Abstract
In recent years, RNA trans-splicing has emerged as a suitable RNA editing tool for the specific replacement of mutated gene regions at the pre-mRNA level. Although the technology has been successfully applied for the restoration of protein function in various genetic diseases, a higher trans-splicing efficiency is still desired to facilitate its clinical application. Here, we describe a modified, easily applicable, fluorescence-based screening system for the generation and analysis of antisense molecules specifically capable of improving the RNA reprogramming efficiency of a selected KRT14-specific RNA trans-splicing molecule. Using this screening procedure, we identified several antisense RNAs and short rationally designed oligonucleotides, which are able to increase the trans-splicing efficiency. Thus, we assume that besides the RNA trans-splicing molecule, short antisense molecules can act as splicing modulators, thereby increasing the trans-splicing efficiency to a level that may be sufficient to overcome the effects of certain genetic predispositions, particularly those associated with dominantly inherited diseases.
1. Introduction
RNA-based therapies constitute elegant options to restore gene function in various genetic diseases. Therapeutic intervention at the posttranscriptional level during the course of naturally occurring mRNA maturation in the cell represents a viable opportunity to manipulate the mRNA sequence prior to protein translation. Examples of such strategies include splice-modulating therapies, such as antisense oligonucleotide (ASO)-mediated exon skipping and spliceosome-mediated RNA trans-splicing (SMaRT), both of which are currently under development for the therapy of several monogenetic diseases [1,2,3].
Others [4,5] and we ourselves [6,7] have applied SMaRT as an emerging RNA editing tool for the specific correction of recessively and dominantly inherited mutations. So far, SMaRT has successfully been implemented in RNA repair studies for genetic disorders such as epidermolysis bullosa (EB) [8,9], muscular dystrophy [4,10], Alzheimer’s disease [11], and Huntington’s disease [12]. SMaRT uses the cell’s own splicing machinery to recombine a rationally designed RNA trans-splicing molecule (RTM) with the target pre-mRNA of interest, generating a new chimeric gene product. This is in contrast to group I intron-based (also referred to as ribozyme-based) trans-splicing RNAs that act without the help of the endogenous spliceosomal machinery. Group I intron ribozymes are mainly found in protists, bacteria, or bacteriophages [13], and similar to SMaRT they can be used for RNA repair via the replacement of the 3′ part of the mRNA with the 3′ exonic sequence provided by the ribozymes [14,15] or by splicing a suicide gene into the target RNA to kill tumor cells [16,17]. In general, however, the efficiency of both these RNA technologies needs to be significantly improved for a prospective clinical application.
The efficiency and specificity of SMaRT mainly relies on the RTM’s composition and binding affinity to the respective target locus of interest. The RTM carries the wild-type coding region to be inserted into the target pre-mRNA, splicing elements for efficient splicing, and a binding domain (BD) that specifically binds an intron of the target pre-mRNA and is critical for initiating RNA recombination. SMaRT is suitable for the specific exchange of 5′-, 3′-, or internal parts of a given transcript, referred to as 5′, 3′, or double RNA trans-splicing [18,19]. Thereby, any pathogenic mutation within the respective gene region can be replaced with a single type of RTM. For EB, a severe skin blistering disease [20], different trans-splicing strategies have been described to correct EB-associated mutations in COL7A1 [7,19], COL17A1 [9], KRT14 [6], and PLEC [8]. Although important progress in RTM design has been achieved, particularly in the selection of the BD sequence [21,22], the low efficiency of the technology remains an obstacle to its broad in vivo application. However, our first ex vivo and in vivo studies in recessive dystrophic epidermolysis bullosa (RDEB) mouse models showed that a low RNA reprogramming efficiency can be sufficient to restore protein function [7,23]. Via transduction of a COL7A1-specific RTM into RDEB keratinocytes using a lentiviral SIN vector, we achieved an RNA repair efficiency of ~2% in an isolated single cell clone, which resulted in restoration of normal levels of type VII collagen [7,24]. As type VII collagen has a reported half-life of two months, at least in murine skin [25], it can be assumed that a low percentage of COL7A1 repair at the RNA level may be sufficient to restore a wild-type phenotype in DEB (dystrophic EB) skin.
In general, a higher trans-splicing efficiency is probably needed for dominant negative mutations which also contribute to the etiology of other EB subtypes, as well as other genodermatoses such as epidermolytic ichthyosis [26,27]. In this regard, Wally et al. demonstrated that RNA trans-splicing-mediated repair of a dominant negative KRT14 mutation in EB simplex (EBS) keratinocytes resulted only in a partial reversion of the phenotype [6]. Thus, a higher reprogramming efficiency is still required for the treatment of dominantly inherited diseases and, certainly for any trans-splicing-based in vivo RNA repair strategy, which can be achieved for instance via splicing modulation using short antisense molecules [4,28].
ASOs can be designed to bind mutation-harboring exons during the pre-mRNA splicing process in order to promote their skipping [1,29,30]. In the optimal case, the resulting mRNA, while shorter, is translated into a fully functional protein. The feasibility of this strategy was recently shown in a mouse model, wherein skipping of the mutation-harboring exon 80 resulted in restoration of COL7A1 function [31]. In this study, however, the outcome of ASO application we explored was not the skipping of a disease-associated exon, but rather the enhancement of trans-splicing efficiency due to inhibition of competitive cis-splicing events within the target region. Studies in spinal muscular atrophy showed that co-expression of a therapeutic RTM engineered for SMN2 correction, together with an antisense RNA molecule that specifically blocked the competing cis-splice-site, led to an improvement in trans-splicing efficiencies both in vitro and in vivo [4,32]. Furthermore, our group recently demonstrated the possibility of enhancing the trans-splicing efficiency of a given RTM via antisense RNAs (asRNAs) in the dystrophic subtype of epidermolysis bullosa (DEB) using a GFP-split model system [28].
The aim of this study was to improve the RNA trans-splicing efficiency of a previously described therapeutic KRT14-RTM [6] by the co-administration of randomly generated asRNAs expressed via a plasmid and rationally designed short ASOs, applied as oligonucleotides, that bind potential splicing elements within the KRT14 target region. Dominant mutations in KRT14 are responsible for the generalized severe form of epidermolysis bullosa simplex (EBS-gen sev). Incorporation of the mutant K14 into the intermediate filament (IF) network compromises its integrity, leading to its collapse into protein aggregates under conditions of stress [6,33,34]. Using our established fluorescence-based screening system [21,22] we investigated the impact of KRT14-specific antisense molecules on the trans-splicing efficiency of our KRT14-RTM and on the general cis-splicing pattern within the KRT14 target region. We achieved increased RNA repair levels after the addition of specific antisense molecules of varying lengths in our screening system. Thus, we were able to identify potential splicing modulators that may further improve the RNA trans-splicing technology to a level sufficient to support its implementation in a clinical setting.
