Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology



Abstract
:
1. Introduction
1.1. A Brief Introduction to Parkinson’s Disease and Pathological Roles of α-Synuclein
- MDVFMKGLSKAKEGVVAAAEKTKQGVAEAAGKTKEGVLYVGSKTKEGVVHGVATVAEKTKEQVTNVGGAVVTGVTAVAQKTVEGAGSIAAATGFVKKDQLGKNEEGAPQEGILEDMPVDPDNEAYEMPSEEGYQDYEPEA
1.2. α-Synuclein Mutations and Parkinson’s Disease
1.3. A53T Mutation
1.4. A30P Mutation
1.5. E46K Mutation
1.6. H50Q and G51D Mutations
1.7. Some Approaches to the PD Treatment
1.8. A Brief Introduction to Alzheimer’s Disease and Amyloid-β (Aβ)
- DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA
1.9. Aβ Mutations and Alzheimer’s Disease
Mutations in N-Terminal Region
1.10. Mutations in the Middle Part of Aβ
1.11. Some Approaches to the AD Treatment
1.12. Current Challenges in Designing Dugs for PD and AD Treatment
1.13. Experimental and Computational Approaches for the Analysis of αS and Aβ Structures
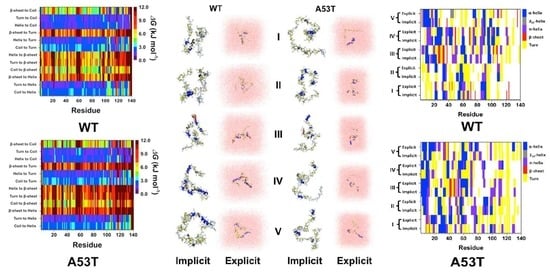
2. Artificial and Pathological Mutations in α-Synuclein: Insights from Molecular Dynamics Simulations
3. Understanding the Outputs of Artificial and Pathological Mutations in Aβ: Insights from the Molecular Dynamics Simulations
4. Aβ and αS Interactions: Insights from the Molecular Dynamics Simulations
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Lim, K.L.; Dawson, V.L.; Dawson, T.M. The cast of molecular characters in Parkinson’s disease: Felons, conspirators, and suspects. Ann. N. Y. Acad. Sci. 2003, 991, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Siderowf, A.; Stern, M. Ubetapdate on Parkinson disease. Ann. Intern. Med. 2003, 138, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.T.; Schapira, A.H. Genetic and environmental factors in the cause of Parkinson’s disease. Ann. Neurol. 2003, 53, S16–S23. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Deleersnijder, A.; Gerard, M.; Debyser, Z.; Baekelandt, V. The remarkable conformational plasticity of α-synuclein: Blessing or curse? Trends Mol. Med. 2013, 19, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Hot, hotter, and hottest trends in α-synuclein research. Curr. Protein Pept. Sci. 2015, 16, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, C.; Li, D.; Tian, Z.; Lai, Y.; Diao, J.; Liu, C. Versatile structures of α-synuclein. Front. Mol. Neurosci. 2016, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Looking at the recent advances in understanding α-synuclein and its aggregation through the proteoform prism. F1000Research 2017, 6, 525. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [PubMed]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [PubMed]
- Munishkina, L.A.; Phelan, C.; Uversky, V.N.; Fink, A.L. Conformational behavior and aggregation of α-synuclein in organic solvents: Modeling the effects of membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Fink, A.L. Lipid binding inhibits α-synuclein fibril formation. J. Biol. Chem. 2003, 278, 16873–16877. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Li, J.; Fink, A.L. The association of α-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem. 2003, 278, 40186–40197. [Google Scholar] [CrossRef] [PubMed]
- Bussell, R., Jr.; Eliezer, D. Effects of Parkinson’s disease-linked mutations on the structure of lipid-associated α-synuclein. Biochemistry 2004, 43, 4810–4818. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D.L.; Nemani, V.M.; Voglmaier, S.M.; Anthony, M.D.; Ryan, T.A.; Edwards, R.H. Neural activity controls the synaptic accumulation of α-synuclein. J. Neurosci. 2005, 25, 10913–10921. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, E.R.; Ramlall, T.F.; Borbat, P.P.; Freed, J.H.; Eliezer, D. Membrane-bound α-synuclein forms an extended helix: Long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 2008, 130, 12856–12857. [Google Scholar] [CrossRef] [PubMed]
- Dikiy, I.; Eliezer, D. Folding and misfolding of α-synuclein on membranes. Biochim. Biophys. Acta 2012, 1818, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Snead, D.; Eliezer, D. α-synuclein function and dysfunction on cellular membranes. Exp. Neurobiol. 2014, 23, 292–313. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured α-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Souillac, P.; Millett, I.S.; Doniach, S.; Jakes, R.; Goedert, M.; Fink, A.L. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of α-synuclein assembly by β- and γ-synucleins. J. Biol. Chem. 2002, 277, 11970–11978. [Google Scholar] [CrossRef] [PubMed]
- Bernado, P.; Bertoncini, C.W.; Griesinger, C.; Zweckstetter, M.; Blackledge, M. Defining long-range order and local disorder in native α-synuclein using residual dipolar couplings. J. Am. Chem. Soc. 2005, 127, 17968–17969. [Google Scholar] [CrossRef] [PubMed]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodoulou, J.; Vendruscolo, M.; Dobson, C.M. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005, 127, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.K.; Kim, H.Y.; Bernado, P.; Fernandez, C.O.; Blackledge, M.; Zweckstetter, M. Amino acid bulkiness defines the local conformations and dynamics of natively unfolded α-synuclein and tau. J. Am. Chem. Soc. 2007, 129, 3032–3033. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K. α-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 2006, 112, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, L.; Giehm, L.; Heimburg, T.; Otzen, D. The influence of vesicle size and composition on α-synuclein structure and stability. Biophys. J. 2009, 96, 2857–2870. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. Synucleins: Are they two-edged swords? J. Neurosci. Res. 2013, 91, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Ducas, V.C.; Rhoades, E. Investigation of intramolecular dynamics and conformations of α-, β- and γ-synuclein. PLoS ONE 2014, 9, e86983. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N. α-synuclein structure, functions, and interactions. J. Res. Med. Sci. 2016, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Eliezer, D. Biophysics of Parkinson’s disease: Structure and aggregation of α-synuclein. Curr. Protein Pept. Sci. 2009, 10, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular nk between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [PubMed]
