Aromatase Activity In vivo: Evidence for a Cyclooxygenase-2-Dependent Mechanism. Cancer Res. 2006, 66, 5504–5511. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Chang, S.; Tolle, K.C.; Dillon, R.; Young, L.J.T.; Cardiff, R.D.; Newman, R.A.; Yang, P.; Thaler, H.T.; Muller, W.J.; et al. HER2/neu-Induced Mammary Tumorigenesis and Angiogenesis Are Reduced in Cyclooxygenase-2 Knock-out Mice. Cancer Res. 2005, 65, 10113–10119. [Google Scholar] [CrossRef] [PubMed]
- Simeone, A.; Li, Y.; Broemeling, L.D.; Johnson, M.M.; Tuna, M.; Tari, A.M. Cyclooxygenase-2 Is Essential for HER2/neu to Suppress N-(4-Hydroxyphenyl)retinamide Apoptotic Effects in Breast Cancer Cells. Cancer Res. 2004, 64, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Croker, A.K.; Goodale, D.; Chu, J.; Postenka, C.; Hedley, B.D.; Hess, D.A.; Allan, A.L. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med. 2009, 13, 2236–2252. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Robertson, F.M.; Simeone, A.-M.; Lucci, A.; McMurray, J.S.; Ghosh, S.; Cristofanilli, M. Differential Regulation of The Aggressive Phenotype of Inflammatory Breast Cancer Cells by Prostanoid Receptors EP3 and EP4. Cancer 2010, 116, 2806–2814. [Google Scholar] [CrossRef] [PubMed]
- Homo Sapiens miRNAs (1917 Sequences). Available online: http://www.mirbase.org/cgi-bin/mirna_summary.pl?org=hsa (accessed on 28 March 2018).
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092. [Google Scholar] [CrossRef] [PubMed]
- Heneghan, H.M.; Miller, N.; Lowery, A.J.; Sweeney, K.J.; Newell, J.; Kerin, M.J. Circulating microRNAs as novel minimally invasive biomarkers for breast cancer. Ann. Surg. 2010, 251, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Landman, E.; Liu, L.; Hess, D.; Lala, P.K. COX-2 Elevates Oncogenic miR-526b in Breast Cancer by EP4 Activation. Mol. Cancer Res. 2015, 13, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Dunn, L.; Liu, L.; Hasan, A.; Vincent, K.; Brackstone, M.; Hess, D.; Lala, P.K. COX-2 induces oncogenic micro RNA miR655 in human breast cancer. Sci. Rep. 2018, 8, 327. [Google Scholar] [CrossRef] [PubMed]
- Kochel, T.J.; Goloubeva, O.G.; Fulton, A.M. Upregulation of Cyclooxygenase-2/Prostaglandin E2 (COX-2/PGE2) Pathway Member Multiple Drug Resistance-Associated Protein 4 (MRP4) and Downregulation of Prostaglandin Transporter (PGT) and 15-Prostaglandin Dehydrogenase (15-PGDH) in Triple-Negative Breast Cancer. Breast Cancer Basic Clin. Res. 2016, 10, 61–70. [Google Scholar] [CrossRef]
- Mosalpuria, K.; Hall, C.; Krishnamurthy, S.; Lodhi, A.; Hallman, D.M.; Baraniuk, M.S.; Bhattacharyya, A.; Lucci, A. Cyclooxygenase-2 expression in non-metastatic triple-negative breast cancer patients. Mol. Clin. Oncol. 2014, 2, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hu, Y.; Hu, M.; Li, B. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Sci. Rep. 2015, 5, 13110. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Forde, P.M.; Hammers, H.; Emens, L.A.; Taube, J.M.; Topalian, S.L. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin. Oncol. 2015, 42, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Gangadhar, T.C.; Salama, A.K. Clinical applications of PD-1-based therapy: A focus on pembrolizumab (MK-3475) in the management of melanoma and other tumor types. OncoTargets Ther. 2015, 8, 929–937. [Google Scholar]
- Bertucci, F.; Gonçalves, A. Immunotherapy in Breast Cancer: The Emerging Role of PD-1 and PD-L1. Curr. Oncol. Rep. 2017, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Albu, D.; Huang, K.; Wu, J.; Twine, N.; Nomoto, K.; Woodall-Jappe, M. Combination of EP4 antagonist and checkpoint inhibitors promotes anti-tumor effector T cells in preclinical tumor models. J. Immunother. Cancer 2015, 3, P350. [Google Scholar] [CrossRef]
- Takitaa, M.; Inadaa, M.; Maruyama, T.; Miyaura, C. Prostaglandin E receptor EP4 antagonist suppresses osteolysis due to bone metastasis of mouse malignant melanoma cells. FEBS Lett. 2007, 581, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Okumura, Y.; Yamagishi, T.; Nukui, S.; Nakao, K. Discovery of AAT-008, a novel, potent, and selective prostaglandin EP4 receptor antagonist. Bioorg. Med. Chem. Lett. 2017, 27, 1186–1192. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majumder, M.; Nandi, P.; Omar, A.; Ugwuagbo, K.C.; Lala, P.K. EP4 as a Therapeutic Target for Aggressive Human Breast Cancer. Int. J. Mol. Sci. 2018, 19, 1019. https://doi.org/10.3390/ijms19041019
Majumder M, Nandi P, Omar A, Ugwuagbo KC, Lala PK. EP4 as a Therapeutic Target for Aggressive Human Breast Cancer. International Journal of Molecular Sciences. 2018; 19(4):1019. https://doi.org/10.3390/ijms19041019
Chicago/Turabian StyleMajumder, Mousumi, Pinki Nandi, Ahmed Omar, Kingsley Chukwunonso Ugwuagbo, and Peeyush K. Lala. 2018. "EP4 as a Therapeutic Target for Aggressive Human Breast Cancer" International Journal of Molecular Sciences 19, no. 4: 1019. https://doi.org/10.3390/ijms19041019
APA StyleMajumder, M., Nandi, P., Omar, A., Ugwuagbo, K. C., & Lala, P. K. (2018). EP4 as a Therapeutic Target for Aggressive Human Breast Cancer. International Journal of Molecular Sciences, 19(4), 1019. https://doi.org/10.3390/ijms19041019