2. Results
2.1. Generation of Antisense Molecules for Trans-Splicing Enhancement
Aiming to increase the KRT14 repair efficiency via RNA trans-splicing in vitro, we adapted our well established fluorescence-based screening system [21] for the selection of randomly and rationally generated antisense molecules capable of directing the splicing reaction from cis to trans in the presence of an RTM (Figure 1A). We compared the functionality of asRNAs, some covering the majority of the respective KRT14 targeting region, and rationally designed ASOs that bind specifically to selected exon–intron boundaries in close proximity to the RTM binding site located within intron 7 of KRT14 (Figure 1A). Both antisense molecule variants are expected to increase the trans-splicing efficiency of the previously described and generated KRT14-specific RTM [6] via blocking or interfering with competitive, naturally occurring cis-splicing events within the KRT14 target region. The RTM was designed to replace the first seven exons of KRT14, covering all known EBS-associated mutations in this gene. The RTM was previously selected via our fluorescence-based screening system and confirmed to be functional in an EBS patient-derived keratinocyte line [6], although its efficiency warranted improvement to achieve a full phenotypic reversion in these cells.
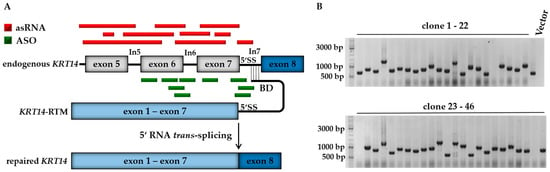
Figure 1.
The combined application of antisense strategies. (A) The RTM, the main component of the SMaRT system, carries the wild-type coding sequence of KRT14 spanning from exon 1 to exon 7, splicing elements for efficient splicing and a binding domain specific for intron 7 of KRT14. Antisense molecules were randomly (red) and rationally (green) generated in order to block competitive cis-splicing elements within the KRT14 target region, thereby facilitating the trans-splicing-induced RNA repair of our pre-selected RTM; (B) for asRNA library generation the PCR-amplified KRT14 target region (1142 bp), spanning from exon 5 to intron 7, was fragmented by sonication and the resulting fragments were randomly cloned into a pcDNA 4.0 expression plasmid. Colony PCR and sequence analysis of individual bacterial clones revealed the presence of various target sequences in sense and antisense orientation. Vector: pcDNA 4.0 expression vector without KRT14 sequence; RTM, RNA trans-splicing molecule; 5′ SS, 5′ splice site.
To generate the asRNA library, the KRT14 target region encompassing 1144 nt spanning exon 5 to intron 7 immediately 5′ proximal to the BD target site, was PCR-amplified and fragmented via sonication. The resulting fragments were randomly cloned into the pcDNA 4.0 vector (Invitrogen, Carlsbad, CA, USA) downstream to the human cytomegalovirus (CMV) promotor and maintained as individual plasmids expressing KRT14 sequences in either the sense or antisense orientation with respect to the target region (Figure 1B).
We obtained a library of 136 asRNAs of varying lengths from 19 nt to 620 nt. Using this library, we initially performed triple transfection experiments, introducing an artificial KRT14 screening minigene (KRT14-scMG; 0.5 µg) and the RTM screening vector (KRT14-scRTM; 0.5 µg) together with the individual asRNA plasmids (3 µg). The engineered KRT14-scMG consists of the KRT14 target region from exon 5 to the end of intron 7 (KRT14 nt: 3320–4462, NCBI Gene ID: 3861) fused to the 3′ half (nt: 337–720) of the GFP coding sequence. The screening RTM is comprised of the dsRed fluorescent reporter molecule sequence, a short linker sequence, the 5′ (nt: 1–337) half of the GFP-coding sequence, a functional 5′ splice site (5′ SS) for efficient splicing, and the BD specific for intron 7 of KRT14 (Figure 2) [6]. Successful and accurate trans-splicing between the pre-mRNA transcribed from the KRT14-scMG and the KRT14-scRTM resulted in the reconstitution of full-length GFP and translation of a functional protein in treated HEK293 cells detectable by flow cytometry and fluorescence microscopy (Figure 2). The intensity of the GFP signal correlates with the efficiency of the trans-splicing reaction. As such, changes in the GFP signal intensity in the presence of the different asRNAs would reflect the positive or negative impact of asRNAs on the trans-splicing efficiency [21,22].
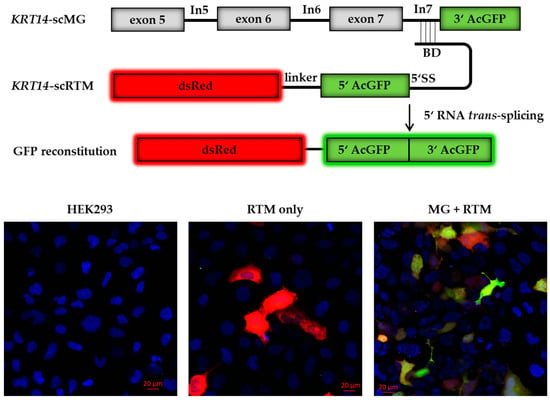
Figure 2.
Screening constructs for antisense molecule selection. The reporter-based screening system consists of an artificial KRT14 screening minigene (KRT14-scMG), containing the KRT14 target region spanning from exon 5 to intron 7 and the 3′ part of GFP, and the RTM screening vector (KRT14-scRTM) carrying a dsRed reporter molecule and the missing 5′ GFP part, splicing elements for efficient splicing and a binding domain specific for intron 7 of KRT14. Accurate trans-splicing between the KRT14-scMG and the KRT14-scRTM (red) restores the GFP expression (green) in the cell manifested in a yellow signal. Cell nuclei were 4′,6-diamidin-2-phenylindol (DAPI) stained (blue). BD, binding domain; 5′ SS, 5′ splice site.
From the original asRNA library, we initially tested 74 asRNAs (Table 1), varying in length and binding position within the KRT14 target region (Figure 3A), excluding those that targeted intron 7 in order to avoid direct binding competition between asRNA and RTM to the target pre-mRNA. Flow cytometric analysis revealed an increase in GFP expression levels from three-fold to 20-fold depending on the delivered asRNA (Figure 3B). Based on the initial results, achieved in two rounds of screening (HEK 1 and HEK 2; Figure 3B), we selected asRNA34, one of the most efficient asRNAs, for further investigation.

Table 1.
Randomly generated asRNAs.