- Latourelle, J.C.; Pankratz, N.; Dumitriu, A.; Wilk, J.B.; Goldwurm, S.; Pezzoli, G.; Mariani, C.B.; DeStefano, A.L.; Halter, C.; Gusella, J.F.; et al. Genomewide association study for onset age in Parkinson disease. BMC Med. Genet. 2009, 10, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankratz, N.; Wilk, J.B.; Latourelle, J.C.; DeStefano, A.L.; Halter, C.; Pugh, E.W.; Doheny, K.F.; Gusella, J.F.; Nichols, W.C.; Foroud, T.; et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 2009, 124, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Mjoenes, H. Hereditary extrapyramidal diseases. Acta Neurol. Scand. 1963, 39, 108–118. [Google Scholar] [CrossRef]
- Sjoougren, T. Investigations of the heredity of psychoses and mental deficiency in two north Swedish parishes. Ann. Hum. Genet. 1935, 6, 253–315. [Google Scholar] [CrossRef]
- Golbe, L.I.; Di Iorio, G.; Sanges, G.; Lazzarini, A.M.; La Sala, S.; Bonavita, V.; Duvoisin, R.C. Clinical genetic analysis of Parkinson’s disease in the contursi kindred. Ann. Neurol. 1996, 40, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Markopoulou, K.; Wszolek, Z.K.; Pfeiffer, R.F. A Greek-American kindred with autosomal dominant, levodopa-responsive Parkinsonism and anticipation. Ann. Neurol. 1995, 38, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Sastry, S.; Chan, P.; Forno, L.S.; Bolin, L.M.; Di Monte, D.A. Novel α-synuclein-immunoreactive proteins in brain samples from the contursi kindred, Parkinson’s, and alzheimer’s disease. Exp. Neurol. 1998, 154, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Mezey, E.; Dehejia, A.M.; Harta, G.; Tresser, N.; Suchy, S.F.; Nussbaum, R.L.; Brownstein, M.J.; Polymeropoulos, M.H. α-synuclein is present in lewy bodies in sporadic Parkinson’s disease. Mol. Psychiatry 1998, 3, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, A.; Veletza, V.; Hadjigeorgiou, G.M.; Patrikiou, A.; Hirano, M.; Anastasopoulos, I. Mutated α-synuclein gene in two Greek kindreds with familial PD: Incomplete penetrance? Neurology 1999, 52, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Veletz, S.; Bostatzopoulou, S.; Hantzigeorgiou, G.; Kazis, A.; Papadimitriou, A. A-synuclein mutation associated with familial Parkinson’s disease in two new greek kindreds. J. Neurol. 1999, 246, 1–43. [Google Scholar]
- Bostantjopoulou, S.; Katsarou, Z.; Papadimitriou, A.; Veletza, V.; Hatzigeorgiou, G.; Lees, A. Clinical features of Parkinsonian patients with the α-synuclein (g209a) mutation. Mov. Disord. 2001, 16, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Spira, P.J.; Sharpe, D.M.; Halliday, G.; Cavanagh, J.; Nicholson, G.A. Clinical and pathological features of a Parkinsonian syndrome in a family with an ala53thr α-synuclein mutation. Ann. Neurol. 2001, 49, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Niwar, M.; Maass, S.; Zimprich, A.; Moller, J.C.; Wuellner, U.; Schmitz-Hubsch, T.; Klein, C.; Tan, E.K.; Schols, L.; et al. α-synuclein and Parkinson’s disease: Implications from the screening of more than 1900 patients. Mov. Disord. 2005, 20, 1191–1194. [Google Scholar] [CrossRef] [PubMed]
- Michell, A.W.; Barker, R.A.; Raha, S.K.; Raha-Chowdhury, R. A case of late onset sporadic Parkinson’s disease with an a53t mutation in α-synuclein. J. Neurol. Neurosurg. Psychiatry 2005, 76, 596–597. [Google Scholar] [CrossRef] [PubMed]
- Ki, C.S.; Stavrou, E.F.; Davanos, N.; Lee, W.Y.; Chung, E.J.; Kim, J.Y.; Athanassiadou, A. The ala53thr mutation in the α-synuclein gene in a Korean family with Parkinson disease. Clin. Genet. 2007, 71, 471–473. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A.; Ross, O.A.; Vilarino-Guell, C.; Lincoln, S.J.; Kachergus, J.M.; Cobb, S.A.; Lindquist, S.G.; Nielsen, J.E.; Wszolek, Z.K.; Farrer, M.; et al. A Swedish family with de novo α-synuclein a53t mutation: Evidence for early cortical dysfunction. Parkinsonism Relat. Disord. 2009, 15, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Uversky, V.N.; Fink, A.L. Effect of familial Parkinson’s disease point mutations a30p and a53t on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 2001, 40, 11604–11613. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Uversky, V.N.; Fink, A.L. Conformational behavior of human α-synuclein is modulated by familial Parkinson’s disease point mutations a30p and a53t. Neurotoxicology 2002, 23, 553–567. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, e46k, of α-synuclein causes Parkinson and lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51d α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDowell, F.H.; Lee, J.E. Levodopa, Parkinson’s disease, and hypotension. Ann. Intern. Med. 1970, 72, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Mohl, B.; Berman, B.D.; Shelton, E.; Tanabe, J. Levodopa response differs in Parkinson’s motor subtypes: A task-based effective connectivity study. J. Comp. Neurol. 2017, 525, 2192–2201. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.C.; August, T.F.; Bush, D.F.; Lasseter, K.C.; Musson, D.G.; Schwartz, S.; Smith, M.E.; Titus, D.C. Pharmacokinetics and bioavailability of sinemet cr: A summary of human studies. Neurology 1989, 39, 25–38. [Google Scholar] [PubMed]
- Rascol, O.; Brooks, D.J.; Korczyn, A.D.; De Deyn, P.P.; Clarke, C.E.; Lang, A.E. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. N. Engl. J. Med. 2000, 342, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Holloway, R.G.; Shoulson, I.; Fahn, S.; Kieburtz, K.; Lang, A.; Marek, K.; McDermott, M.; Seibyl, J.; Weiner, W.; Musch, B.; et al. Pramipexole vs. levodopa as initial treatment for Parkinson disease: A 4-year randomized controlled trial. Arch. Neurol. 2004, 61, 1044–1053. [Google Scholar] [PubMed]
- Seiple, W.; Jennings, D.; Rosen, R.B.; Borchert, L.; Canale, L.; Fagan, N.; Gordon, M.F. Ophthalmologic baseline characteristics and 2-year ophthalmologic safety profile of pramipexole ir compared with ropinirole ir in patients with early Parkinson’s disease. Parkinsons Dis. 2016, 2016, 8298503. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Kieburtz, K.; Leinonen, M.; Elmer, L.; Giladi, N.; Hauser, R.A.; Klepiskaya, O.S.; Kreitzman, D.L.; Lew, M.F.; Russell, D.S.; et al. A randomized trial of a low-dose rasagiline and pramipexole combination (p2b001) in early Parkinson’s disease. Mov. Disord. 2017, 32, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.A.; Olanow, C.W.; Dzyngel, B.; Bilbault, T.; Shill, H.; Isaacson, S.; Dubow, J.; Agro, A. Sublingual apomorphine (apl-130277) for the acute conversion of off to on in Parkinson’s disease. Mov. Disord. 2016, 31, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- De Smet, Y.; Ruberg, M.; Serdaru, M.; Dubois, B.; Lhermitte, F.; Agid, Y. Confusion, dementia and anticholinergics in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1982, 45, 1161–1164. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.A.; Li, R.; Perez, A.; Ren, X.; Weintraub, D.; Elm, J.; Goudreau, J.L.; Morgan, J.C.; Fang, J.Y.; Aminoff, M.J.; et al. Longer duration of mao-b inhibitor exposure is associated with less clinical decline in Parkinson’s disease: An analysis of net-pd ls1. J. Parkinsons Dis. 2017, 7, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.H.; Deuschl, G.; Gordin, A.; Kultalahti, E.R.; Leinonen, M.; Celomen Study, G. Efficacy and safety of entacapone in Parkinson’s disease patients with suboptimal levodopa response: A 6-month randomized placebo-controlled double-blind study in Germany and Austria (celomen study). Acta Neurol. Scand. 2002, 105, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Schwab, R.S.; England, A.C., Jr.; Poskanzer, D.C.; Young, R.R. Amantadine in the treatment of Parkinson’s disease. JAMA 1969, 208, 1168–1170. [Google Scholar] [CrossRef] [PubMed]
- Oertel, W.; Eggert, K.; Pahwa, R.; Tanner, C.M.; Hauser, R.A.; Trenkwalder, C.; Ehret, R.; Azulay, J.P.; Isaacson, S.; Felt, L.; et al. Randomized, placebo-controlled trial of ads-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (ease lid 3). Mov. Disord. 2017, 32, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Henderson, E.J.; Lord, S.R.; Brodie, M.A.; Gaunt, D.M.; Lawrence, A.D.; Close, J.C.; Whone, A.L.; Ben-Shlomo, Y. Rivastigmine for gait stability in patients with Parkinson’s disease (respond): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 249–258. [Google Scholar] [CrossRef]
- Pham, T.; Bronstein, J.M. Neuropsychological outcomes from deep brain stimulation-stimulation versus micro-lesion. Ann. Transl. Med. 2017, 5, 217. [Google Scholar] [CrossRef] [PubMed]
- Bot, M.; van den Munckhof, P.; Bakay, R.; Stebbins, G.; Verhagen Metman, L. Accuracy of intraoperative computed tomography during deep brain stimulation procedures: Comparison with postoperative magnetic resonance imaging. Stereotact. Funct. Neurosurg. 2017, 95, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.-W.; Weissfeld, T.A.; Bober, T. New developments in the treatment of Parkinson’s disease: Bridging the gap between medical and surgical therapy for pd. JHN J. 2016, 11. [Google Scholar]
- Halasz, L.; Kis, D.; Entz, G.; Tamas, G.; Klivenyi, P.; Fabo, D.; Barzo, P.; Eross, L. Ep 77. Target identification in deep brain stimulation for Parkinson’s disease: The role of probabilistic tractography. Clin. Neurophys. 2016, 127, e206–e207. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A structural model for alzheimer’s β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742–16747. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Annamalai, K.; Schmidt, M.; Grigorieff, N.; Fandrich, M. Cryo-em reveals the steric zipper structure of a light chain-derived amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, 6200–6205. [Google Scholar] [CrossRef] [PubMed]
- Walti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Bockmann, A.; Guntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Yau, W.M.; Lu, J.X.; Collinge, J.; Tycko, R. Structural variation in amyloid-β fibrils from alzheimer’s disease clinical subtypes. Nature 2017, 541, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Wise-Scira, O.; Xu, L.; Kitahara, T.; Perry, G.; Coskuner, O. Amyloid-β peptide structure in aqueous solution varies with fragment size. J. Chem. Phys. 2011, 135, 205101. [Google Scholar] [CrossRef] [PubMed]
- Baldassarre, M.; Bennett, M.; Barth, A. Simultaneous acquisition of infrared, fluorescence and light scattering spectra of proteins: Direct evidence for pre-fibrillar species in amyloid fibril formation. Analyst 2016, 141, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, P.; Fatima, M.T.; Abdelhameed, A.S.; Nusrat, S.; Khan, R.H. Structure of amyloid oligomers and their mechanisms of toxicities: Targeting amyloid oligomers using novel therapeutic approaches. Eur. J. Med. Chem. 2016, 114, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-β oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Kirkitadze, M.D.; Condron, M.M.; Teplow, D.B. Identification and characterization of key kinetic intermediates in amyloid β-protein fibrillogenesis. J. Mol. Biol. 2001, 312, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Shao, H.; Zhang, Y.; Li, H.; Menon, N.K.; Neuhaus, E.B.; Brewer, J.M.; Byeon, I.J.; Ray, D.G.; Vitek, M.P.; et al. Solution NMR studies of the Aβ(1–40) and Aβ(1–42) peptides establish that the met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004, 126, 1992–2005. [Google Scholar] [CrossRef] [PubMed]
- Goldsbury, C.; Baxa, U.; Simon, M.N.; Steven, A.C.; Engel, A.; Wall, J.S.; Aebi, U.; Muller, S.A. Amyloid structure and assembly: Insights from scanning transmission electron microscopy. J. Struct. Biol. 2011, 173, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Enache, T.A.; Chiorcea-Paquim, A.M.; Oliveira-Brett, A.M. Amyloid-β peptides time-dependent structural modifications: Afm and voltammetric characterization. Anal. Chim. Acta 2016, 926, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Watanabe-Nakayama, T.; Ono, K.; Itami, M.; Takahashi, R.; Teplow, D.B.; Yamada, M. High-speed atomic force microscopy reveals structural dynamics of amyloid β1–42 aggregates. Proc. Natl. Acad. Sci. USA 2016, 113, 5835–5840. [Google Scholar] [CrossRef] [PubMed]