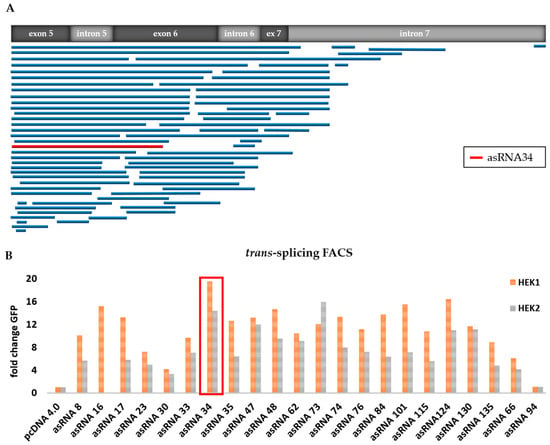
Figure 3.
Functional analysis of selected asRNAs. (A) Binding position of sequence-analysed individual asRNAs, expressed from the pcDNA 4.0 plasmid, within the KRT14 target region spanning from exon 5 to intron 7; (B) flow cytometric analysis of HEK293 cells upon triple-transfection with the KRT14-scRTM, the KRT14-scMG and individual asRNAs increased the trans-splicing efficiency in comparison to that detectable in HEK293 cells transfected with the screening molecules and an empty (without asRNA sequence) pcDNA4.0 expression plasmid in all transfection experiments. The level of accurate trans-splicing was quantified by flow cytometric counting of GFP-expressing cells two days post treatment and referred to the pcDNA4.0 control (set to 1). Two independent transfection experiments (HEK1 and HEK2) are shown.
2.2. asRNA34 Facilitates RTM-Mediated RNA Editing
We performed further triple transfection experiments in the HEK293 system using asRNA34 in order to confirm the results from our library screen (Figure 4A). In all described experiments, the total amount of DNA transfected was the same in each setting to exclude variabilities that could arise from transfection efficiency and cell toxicity when using different amounts of DNA. We used pcDNA 4.0 empty vector where needed to ensure equal levels of transfected plasmids. Combining the results of all experiments, we were able to achieve full-length GFP expression in ~42% of HEK293 cells treated with the KRT14-scMG (0.5 µg), KRT14-scRTM (0.5 µg), and asRNA34 (3 µg). In contrast, in cells receiving pcDNA4.0 (3 µg) instead of asRNA34, only ~9% of treated cells were GFP-positive by flow cytometric analysis (Figure 4B). The geometric mean of GFP-positive cells was 3.7 in the absence of asRNA34, and rose to 13.2 in the presence of asRNA34, representing a 3.6-fold increase in GFP intensity.
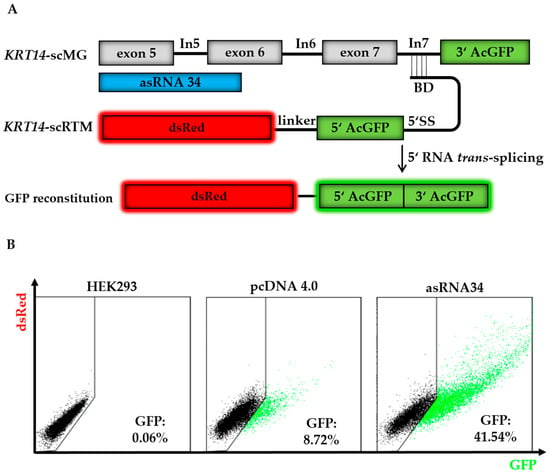
Figure 4.
asRNA34 increases trans-splicing efficiency in GFP-split model. (A) The co-delivery of the screening molecules (KRT14-scRTM and KRT14-scMG), carrying the respective GFP parts, results in full-length GFP expression upon accurate RNA trans-splicing. The addition of asRNA34, specific for KRT14 exon 5 to exon 6, into HEK293 cells is expected to increase the level of RNA trans-splicing represented by the amount of restored GFP in the cells; (B) triple-transfection experiments with the KRT14-scMG, the KRT14-scRTM, and asRNA34 in HEK293 cells led to an increase in GFP-expressing cells from ~9% (empty pcDNA4.0) to ~42%, measured by flow cytometry. BD, binding domain; 5′ SS, 5′ splice site.
Additionally, asRNA34 had a dose-dependent effect on GFP expression on the protein level, as assayed by flow cytometry and on RNA expression, evaluated by semi-quantitative real time PCR (sqRT-PCR) (Figure 5A,B). Together, these results revealed an increase in trans-splicing efficiency of up to 60-fold in the presence of 3 µg asRNA34 instead of 1 µg (Figure 5B). asRNA34 has a length of 324 nt and is predicted to be complementary to a region of KRT14 spanning the exon 5–intron 5 and intron 5–exon 6 junctions (Figure 4A). As such, asRNA34 is expected to impact cis-splicing of intron 5 and not intron 7. To understand how interfering with cis-splicing of intron 5 could positively impact trans-splicing, we first investigated the impact of increasing asRNA34 concentrations on cis-splicing levels at all exon–exon junctions within the KRT14-scMG pre-mRNA in the experiments described above. Towards this end, primers spanning the exon–exon junctions were designed to assess the number of mRNA molecules harbouring correctly spliced exons by sqRT-PCR. The analysis of competitive cis-splicing events within the KRT14-scMG upon co-delivery of different levels of asRNA34-expressing plasmids indicated a potential stabilizing effect of the asRNA on the target pre-mRNA as increased cis-splicing levels were detectable via sqRT-PCR analysis (Figure 5C). This finding was contrary to our initial assumption that blocking potential splicing elements within the KRT14-scMG resulted in reduced cis-splicing rates. In contrast, the amount of transfected RTM primarily impacted only the trans-splicing level, and not the cis-splicing level as shown via flow cytometric analysis (Figure 5D) and sqRT-PCR (Figure 5E,F).
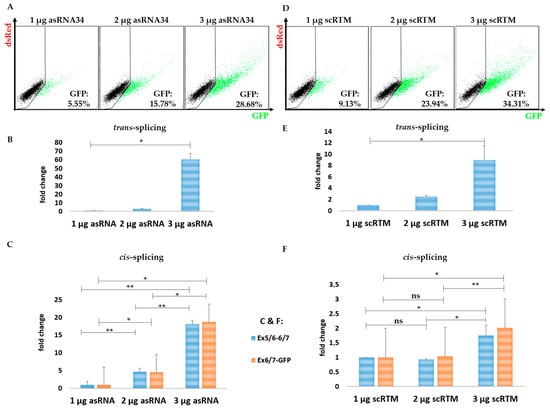
Figure 5.
The impact of asRNA34 on cis-splicing in the fluorescence-based setting. The transfection experiments further revealed an asRNA34 dose-dependent increase of the trans-splicing efficiency confirmed via flow cytometric analysis at protein level (A) and via sqRT-PCR at mRNA level (B). The asRNA34-mediated trans-splicing increment was accompanied by increased cis-splicing levels, detected via sqRT-PCR and normalized to GAPDH mRNA levels (C) (the mean value + SD of one representative experiment is shown), indicating a stabilizing effect on the target pre-mRNA by antisense molecule interference. Empty pcDNA 4.0 expression plasmids were transfected into HEK293 cells to adjust total plasmid levels delivered into the target cells. Increasing the amount of transfected RTMs results in an increased GFP expression as detected via flow cytometry (D) and sqRT-PCR (E), although the cis-splicing rate within the KRT14-scMG remains largely unaffected (F). An unpaired Student’s t-test (two-tailed) was performed with the GraphPad Prism software (GraphPad Software, San Diego, CA, USA) to prove statistical significance (Table 2 and Table 3). Significance values are given in asterisks (ns = not significant; * p-value ≤ 0.05; ** p-value ≤ 0.01).
2.3. Splice Site-Specific Antisense Oligonucleotides Increase RNA Trans-Splicing Levels
Although asRNA34 could confer a significant improvement on the KRT14-scRTM’s RNA trans-splicing efficiency, we also observed increased cis-splicing of intron 7 with increasing concentrations of the asRNA, which would be in direct competition with our desired trans-splicing reaction. To avoid the unwanted effect on cis-splicing, we generated shorter site-specific ASOs (Table 4) designed to directly block competitive 5′ splice sites (ASO1, 2, 3, 6, 7, 8), potential splice site enhancer sequences (ASO4, 9) (Figure S1), or both, within the KRT14-scMG sequence. In total, nine ASOs 20–24 nts in length were rationally designed to cover exon 6 to intron 7 including the exon–intron junctions upstream of the RTM binding site within the KRT14 target region (Figure 6A). Coady et al. (2008) recently showed that the inhibition of cis-splicing of adjacent introns can have a positive impact on the trans-splicing efficiency of a given RTM [32]. Therefore, in addition to ASOs specific for the exon–intron 7 junction, we also tested ASOs 1, 2, and 3, which target the exon–intron 6 junction further upstream to the RTM binding site. A scrambled ASO (scrASO), which was designed not to target any region within the KRT14-scMG, was included as a control in initial triple transfection experiments. Flow cytometric analysis of transfected HEK293 cells revealed a significant increase of GFP-expressing cells from 11% (scrASO) up to 33% depending on the ASO introduced (Figure 6B). ASO7 and ASO9, binding the exon 7–intron 7 junction and the 5′ end of exon 7, respectively, represented the most promising ASOs, resulting in 33% and 30% GFP-positive cells in the screen. We confirmed the results with these two ASOs in additional experiments, obtaining a similar increase in GFP-positive cell numbers from 15% (scrASO) to 30% (ASO7) and 29% (ASO9) (Figure 6C). Furthermore, Western blot analysis of whole cell lysates from triple-transfected HEK293 cells validated the elevation of trans-spliced chimeric products as indicated by an increase in GFP expression (Figure 6D).
Table 4.
Rationally designed ASOs.
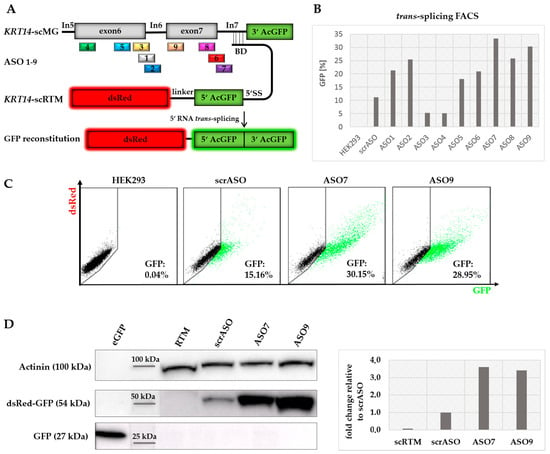
Figure 6.
Comparison of rationally designed ASOs. (A) ASOs were designed specifically for the exon 6 to intron 7 junctions within the KRT14 target region upstream to the RTM binding site; (B) in contrast to the scrASOs, acting as mock control in the experiment, seven ASOs were capable of increasing the trans-splicing activity of the RTM at an ASO concentration of 70 nmol, quantified via flow cytometric analysis; (C) the most promising ASOs (ASO7 and ASO9), specific for exon 7 and the exon 7–intron 7 junction of KRT14, induced the trans-splicing-mediated GFP restoration in 30% (ASO7) and 29% (ASO9) of all analysed cells, whereas in the mock control (scrASO) only 15% of the cell population expressed GFP; (D) Western blot analysis revealed an over three-fold increase of full-length dsRed-GFP (54 kDa) expression at protein level in HEK293 cells treated with ASO7 and ASO9 in comparison to cells treated with the scrASO. eGFP (27 kDa) was used as positive control to confirm the specificity of the antibody. The loading control α-actinin (100 kDa) was used for normalization.
We additionally verified this increase in trans-splicing efficiency at the mRNA level via sqRT-PCR analysis (Figure S2), revealing an up to 2–4-fold increase in the levels of reconstituted GFP mRNA in cells harboring the selected ASOs as compared to scrASO-treated controls (Figure 7A). In contrast to the results obtained with asRNA34, the shorter ASO7 and ASO9 had no significant effect on the general cis-splicing events occurring within the KRT14-scMG. Rather the levels of cis-splicing within the target pre-mRNA were slightly reduced (~2-fold) in the presence of ASO7, whereas ASO9 exhibited no significant impact on these events (Figure 7A). Increasing the concentrations of ASO7 added to the trans-splicing reaction, revealing the highest trans-splicing efficiency at a concentration of 60 nmol (Figure 7B).
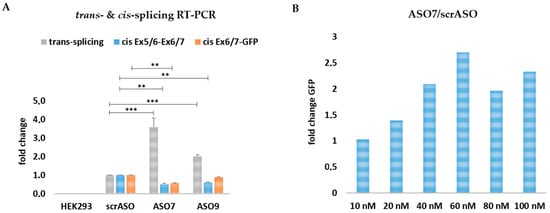
Figure 7.
Comparison of rationally designed ASOs on RNA level. (A) sqRT-PCR analysis revealed a ~4-fold increase in GFP expression in ASO7 and a ~2-fold increase in ASO9-treated cells in comparison to cells treated with the scrambled ASO. An unpaired Student’s t-test (two-tailed) was performed with GraphPad Prism software (GraphPad Software, San Diego, CA, USA) to prove the statistical significance between scrASO and ASO7, as well as scrASO and ASO9 (Table 5). The cis-splicing activity within the Ex5/6-Ex6/7 KRT14-scMG region was significantly decreased for ASO7 and ASO9. Within the Ex6/7-GFP region the cis-splicing activity was decreased for ASO7 but unaffected for ASO9; (B) the co-delivery of the KRT14-scMG, the KRT14-scRTM, and different ASO7 concentrations into HEK232 cells resulted in the most promising trans-splicing efficiency at an ASO concentration of 60 nmol. HEK293 cells treated with the screening constructs and respective scrambled ASO concentrations were set to 1. Significance values are given in asterisks (** p-value ≤ 0.01; *** p-value ≤ 0.001).