- Canale, C.; Oropesa-Nunez, R.; Diaspro, A.; Dante, S. Amyloid and membrane complexity: The toxic interplay revealed by AFM. Semin. Cell Dev. Biol. 2018, 73, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Scholzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β(1–42) by cryo-electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W.; von Helden, G.; Pagel, K. Ion mobility-mass spectrometry and orthogonal gas-phase techniques to study amyloid formation and inhibition. Curr. Opin. Struct. Biol. 2017, 46, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W.M.; Mattson, M.P.; Tycko, R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 2005, 307, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [PubMed]
- Tycko, R. Amyloid polymorphism: Structural basis and neurobiological relevance. Neuron 2015, 86, 632–645. [Google Scholar] [CrossRef] [PubMed]
- Luhrs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H.; Schubert, D.; Riek, R. 3d structure of Alzheimer’s amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.X.; Qiang, W.; Yau, W.M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Kelley, K.; Tycko, R. Polymorph-specific kinetics and thermodynamics of β-amyloid fibril growth. J. Am. Chem. Soc. 2013, 135, 6860–6871. [Google Scholar] [CrossRef] [PubMed]
- Goate, A. Segregation of a missense mutation in the amyloid β-protein precursor gene with familial Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 341–347. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A recessive mutation in the app gene with dominant-negative effect on amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in app protects against Alzheimer’s disease and age-related cognitive d ecline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.C.; Beck, J.A.; Campbell, T.A.; Dickinson, A.; Fox, N.C.; Harvey, R.J.; Houlden, H.; Rossor, M.N.; Collinge, J. Early onset familial Alzheimer’s disease: Mutation frequency in 31 families. Neurology 2003, 60, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Hong, C.J.; Lin, Y.T.; Chang, W.H.; Huang, H.T.; Liao, J.Y.; Chang, Y.J.; Hsieh, Y.F.; Cheng, C.Y.; Liu, H.C.; et al. Amyloid-β (Aβ) d7h mutation increases oligomeric Aβ42 and alters properties of Aβ-zinc/copper assemblies. PLoS ONE 2012, 7, e35807. [Google Scholar] [CrossRef] [PubMed]
- Wakutani, Y.; Watanabe, K.; Adachi, Y.; Wada-Isoe, K.; Urakami, K.; Ninomiya, H.; Saido, T.C.; Hashimoto, T.; Iwatsubo, T.; Nakashima, K. Novel amyloid precursor protein gene missense mutation (d678n) in Probable familial Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Condron, M.M.; Teplow, D.B. Effects of the English (h6r) and Tottori (d7n) familial Alzheimer disease mutations on amyloid β-protein assembly and toxicity. J. Biol. Chem. 2010, 285, 23186–23197. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Hartley, D.M.; Condron, M.M.; Selkoe, D.J.; Teplow, D.B. In vitro studies of amyloid β-protein fibril assembly and toxicity provide clues to the Aetiology of Flemish variant (ala692→gly) Alzheimer’s disease. Biochem. J. 2001, 355, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Tsubuki, S.; Takaki, Y.; Saido, T.C. Dutch, Flemish, Italian, and Arctic mutations of app and resistance of Aβ to physiologically relevant proteolytic degradation. Lancet 2003, 361, 1957–1958. [Google Scholar] [CrossRef]
- Morimoto, A.; Irie, K.; Murakami, K.; Ohigashi, H.; Shindo, M.; Nagao, M.; Shimizu, T.; Shirasawa, T. Aggregation and neurotoxicity of mutant amyloid β (Aβ) peptides with proline replacement: Importance of turn formation at positions 22 and 23. Biochem. Biophys. Res. Commun. 2002, 295, 306–311. [Google Scholar] [CrossRef]
- Murakami, K.; Irie, K.; Morimoto, A.; Ohigashi, H.; Shindo, M.; Nagao, M.; Shimizu, T.; Shirasawa, T. Synthesis, aggregation, neurotoxicity, and secondary structure of various Aβ 1-42 mutants of familial Alzheimer’s disease at positions 21–23. Biochem. Biophys. Res. Commun. 2002, 294, 5–10. [Google Scholar] [CrossRef]
- Murakami, K.; Irie, K.; Morimoto, A.; Ohigashi, H.; Shindo, M.; Nagao, M.; Shimizu, T.; Shirasawa, T. Neurotoxicity and physicochemical properties of Aβ mutant peptides from cerebral amyloid angiopathy: Implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer’s disease. J. Biol. Chem. 2003, 278, 46179–46187. [Google Scholar] [CrossRef] [PubMed]
- Miravalle, L.; Tokuda, T.; Chiarle, R.; Giaccone, G.; Bugiani, O.; Tagliavini, F.; Frangione, B.; Ghiso, J. Substitutions at codon 22 of Alzheimer’s Aβ peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J. Biol. Chem. 2000, 275, 27110–27116. [Google Scholar] [PubMed]
- Van Nostrand, W.E.; Melchor, J.P.; Cho, H.S.; Greenberg, S.M.; Rebeck, G.W. Pathogenic effects of d23n iowa mutant amyloid β -protein. J. Biol. Chem. 2001, 276, 32860–32866. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Hartley, D.M.; Petre, B.M.; Wall, J.S.; Simon, M.N.; Walz, T.; Lansbury, P.T., Jr. Mixtures of WT and a pathogenic (e22g) form of Aβ40 in vitro accumulate protofibrils, including amyloid pores. J. Mol. Biol. 2003, 332, 795–808. [Google Scholar] [CrossRef]