3. Discussion
In recent years, RNA trans-splicing has been predominantly applied in RNA repair studies for various genetic diseases or for the RTM-mediated delivery of “suicide genes” into tumour cells [35,36]. Reprogramming at the pre-mRNA level represents an elegant approach for the editing of aberrant RNA molecules that maintains endogenous regulation of the target transcript, making the technology potentially safe and well suited for the therapy of certain genetic conditions. These include disorders caused by mutations affecting large genes or dominantly inherited mutations, difficult to correct via conventional full-length cDNA replacement strategies. In this respect, we have already demonstrated promising results with a COL7A1-targeting RTM capable of correcting ~40% of all RDEB-associated mutations encoded within the ~3300-nucleotide long target pre-mRNA [7]. In the case of dominant negative mutations in EBS-gen sev, Cao et al. showed with an inducible EBS mtK14 mouse model, that dominance of the mutation is dependent on the ratio of wild-type to mutated alleles. A reduction in expression of the mutated allele by ~50% appeared sufficient to overcome the EBS phenotype [37]. In this respect, RNA trans-splicing-mediated correction of the mutant RNA is expected to not only increase the amount of wild-type KRT14 transcripts, but also simultaneously decrease the level of mutant transcripts by competing with the normal cis-splicing events. Indeed, we could achieve partial reversion of the phenotype of EBS keratinocytes associated with a dominant-negative mutation in KRT14 with an appropriate KRT14-targeting RTM [6].
With the recent advances in the identification and selection of the BD for the RTM [18,21,22], the main obstacle that remains is the low efficiency of the technique, which limits its potential for future in vivo or clinical application. Therefore, we have developed a fluorescence-based screening system aimed at identifying factors with the potential to increase the trans-splicing efficiency, such as antisense RNA molecules [28]. In the course of this study, we could identify antisense RNA molecules capable of enhancing the trans-splicing efficiency induced by the KRT14-targeting RTM by up to 20-fold. The system proved practical for the screening of an unbiased library of randomly generated sequences of varying lengths and binding positions along the target pre-mRNA, as well as short, site-specific ASOs rationally designed to block splice sites and splicing enhancer elements. Our results revealed both antisense strategies to be attractive for future RNA repair studies in patient cells, but also highlight several points for consideration.
In the described screening system, the KRT14-scRTM induced a trans-splicing reaction between the 5′ GFP coding sequence on the RTM and the 3′ GFP sequence located immediately after intron 7 on the target pre-mRNA molecule. Notably, some of the best-acting antisense molecules identified (asRNA34 and ASO9) were not expected to directly inhibit the competing cis-splicing of intron 7. ASO9 was expected to bind to the 5′ region of exon 7, whereas asRNA34 bound a 324-nt region spanning all of exon 5, intron 5, and half of exon 6. As such, asRNA34 was rather expected to interfere with cis-splicing of intron 5, whereas ASO9 may or may not interfere with cis-splicing of intron 6. The mechanisms by which these molecules enhanced trans-splicing efficiency are not yet completely understood. However, blocking potential splicing enhancer and silencer sequences located within the KRT14-scMG is likely to impact both types of splicing reactions (cis and trans). asRNA34, in particular, would block many predicted enhancer and silencer sequences, and thereby affect the general spliceosome-mediated splicing efficiency (Figure S3) in an unpredictable manner. In this respect, increasing amounts of asRNA34 added to the cells led to a significant enhancement of both cis- and trans-splicing reactions. Besides blocking potential splicing motifs within the MG sequence, asRNA34 may also exert a general stabilizing effect on the target pre-mRNA that promotes both cis- and trans-splicing reactions. Thus, while we achieved increased trans-splicing rates by the addition of asRNA34, the concurrent upregulation of cis-splicing in the target pre-mRNA would be expected to negate any of its associated benefits, as no alteration in the ratios of wild-type (corrected) transcripts to mutant transcripts can be expected.
In contrast to long asRNA sequences, the mechanisms of action by which short ASOs exert their effects are more clearly defined, as they are designed to inhibit splicing either by directly blocking the splice donor/acceptor sites located within the exon–intron junctions (as in the case of ASO7), or by blocking potential splicing enhancer motifs adjacent to the intron–exon junction (ASO9). Both ASO7 and ASO9 resulted in enhanced trans-splicing of the RTM without any detectable increase in cis-splicing events within the target-pre-mRNA, suggesting that these shorter ASOs are better suited for our purposes. The most auspicious ASO7, targeting the exon 7–intron 7 boundary, additionally exhibited an inhibitory effect on the competitive cis-splicing of intron 7 as expected. The observed 2-fold inhibition in cis-splicing of intron 7, combined with the 4-fold increase in trans-splicing will not only reduce the level of mutated transcripts in the patient cells but also shift the ratio of wild-type to mutant transcripts further towards wild-type. This is expected to decrease the dominant negative effect of the disease-causing mutation to a level that may be sufficient to fully restore the wild-type phenotype in patient cells.
This study describes a straightforward and practical protocol that can be adapted to the screening of any type of splicing modulator, including pharmacological compounds. Our results also demonstrate that there are different mechanisms by which antisense technologies can impact splicing events and further underscore the need to comprehensively evaluate the effects of such molecules on different processes and outcomes including, for example, RNA stability. To this end, future studies will not only focus on evaluation of this therapeutic strategy in EBS patient-derived keratinocytes, but also on the investigation of mechanisms of action and precise quantification of corrected and mutant transcripts using next-generation sequencing platforms.
4. Materials and Methods
4.1. Construction of asRNA Library and Antisense Oligonucleotides
An asRNA library was constructed by the fragmentation of the PCR-amplified KRT14 target region spanning from exon 5 to intron 7 by sonication according to an already established protocol [21,28]. PCR amplification was performed using genomic DNA from a healthy donor and the GoTaq DNA polymerase (Promega, Madison, MI, USA). The resulting fragments were treated with a DNA Terminator End Repair Kit (Lucigen, Middleton, WI, USA), cloned into a pcDNA 4.0 plasmid (Invitrogen, Carlsbad, CA, USA) using the restriction site for EcoRV, and analyzed for their correct orientation (complementary to the target region) by sequence analysis. PCR products were digested with the corresponding restriction enzymes for 1 h at 37 °C, purified with a Illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, Chalfont St. Giles, UK) and ligated with T4 DNA ligase (Thermo Fisher Scientific, Waltham, MA, USA), all according to the manufacturer’s protocols. The ligation was then transformed into chemically competent SoloPack® Gold Competent Cells (Agilent Technologies, Santa Clara, CA, USA) and plasmid preparations were carried out using a Plasmid Mini Prep Kit (Sigma-Aldrich, Taufkirchen, Germany), according to the manufacturer’s protocol. Sequence analysis of all plasmids and PCR products was performed using a 3500 ABI automated sequence analyzer and ABI PRISM dye terminator cycle sequencing kit (Applied Biosystems, Foster City, CA, USA).
4.2. Screening Constructs
Screening constructs used for detection of cis-splicing and correct trans-splicing were designed and cloned according to the protocol of Bauer et al. [21]. Concisely said, the KRT14-scRTM carries a 5′ split-GFP part consisting of the first nucleotides (nt: 1–336) from a full length acGFP (pacGFP vector, Clontech, Mountain View, CA, USA) and a BD sequence that specifically targets Intron 7 of KRT14 cloned into the pcDNA3.1D/V5-HIS (Invitrogen, Carlsbad, CA, USA) backbone. The KRT14-scMG spanning exon 5 to intron 7 was generated using a KRT14 exon 5 forward primer (5′-GATCAAGCTTCACCACAGAGGAGCTGAACCGCGAGGTGGC-3′) and a KRT14 intron 7 reverse primer (5′-GATCGGATCCGGGGAAGAGGTGGGAAGAGGACGTTACC-3′) for amplification via GoTaq DNA polymerase (Promega, Madison, MI, USA). Afterwards, the amplified PCR product was cloned into the screening vector backbone downstream to the CMV-promoter (pcDNA3.1, Invitrogen, Carlsbad, CA, USA) using HindIII and BamHI restriction sites [21,28]. The KRT14-scMG also carries the 3′ split-GFP portion consisting of the rearward part of acGFP (nt: 337–720) to enable a reconstitution of the full length acGFP upon accurate trans-splicing.
4.3. Cell Culture and Plasmid Transfection
For triple-transfection studies, the human embryonic kidney cell line HEK293 (Stratagene, La Jolla, CA, USA) was used and grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/mL penicillin–streptomycin (Biochrom, Berlin, Germany) at 37 °C and 5% CO2 in a humidified incubator. Passaging of the cells was carried out with Trypsin (0.05%)-EDTA (0.02%) (Biochrom, Berlin, Germany) treatment followed by Trypsin-EDTA inactivation with Solution A (7.25 g HEPES; 1.8 g glucose; 0.22 g NaCl; 0.27 g Na2HPO4; 1 mL phenol red; 900 mL H2O; pH 7.4) supplemented with 10% FBS. Cells were centrifuged at 350× g for 5 min and seeded in fresh tissue culture 6-well plates or 60 mm plates.
Transient transfections of screening plasmids were performed the next day using the jetPEI reagent (Polyplus-transfection SA, Illkirch, France) according to the manufacturer’s protocol. The transfected DNA amounts varied in the different experiments: Section 2.2: 0.5 µg KRT14-MG + 0.5 µg RTM + 3 µg asRNA were transfected into HEK293 cells cultivated in 6-well plates; Section 2.3: 0.5 µg KRT14-MG + 0.5 µg RTM + 70 nM ASO were transfected into HEK293 cells cultivated in 6-well plates.
4.4. Flow Cytometric Analysis
The amount of trans-splicing events, manifested in the expression of GFP, was measured 48–72 h after transfection using a Beckman Coulter FC-500 FACS analyzer (Beckman Coulter, Vienna, Austria). The transfected HEK293 cells were washed once with phosphate buffer saline (Dulbecco’s PBS) and detached from the cell culture plates using Trypsin-EDTA. Trypsin-EDTA was inactivated with Solution A (7.25 g HEPES; 1.8 g glucose; 0.22 g NaCl; 0.27 g NaHPO4; 1 mL phenol red; 900 mL H2O; pH 7.4) supplemented with 10% FBS. After centrifugation for 5 min at 350× g, 10,000 cells were analyzed according to their GFP expression and geometric mean with the Kaluza 1.3 software (Beckman Coulter, Brea, CA, USA).
4.5. RNA Isolation and cDNA Synthesis
HEK293 cells were harvested 48–72 h post transfection and RNA was isolated using the Innuprep RNA Mini Kit (Analytik Jena, Jena, Germany) according to the manufacturer’s protocol. Afterwards cDNA synthesis was performed with 3 µg RNA using the iScript™ cDNA Synthesis Kit (BioRad, Hercules, CA, USA) according to the manufacturer’s protocol.
4.6. sqRT-PCR Analysis of Cis- and Trans-Splicing Levels
sqRT-PCR was performed to detect fused GFP transcripts in treated HEK293 cells. For sqRT-PCR analysis, a 5′ GFP-specific forward primer (5′-GCTGACCCTGAAGTTCATCTG-3′), a 3′ GFP-specific reverse primer (5′-CGCCGATGGGGGTATTCTGCTGG-3′), cDNA of treated cells and GoTaq® qPCR Master Mix (Promega, Madison, WI, USA) was used. The PCR was performed using a Bio-Rad CFX™ system (BioRad, Hercules, CA, USA) with the following conditions: 95 °C for 2 min, and 50 cycles of 20 s at 95 °C, 20 s at 62 °C, and 25 s at 72 °C. Experiments were carried out in duplicates and repeated two times. Correct PCR products were verified by direct sequence analysis.
To analyze differences in cis-splicing events after transfection of HEK293 cells exon–exon junction-specific forward (Exon5/6: 5′-AGCTCAGCATGAAAGCATCCCT-3′, Exon6/7: 5′-AGGACGCCCACCTCTCCTCC-3′) and reverse (Exon6/7 5′-GGAGGAGAGGTGGGCGTCCT-3′, GFP: 5′-GGTCAGCTCGATGCGATTCACC-3′) primers, cDNA of treated cells and GoTaq® qPCR Master Mix (Promega, Madison, WI, USA) was used. The PCR was performed using a Bio-Rad CFX™ system with the following conditions: 95 °C for 2 min, and 50 cycles of 20 s at 95 °C, 20 s at 64 °C, and 25 s at 72 °C. Experiments were carried out in duplicates and repeated two times. Correct PCR products were verified by direct sequence analysis.