- Betts, V.; Leissring, M.A.; Dolios, G.; Wang, R.; Selkoe, D.J.; Walsh, D.M. Aggregation and catabolism of disease-associated intra-Aβ mutations: Reduced proteolysis of Aβa21g by neprilysin. Neurobiol. Dis. 2008, 31, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Hecht, M.H. Mutations enhance the aggregation propensity of the Alzheimer’s a beta peptide. J. Mol. Biol. 2008, 377, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K.; Murugan, N.A.; Langstrom, B.; Nordberg, A.; Agren, H. Effect of Alzheimer familial chromosomal mutations on the amyloid fibril interaction with different pet tracers: Insight from molecular modeling studies. ACS Chem. Neurosci. 2017, 8, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Qiang, W. Solid-state-nmr-structure-based inhibitor design to achieve selective inhibition of the parallel-in-register β-sheet versus antiparallel Iowa mutant β-amyloid fibrils. J. Phys. Chem. B 2017, 121, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- Hatami, A.; Monjazeb, S.; Milton, S.; Glabe, C.G. Familial Alzheimer’s disease mutations within the amyloid precursor protein alter the aggregation and conformation of the amyloid-β peptide. J. Biol. Chem. 2017, 292, 3172–3185. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, K.; Tomiyama, T.; Ishibashi, K.; Ito, K.; Teraoka, R.; Lambert, M.P.; Klein, W.L.; Mori, H. The e693Δ mutation in amyloid precursor protein increases intracellular accumulation of amyloid β oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am. J. Pathol. 2009, 174, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A mouse model of amyloid β oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid β variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Tomiyama, T.; Sakama, N.; Tanaka, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Intraneuronal amyloid β oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J. Neurosci. Res. 2011, 89, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Kulic, L.; McAfoose, J.; Welt, T.; Tackenberg, C.; Spani, C.; Wirth, F.; Finder, V.; Konietzko, U.; Giese, M.; Eckert, A.; et al. Early accumulation of intracellular fibrillar oligomers and late congophilic amyloid angiopathy in mice expressing the Osaka intra-Aβ app mutation. Transl. Psychiatry 2012, 2, e183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galimberti, D.; Scarpini, E. Idalopirdine as a treatment for Alzheimer’s disease. Expert Opin. Investig. Drugs 2015, 24, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, N.; O’Regan, J.; Ruthirakuhan, M.; Kiss, A.; Eryavec, G.; Williams, E.; Lanctot, K.L. A randomized placebo-controlled discontinuation study of cholinesterase inhibitors in institutionalized patients with moderate to severe Alzheimer disease. J. Am. Med. Dir. Assoc. 2016, 17, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lang, M.; Youdim, M.B.H.; Amit, T.; Sun, Y.; Zhang, Z.; Wang, Y.; Weinreb, O. Design, synthesis and evaluation of novel dual monoamine-cholinesterase inhibitors as potential treatment for Alzheimer’s disease. Neuropharmacology 2016, 109, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, K.; Howlett, S.E.; Hoffman, D.; Schindler, R.; Mitnitski, A. Clinical meaningfulness of Alzheimer’s disease assessment scale-cognitive subscale change in relation to goal attainment in patients on cholinesterase inhibitors. Alzheimer’s Dement 2017, 13, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Rosini, M.; Simoni, E.; Caporaso, R.; Minarini, A. Multitarget strategies in Alzheimer’s disease: Benefits and challenges on the road to therapeutics. Future Med. Chem. 2016, 8, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.L.; Farlow, M.R.; Doody, R.S.; Mohs, R.; Friedhoff, L.T.; Donepezil Study Group. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology 1998, 50, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Anand, R.; Messina, J.; Hartman, R.; Veach, J. An efficacy and safety analysis of exelon in Alzheimer’s disease patients with concurrent vascular risk factors. Eur. J. Neurol. 2000, 7, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Bloniecki, V.; Aarsland, D.; Blennow, K.; Cummings, J.; Falahati, F.; Winblad, B.; Freund-Levi, Y. Effects of risperidone and galantamine treatment on Alzheimer’s disease biomarker levels in cerebrospinal fluid. J. Alzheimer’s Dis. 2017, 57, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Patience, A.A.; Sharma, N.; Khurana, N. The present and future of pharmacotherapy of Alzheimer’s disease: A comprehensive review. Eur. J. Pharmacol. 2017, 815, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Jiang, H. Efficacy and adverse effects of memantine treatment for Alzheimer’s disease from randomized controlled trials. Neurol. Sci. 2015, 36, 1633–1641. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Yankner, B.A. Amyloid fibril toxicity in Alzheimer’s disease and diabetes. Ann. N. Y. Acad. Sci. 1996, 777, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Man, V.H.; Nguyen, P.H.; Derreumaux, P. High-resolution structures of the amyloid-β 1-42 dimers from the comparison of four atomistic force fields. J. Phys. Chem. B 2017, 121, 5977–5987. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, G.A.; Silva, J.L. The push-and-pull hypothesis in protein unfolding, misfolding and aggregation. Biophys. Chem. 2017, 231, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K. The nucleation of protein aggregates—From crystals to amyloid fibrils. Int. Rev. Cell Mol. Biol. 2017, 329, 187–226. [Google Scholar] [PubMed]
- Johnson, R.D.; Schauerte, J.A.; Chang, C.C.; Wisser, K.C.; Althaus, J.C.; Carruthers, C.J.; Sutton, M.A.; Steel, D.G.; Gafni, A. Single-molecule imaging reveals Aβ42:Aβ40 ratio-dependent oligomer growth on neuronal processes. Biophys. J. 2013, 104, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Ganzinger, K.A.; McColl, J.; Weimann, L.; Meehan, S.; Qamar, S.; Carver, J.A.; Wilson, M.R.; St George-Hyslop, P.; Dobson, C.M.; et al. Single molecule characterization of the interactions between amyloid-β peptides and the membranes of hippocampal cells. J. Am. Chem. Soc. 2013, 135, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Santi, S.; Musi, V.; Descrovi, E.; Paeder, V.; Di Francesco, J.; Hvozdara, L.; van der Wal, P.; Lashuel, H.A.; Pastore, A.; Neier, R.; et al. Real-time amyloid aggregation monitoring with a photonic crystal-based approach. ChemPhysChem 2013, 14, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rempel, D.L.; Zhang, J.; Sharma, A.K.; Mirica, L.M.; Gross, M.L. Pulsed hydrogen-deuterium exchange mass spectrometry probes conformational changes in amyloid β (Aβ) peptide aggregation. Proc. Natl. Acad. Sci. USA 2013, 110, 14604–14609. [Google Scholar] [CrossRef] [PubMed]