4.7. Western Blot Analysis
Enhancement of asRNA/ASO-induced trans-splicing manifests in an increase in full-length GFP expression, which was quantified at the protein level via Western blot analysis 48–96 h post transfection. HEK293 cells were washed with PBS, harvested, and resuspended in radioimmunoprecipitation assay buffer RIPA (Santa Cruz Biotechnology, Heidelberg, Germany). Samples were denatured for 5 min at 95 °C in 4× SDS loading buffer (0.25 M Tris-HCl; 8% SDS; 30% glycerol; 0.02% bromphenol blue; 0.3 M β-mercaptoethanol; pH 6.8). SDS-gel-electrophoresis was performed at 140 V for a maximum of 2 h using a NuPAGE 12% Bis-Tris gel (Invitrogen, Carlsbad, CA, USA) and NuPAGE MOPS running buffer. Proteins were electro-blotted onto a nitrocellulose membrane (Amersham Hybond-ECL, Amersham, Buckinghamshire, UK) at 0.25 A for 75 min. The membrane was blocked with Western Blocking Reagent (Roche, Basel, Switzerland) in Tris buffered saline (TBS) + 0.2% Tween (TBS-T) for 1 h at RT and incubated with a primary anti-GFP rabbit IgG antibody (MBL-598, MBL International, Woburn, MA, USA) in a dilution of 1:750 in TBS-T at 6 °C overnight. After three washing steps (3 × 10 min) with TBS-T the membrane was incubated with a secondary HRP Envision+ labelled anti rabbit antibody (1:100 in TBS-T, Agilent Technologies, Santa Clara, CA, USA) and incubated for 90 min at room temperature. Finally, GFP expression was visualized using the Luminata™ Forte Western HRP Substrate (Millipore, Burlington, MA, USA). α-actinin (H-2) mouse monoclonal IgG antibody (1:1000 in TBS-T; Santa Cruz. Dallas, TX, USA) and a secondary HRP Envision+ labelled anti mouse antibody (1:100 in TBS-T Tween, Agilent Technologies, Santa Clara, CA, USA) were used to detect α-actinin which served as protein loading control. The relative quantification of GFP expression was measured and calculated using the Image Lab 5.2.1 (Bio-Rad) software.
4.8. Statistical Analysis of SqRT-PCR Data
For statistical analysis, an unpaired Student’s t-test (two-tailed) was performed with the GraphPad Prism 5.03 software (GraphPad Software, San Diego, CA, USA) to prove statistical significance between the values of controls (pcDNA 4.0 & scrASO) and samples (asRNA34, ASO7, ASO9). A sample size of at least n = 4 was analyzed for mean ± SEM to provide a p-value.
4.9. Analysis of Splicing Enhancer and Silencer
DNA sequence motifs, that probably regulate/influence the general splicing mechanism within the KRT14 targeting region, were predicted using the prediction websites http://regrna.mbc.nctu.edu.tw/html/prediction.html and http://www.umd.be/HSF3/HSF.shtml [38].
Supplementary Materials
Supplementary materials can be found at http://www.mdpi.com/1422-0067/19/3/762/s1.
Acknowledgments
The work was funded by Austrian Science Fund (FWF): P 27336-B26, DEBRA Austria, and Paracelsus Medical University, Salzburg, Austria (PMU-FF): A-15/01/016-KOL.
Author Contributions
Ulrich Koller, Josefina Piñón Hofbauer, and Verena Wally conceived and designed the experiments; Bernadette Liemberger, Claudia Arzt, Stefan Hainzl, and Thomas Kocher performed the experiments and analysed the data; Bernadette Liemberger, Josefina Piñón Hofbauer, and Ulrich Koller wrote the paper; Eva M. Murauer and Ulrich Koller established the fluorescence-based screening system; Eva M. Murauer, Verena Wally, Julia Reichelt, and Josefina Piñón Hofbauer were involved in paper editing; Julia Reichelt and Johann W. Bauer were involved in final paper editing and gave final approval for publishing the manuscript.
Conflicts of Interest
Johann W. Bauer is an inventor on US (US8735366) and European (EP2320952) patent “Improved pre-mRNA trans-splicing molecules (RTM) and their uses”.
Abbreviations
| ASO | antisense oligonucleotide |
| asRNA | antisense RNA |
| BD | binding domain |
| EB | epidermolysis bullosa |
| EBS | epidermolysis bullosa simplex |
| DEB | dystrophic epidermolysis bullosa |
| KRT14-scMG | KRT14 screening minigene |
| KRT14-scRTM | KRT14 screening RTM |
| RDEB | recessive dystrophic epidermolysis bullosa |
| RTM | RNA trans-splicing molecule |
| SMaRT | Spliceosome mediated RNA trans-splicing |
References
- Bornert, O.; Peking, P.; Bremer, J.; Koller, U.; van den Akker, P.C.; Aartsma-Rus, A.; Pasmooij, A.M.G.; Murauer, E.M.; Nyström, A. RNA-based therapies for genodermatoses. Exp. Dermatol. 2017, 26, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Popplewell, L.; Athanasopoulos, T.; Dickson, G. Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Hum. Gene Ther. 2014, 25, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Coady, T.H.; Lorson, C.L. Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J. Neurosci. 2010, 30, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Lorain, S.; Peccate, C.; Le Hir, M.; Garcia, L. Exon Exchange Approach to Repair Duchenne Dystrophin Transcripts. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Brunner, M.; Lettner, T.; Wagner, M.; Koller, U.; Trost, A.; Murauer, E.M.; Hainzl, S.; Hintner, H.; Bauer, J.W. K14 mRNA reprogramming for dominant epidermolysis bullosa simplex. Hum. Mol. Genet. 2010, 19, 4715–4725. [Google Scholar] [CrossRef] [PubMed]
- Peking, P.; Koller, U.; Duarte, B.; Murillas, R.; Wolf, S.; Maetzig, T.; Rothe, M.; Kocher, T.; García, M.; Brachtl, G.; et al. An RNA-targeted therapy for dystrophic epidermolysis bullosa. Nucleic Acids Res. 2017, 45, 10259–10269. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Klausegger, A.; Koller, U.; Lochmüller, H.; Krause, S.; Wiche, G.; Mitchell, L.G.; Hintner, H.; Bauer, J.W. 5′ trans-splicing repair of the PLEC1 gene. J. Investig. Dermatol. 2008, 128, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Koller, U.; Wally, V.; Mitchell, L.G.; Klausegger, A.; Murauer, E.M.; Mayr, E.; Gruber, C.; Hainzl, S.; Hintner, H.; Bauer, J.W. A novel screening system improves genetic correction by internal exon replacement. Nucleic Acids Res. 2011, 39, E108. [Google Scholar] [CrossRef] [PubMed]