- Crespo, R.; Villar-Alvarez, E.; Taboada, P.; Rocha, F.A.; Damas, A.M.; Martins, P.M. What can the kinetics of amyloid fibril formation tell about off-pathway aggregation? J. Biol. Chem. 2016, 291, 2018–2032. [Google Scholar] [CrossRef] [PubMed]
- Drew, S.C.; Noble, C.J.; Masters, C.L.; Hanson, G.R.; Barnham, K.J. Pleomorphic copper coordination by Alzheimer’s disease amyloid-β peptide. J. Am. Chem. Soc. 2009, 131, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Liu, C.; Guo, Z. Structural insights into Aβ42 oligomers using site-directed spin labeling. J. Biol. Chem. 2013, 288, 18673–18683. [Google Scholar] [CrossRef] [PubMed]
- Wise-Scira, O.; Xu, L.; Perry, G.; Coskuner, O. Structures and free energy landscapes of aqueous zinc(ii)-bound amyloid-beta(1-40) and zinc(ii)-bound amyloid-β(1–42) with dynamics. J. Biol. Inorg. Chem. 2012, 17, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Wise, O.; Coskuner, O. New force field parameters for metalloproteins i: Divalent copper ion centers including three histidine residues and an oxygen-ligated amino acid residue. J. Comput. Chem. 2014, 35, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Coskuner, O. Divalent copper ion bound amyloid-β(40) and amyloid-β(42) alloforms are less preferred than divalent zinc ion bound amyloid-β(40) and amyloid-β(42) alloforms. J. Biol. Inorg. Chem. 2016, 21, 957–973. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Skelton, A.A. 12-crown-4 ether disrupts the patient brain-derived amyloid-β-fibril trimer: Insight from all-atom molecular dynamics simulations. ACS Chem. Neurosci. 2016, 7, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Jakova, E.; Lee, J.S. Behavior of α-synuclein-drug complexes during nanopore analysis with a superimposed ac field. Electrophoresis 2017, 38, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Stark, L.; Musgrave, I.F.; Pukala, T.; Smid, S.D. Bioactive polyphenol interactions with β amyloid: A comparison of binding modelling, effects on fibril and aggregate formation and neuroprotective capacity. Food Funct. 2016, 7, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Rinne, J.O.; Brooks, D.J.; Rossor, M.N.; Fox, N.C.; Bullock, R.; Klunk, W.E.; Mathis, C.A.; Blennow, K.; Barakos, J.; Okello, A.A.; et al. 11c-pib pet assessment of change in fibrillar amyloid-β load in patients with Alzheimer’s disease treated with bapineuzumab: A phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010, 9, 363–372. [Google Scholar] [CrossRef]
- Hirohata, M.; Ono, K.; Morinaga, A.; Yamada, M. Non-steroidal anti-inflammatory drugs have potent anti-fibrillogenic and fibril-destabilizing effects for α-synuclein fibrils in vitro. Neuropharmacology 2008, 54, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Porat, Y.; Abramowitz, A.; Gazit, E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 2006, 67, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, M.; Ono, K.; Naiki, H.; Yamada, M. Non-steroidal anti-inflammatory drugs have anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. Neuropharmacology 2005, 49, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, M.; Rajamani, S.; Uversky, V.N.; Fink, A.L. Rifampicin inhibits α-synuclein fibrillation and disaggregates fibrils. Chem. Biol. 2004, 11, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Rochet, J.C.; Bieganski, R.M.; Lansbury, P.T., Jr. Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, L.O.; Naslund, J.; Lindqvist, F.; Johansson, J.; Karlstrom, A.R.; Thyberg, J.; Terenius, L.; Nordstedt, C. Arrest of β-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 1996, 271, 8545–8548. [Google Scholar] [CrossRef] [PubMed]
- Norris, E.H.; Giasson, B.I.; Ischiropoulos, H.; Lee, V.M. Effects of oxidative and nitrative challenges on α-synuclein fibrillogenesis involve distinct mechanisms of protein modifications. J. Biol. Chem. 2003, 278, 27230–27240. [Google Scholar] [CrossRef] [PubMed]
- Dorval, V.; Fraser, P.E. Small ubiquitin-like modifier (sumo) modification of natively unfolded proteins tau and α-synuclein. J. Biol. Chem. 2006, 281, 9919–9924. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Hu, D.; Han, S.; Reaney, S.H.; Di Monte, D.A.; Fink, A.L. Effect of 4-hydroxy-2-nonenal modification on α-synuclein aggregation. J. Biol. Chem. 2007, 282, 5862–5870. [Google Scholar] [CrossRef] [PubMed]
- Trexler, A.J.; Rhoades, E. N-terminal acetylation is critical for forming α-helical oligomer of α-synuclein. Protein Sci. 2012, 21, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Bondalapati, S.; Jbara, M.; Brik, A. Expanding the chemical toolbox for the synthesis of large and uniquely modified proteins. Nat. Chem. 2016, 8, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Brown, J.W.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, G.; Fecchio, C.; Sharon, R.; Schapira, A.H.V.; Proukakis, C.; Bellotti, V.; de Laureto, P.P. α-synuclein structural features inhibit harmful polyunsaturated fatty acid oxidation, suggesting roles in neuroprotection. J. Biol. Chem. 2017, 292, 6927–6937. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Jangir, D.K.; Verma, G.; Shekhar, S.; Hanpude, P.; Kumar, S.; Kumari, R.; Singh, N.; Sarovar Bhavesh, N.; Ranjan Jana, N.; et al. S-nitrosylation of uchl1 induces its structural instability and promotes α-synuclein aggregation. Sci. Rep. 2017, 7, 44558. [Google Scholar] [CrossRef] [PubMed]
- Szenasi, T.; Olah, J.; Szabo, A.; Szunyogh, S.; Lang, A.; Perczel, A.; Lehotzky, A.; Uversky, V.N.; Ovadi, J. Challenging drug target for Parkinson’s disease: Pathological complex of the chameleon tppp/p25 and α-synuclein proteins. Biochim. Biophys. Acta 2017, 1863, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Bar-On, P.; Sharikov, Y.; Crews, L.; Hashimoto, M.; Miller, M.A.; Keller, S.H.; Platoshyn, O.; Yuan, J.X.; Masliah, E. Dynamics of α-synuclein aggregation and inhibition of pore-like oligomer development by β-synuclein. FEBS J. 2007, 274, 1862–1877. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.P.; Weinstock, D.S.; Narayanan, C.; Levy, R.M.; Baum, J. Structural reorganization of α-synuclein at low ph observed by NMR and remd simulations. J. Mol. Biol. 2009, 391, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Jang, S.; Lee, K.; Shin, S. Simulation studies on the stabilities of aggregates formed by fibril-forming segments of α-synuclein. J. Biomol. Struct. Dyn. 2009, 27, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Sammalkorpi, M.; DeWitt, D.C.; Trexler, A.J.; Elbaum-Garfinkle, S.; O’Hern, C.S.; Rhoades, E. The conformational ensembles of α-synuclein and tau: Combining single-molecule fret and simulations. Biophys. J. 2012, 103, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Yoon, J.; Jang, S.; Lee, K.; Shin, S. The role of the acidic domain of α-synuclein in amyloid fibril formation: A molecular dynamics study. J. Biomol. Struct. Dyn. 2016, 34, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M.; Rao, F.; Seeber, M.; Caflisch, A. Replica exchange molecular dynamics simulations of amyloid peptide aggregation. J. Chem. Phys. 2004, 121, 10748–10756. [Google Scholar] [CrossRef] [PubMed]
- Rao, F.; Caflisch, A. Replica exchange molecular dynamics simulations of reversible folding. J. Chem. Phys. 2003, 119, 4035–4042. [Google Scholar] [CrossRef]
- Petraglia, R.; Nicolai, A.; Wodrich, M.D.; Ceriotti, M.; Corminboeuf, C. Beyond static structures: Putting forth remd as a tool to solve problems in computational organic chemistry. J. Comput. Chem. 2016, 37, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Bekker, G.J.; Kamiya, N.; Araki, M.; Fukuda, I.; Okuno, Y.; Nakamura, H. Accurate prediction of complex structure and affinity for a flexible protein receptor and its inhibitor. J. Chem. Theory Comput. 2017, 13, 2389–2399. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.C.; Melo, M.C.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.A.C.; White, G.W.N.; Daggett, V. Exploring the energy landscape of protein folding using replica-exchange and conventional molecular dynamics simulations. J. Struct. Biol. 2007, 157, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Rhee, Y.M.; Pande, V.S. Multiplexed-replica exchange molecular dynamics method for protein folding simulation. Biophys. J. 2003, 84, 775–786. [Google Scholar] [CrossRef]
- Coskuner, O.; Uversky, V.N. How accurate are your simulations? Effects of confined aqueous volume and amberff99sb and charmm22/cmap force field parameters on structural ensembles of intrinsically disordered proteins: Amyloid-β42 in water. Intirinsically Disord. Proteins 2017, 5, e1377813. [Google Scholar]
- Carballo-Pacheco, M.; Strodel, B. Comparison of force fields for Alzheimer’s Aβ42: A case study for intrinsically disordered proteins. Protein Sci. 2017, 26, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Li, M.S.; Derreumaux, P. Effects of all-atom force fields on amyloid oligomerization: Replica exchange molecular dynamics simulations of the Aβ16–22 dimer and trimer. Phys. Chem. Chem. Phys. 2011, 13, 9778–9788. [Google Scholar] [CrossRef] [PubMed]
- Somavarapu, A.K.; Kepp, K.P. The dependence of amyloid-β dynamics on protein force fields and water models. Chemphyschem 2015, 16, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Gerben, S.R.; Lemkul, J.A.; Brown, A.M.; Bevan, D.R. Comparing atomistic molecular mechanics force fields for a difficult target: A case study on the Alzheimer’s amyloid β-peptide. J. Biomol. Struct. Dyn. 2014, 32, 1817–1832. [Google Scholar] [CrossRef] [PubMed]
- Ullman, O.; Fisher, C.K.; Stultz, C.M. Explaining the structural plasticity of α-synuclein. J. Am. Chem. Soc. 2011, 133, 19536–19546. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Ulrih, N.P.; Barry, C.H.; Fink, A.L. Impact of TYR to ala mutations on α-synuclein fibrillation and structural properties. Biochim. Biophys. Acta 2008, 1782, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Izawa, Y.; Tateno, H.; Kameda, H.; Hirakawa, K.; Hato, K.; Yagi, H.; Hongo, K.; Mizobata, T.; Kawata, Y. Role of c-terminal negative charges and tyrosine residues in fibril formation of α-synuclein. Brain Behav. 2012, 2, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Lamberto, G.R.; Binolfi, A.; Orcellet, M.L.; Bertoncini, C.W.; Zweckstetter, M.; Griesinger, C.; Fernandez, C.O. Structural and mechanistic basis behind the inhibitory interaction of pcts on α-synuclein amyloid fibril formation. Proc. Natl. Acad. Sci. USA 2009, 106, 21057–21062. [Google Scholar] [CrossRef] [PubMed]
- Sanjeev, A.; Mattaparthi, V.S.K. Computational investigation on tyrosine to alanine mutations delaying the early stage of α-synuclein aggregation. Curr. Proteom. 2017, 14, 31–41. [Google Scholar] [CrossRef]
- Coskuner, O.; Wise-Scira, O. Structures and free energy landscapes of the a53t mutant α-synuclein protein and impact of a53t mutation on the structures of the WT α-synuclein protein with dynamics. ACS Chem. Neurosci. 2013, 4, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Wise-Scira, O.; Dunn, A.; Aloglu, A.K.; Sakallioglu, I.T.; Coskuner, O. Structures of the e46k mutant α-synuclein protein and impact of e46k mutation on the structures of the WT α-synuclein protein. ACS Chem. Neurosci. 2013, 4, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Wise-Scira, O.; Aloglu, A.K.; Dunn, A.; Sakallioglu, I.T.; Coskuner, O. Structures and free energy landscapes of the WT and a30p mutant α-synuclein proteins with dynamics. ACS Chem. Neurosci. 2013, 4, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, C.; Duan, Y. Convergence of replica exchange molecular dynamics. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Patriksson, A.; van der Spoel, D. A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 2008, 10, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Clarendon Press: Gloucestershire, UK, 1999. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications; Academic Press: Cambridge, MA, USA, 2002; Volume 1. [Google Scholar]
- Allison, T.C.; Coskuner, O.; Gonzalez, C.A. Metallic Systems: A Quantum Chemist’s Perspective; CRC Press: Boca Raton, FL, USA; Taylor & Francis: Abingdon, UK, 2011. [Google Scholar]
- Coskuner, O.; Deiters, U.K. Hydrophobic interactions of xenon by Monte Carlo simulations. Z. Phys. Chem. 2007, 221, 785–799. [Google Scholar] [CrossRef]