- Monjaret, F.; Bourg, N.; Suel, L.; Roudaut, C.; Le Roy, F.; Richard, I.; Charton, K. Cis-splicing and Translation of the Pre-Trans-splicing Molecule Combine With Efficiency in Spliceosome-mediated RNA Trans-splicing. Mol. Ther. 2014, 22, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Avale, M.E.; Rodríguez-Martín, T.; Gallo, J.-M. Trans-splicing correction of tau isoform imbalance in a mouse model of tau mis-splicing. Hum. Mol. Genet. 2013, 22, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Rindt, H.; Yen, P.-F.; Thebeau, C.N.; Peterson, T.S.; Weisman, G.A.; Lorson, C.L. Replacement of huntingtin exon 1 by trans-splicing. Cell. Mol. Life Sci. 2012, 69. [Google Scholar] [CrossRef] [PubMed]
- Haugen, P.; Simon, D.M.; Bhattacharya, M. The natural history of group I introns. Trends Genet. 2005, 21, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.E.; Müller, U.F. An in vivo selection method to optimize trans-splicing ribozymes. RNA 2012, 18, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Lan, N.; Howrey, R.P.; Lee, S.W.; Smith, C.A.; Sullenger, B.A. Ribozyme-mediated repair of sickle β-globin mRNAs in erythrocyte precursors. Science 1998, 280, 1593–1596. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.-S.; Jung, H.-S.; Song, M.-S.; Cho, K.S.; Kim, S.-C.; Kimm, K.; Jeong, J.S.; Kim, I.-H.; Lee, S.-W. Specific regression of human cancer cells by ribozyme-mediated targeted replacement of tumor-specific transcript. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Kim, K.T.; Lee, S.-J.; Hong, S.-H.; Moon, J.Y.; Yoon, E.K.; Kim, S.; Kim, E.O.; Kang, S.H.; Kim, S.K.; et al. Image-aided Suicide Gene Therapy Utilizing Multifunctional hTERT-targeting Adenovirus for Clinical Translation in Hepatocellular Carcinoma. Theranostics 2016, 6, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Koller, U.; Wally, V.; Bauer, J.W.; Murauer, E.M. Considerations for a Successful RNA Trans-splicing Repair of Genetic Disorders. Mol. Ther. Nucleic Acids 2014, 3, E157. [Google Scholar] [CrossRef] [PubMed]
- Hüttner, C.; Murauer, E.M.; Hainzl, S.; Kocher, T.; Neumayer, A.; Reichelt, J.; Bauer, J.W.; Koller, U. Designing Efficient Double RNA trans-Splicing Molecules for Targeted RNA Repair. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.-D.; Bruckner-Tuderman, L.; Eady, R.A.J.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.W.; Murauer, E.M.; Wally, V.; Koller, U. RNA trans-splicing for genodermatoses. Methods Mol. Biol. 2013, 961, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Murauer, E.M.; Koller, U.; Hainzl, S.; Wally, V.; Bauer, J.W. A reporter-based screen to identify potent 3’ trans-splicing molecules for endogenous RNA repair. Hum. Gene Ther. Methods 2013, 24, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Peking, P.; Koller, U.; Hainzl, S.; Kitzmueller, S.; Kocher, T.; Mayr, E.; Nyström, A.; Lener, T.; Reichelt, J.; Bauer, J.W.; et al. A Gene Gun-mediated Nonviral RNA trans-splicing Strategy for COL7A1 Repair. Mol. Ther. Nucleic Acids 2016, 5, E287. [Google Scholar] [CrossRef] [PubMed]
- Tockner, B.; Kocher, T.; Hainzl, S.; Reichelt, J.; Bauer, J.W.; Koller, U.; Murauer, E.M. Construction and validation of an RNA trans-splicing molecule suitable to repair a large number of COL7A1 mutations. Gene Ther. 2016, 23, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Remington, J.; Wang, X.; Hou, Y.; Zhou, H.; Burnett, J.; Muirhead, T.; Uitto, J.; Keene, D.R.; Woodley, D.T.; Chen, M. Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Syder, A.J.; Yu, Q.C.; Letai, A.; Paller, A.S.; Fuchs, E. The genetic basis of epidermolytic hyperkeratosis: A disorder of differentiation-specific epidermal keratin genes. Cell 1992, 70, 811–819. [Google Scholar] [CrossRef]
- Chipev, C.C.; Korge, B.P.; Markova, N.; Bale, S.J.; DiGiovanna, J.J.; Compton, J.G.; Steinert, P.M. A leucine—Proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell 1992, 70, 821–828. [Google Scholar] [CrossRef]
- Koller, U.; Hainzl, S.; Kocher, T.; Hüttner, C.; Klausegger, A.; Gruber, C.; Mayr, E.; Wally, V.; Bauer, J.W.; Murauer, E.M. Trans-Splicing Improvement by the Combined Application of Antisense Strategies. Int. J. Mol. Sci. 2015, 16, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Turczynski, S.; Titeux, M.; Pironon, N.; Hovnanian, A. Antisense-mediated exon skipping to reframe transcripts. Methods Mol. Biol. 2012, 867, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Sawamura, D.; Nishie, W.; Sakai, K.; McMillan, J.R.; Akiyama, M.; Shimizu, H. Targeted skipping of a single exon harboring a premature termination codon mutation: Implications and potential for gene correction therapy for selective dystrophic epidermolysis bullosa patients. J. Investig. Dermatol. 2006, 126, 2614–2620. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Lu, Z.; Li, F.; Wang, W.; Qian, N.; Duan, J.; Zhang, Y.; Wang, F.; Chen, T. Efficient in vivo gene editing using ribonucleoproteins in skin stem cells of recessive dystrophic epidermolysis bullosa mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Coady, T.H.; Baughan, T.D.; Shababi, M.; Passini, M.A.; Lorson, C.L. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS ONE 2008, 3, E3468. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.-D. Inherited epidermolysis bullosa. Orphanet J. Rare Dis. 2010, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.; Peking, P.; Klausegger, A.; Murauer, E.M.; Hofbauer, J.P.; Wally, V.; Lettner, T.; Hainzl, S.; Ablinger, M.; Bauer, J.W.; et al. Cut and Paste: Efficient Homology-Directed Repair of a Dominant Negative KRT14 Mutation via CRISPR/Cas9 Nickases. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.; Gratz, I.K.; Murauer, E.M.; Mayr, E.; Koller, U.; Bruckner-Tuderman, L.; Meneguzzi, G.; Hintner, H.; Bauer, J.W. Spliceosome-mediated RNA trans-splicing facilitates targeted delivery of suicide genes to cancer cells. Mol. Cancer Ther. 2011, 10, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.; Koller, U.; Murauer, E.M.; Hainzl, S.; Hüttner, C.; Kocher, T.; South, A.P.; Hintner, H.; Bauer, J.W. The design and optimization of RNA trans-splicing molecules for skin cancer therapy. Mol. Oncol. 2013, 7, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Longley, M.A.; Wang, X.-J.; Roop, D.R. An Inducible Mouse Model for Epidermolysis Bullosa Simplex. J. Cell Biol. 2001, 152, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, E67. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).