- Coskuner, O.; Deiters, U.K. Hydrophobic interactions by Monte Carlo simulations. Z. Phys. Chem. 2006, 220, 349–369. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure - pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Coskuner, O.; Wise-Scira, O.; Perry, G.; Kitahara, T. The structures of the e22Δ mutant amyloid-β alloforms and the impact of e22Δ mutation on the structures of the WT amyloid-β alloforms. ACS Chem. Neurosci. 2013, 4, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Fawver, J.N.; Duong, K.T.; Wise-Scira, O.; Chapa, R.P.; Schall, H.E.; Coskuner, O.; Zhu, X.W.; Colom, L.V.; Murray, I.V.J. Probing and trapping a sensitive conformation: Amyloid-β fibrils, oligomers, and dimers. J. Alzheimer’s Dis. 2012, 32, 197–215. [Google Scholar]
- Case, D.A. Normal-mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 1994, 4, 285–290. [Google Scholar] [CrossRef]
- Schlitter, J. Estimation of absolute and relative entropies of macromolecules using the covariance-matrix. Chem. Phys. Lett. 1993, 215, 617–621. [Google Scholar] [CrossRef]
- Choong, C.J.; Say, Y.H. Neuroprotection of α-synuclein under acute and chronic rotenone and maneb treatment is abolished by its familial Parkinson’s disease mutations a30p, a53t and e46k. Neurotoxicology 2011, 32, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Zibaee, S.; Jakes, R.; Serpell, L.C.; Davletov, B.; Crowther, R.A.; Goedert, M. Mutation e46k increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 2004, 576, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Moussa, C.E.; Wersinger, C.; Tomita, Y.; Sidhu, A. Differential cytotoxicity of human wild type and mutant α-synuclein in human neuroblastoma sh-sy5y cells in the presence of dopamine. Biochemistry 2004, 43, 5539–5550. [Google Scholar] [CrossRef] [PubMed]
- Wersinger, C.; Sidhu, A. Differential cytotoxicity of dopamine and h2o2 in a human neuroblastoma divided cell line transfected with α-synuclein and its familial Parkinson’s disease-linked mutants. Neurosci. Lett. 2003, 342, 124–128. [Google Scholar] [CrossRef]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson’s disease mutant a30p α-synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Interaction of human α-synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J. Biol. Chem. 2000, 275, 34393–34398. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999, 274, 7619–7622. [Google Scholar] [CrossRef] [PubMed]
- Sahay, S.; Ghosh, D.; Singh, P.K.; Maji, S.K. Alteration of structure and aggregation of α-synuclein by familial Parkinson’s disease associated mutations. Curr. Protein Pept. Sci. 2017, 18, 656–676. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Ikeda, T.; Takasaki, J.; Yamada, M. Familial Parkinson disease mutations influence α-synuclein assembly. Neurobiol. Dis. 2011, 43, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Brucale, M.; Sandal, M.; Di Maio, S.; Rampioni, A.; Tessari, I.; Tosatto, L.; Bisaglia, M.; Bubacco, L.; Samori, B. Pathogenic mutations shift the equilibria of α-synuclein single molecules towards structured conformers. ChemBioChem 2009, 10, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Heise, H.; Celej, M.S.; Becker, S.; Riedel, D.; Pelah, A.; Kumar, A.; Jovin, T.M.; Baldus, M. Solid-state NMR reveals structural differences between fibrils of WT and disease-related a53t mutant α-synuclein. J. Mol. Biol. 2008, 380, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Kamiyoshihara, T.; Kojima, M.; Ueda, K.; Tashiro, M.; Shimotakahara, S. Observation of multiple intermediates in α-synuclein fibril formation by singular value decomposition analysis. Biochem. Biophys. Res. Commun. 2007, 355, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T., Jr. α-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef]
- Bussell, R., Jr.; Eliezer, D. Residual structure and dynamics in Parkinson’s disease-associated mutants of α-synuclein. J. Biol. Chem. 2001, 276, 45996–46003. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate α-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, S.A.; Mohanty, S.; Irback, A. Distinct phases of free α-synuclein--a Monte Carlo study. Proteins 2012, 80, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Volles, M.J.; Lee, S.J.; Rochet, J.C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar α-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; McLaurin, J.; Yip, C.M.; St George-Hyslop, P.; Fraser, P.E. α-synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254. [Google Scholar] [CrossRef]
- Balesh, D.; Ramjan, Z.; Floriano, W.B. Unfolded annealing molecular dynamics conformers for WT and disease-associated variants of α-synuclein show no propensity for β-sheet formation. J. Biophys. Chem. 2011, 2, 124–134. [Google Scholar] [CrossRef]
- Losasso, V.; Pietropaolo, A.; Zannoni, C.; Gustincich, S.; Carloni, P. Structural role of compensatory amino acid replacements in the α-synuclein protein. Biochemistry 2011, 50, 6994–7001. [Google Scholar] [CrossRef] [PubMed]
- Hazy, E.; Bokor, M.; Kalmar, L.; Gelencser, A.; Kamasa, P.; Han, K.H.; Tompa, K.; Tompa, P. Distinct hydration properties of WT and familial point mutant A53t of α-synuclein associated with Parkinson’s disease. Biophys. J. 2011, 101, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Fernandez, C.O.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Familial mutants of α-synuclein with increased neurotoxicity have a destabilized conformation. J. Biol. Chem. 2005, 280, 30649–30652. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Sengupta, N. Effect of the A30p mutation on the structural dynamics of micelle-bound αSynuclein released in water: A molecular dynamics study. Eur. Biophys. J. 2012, 41, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.; Moran, C.R.; Ferreon, J.C.; Deniz, A.A. Alteration of the α-synuclein folding landscape by a mutation related to Parkinson’s disease. Angew. Chem. Int. Ed. Engl. 2010, 49, 3469–3472. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Langen, R.; Hummel, P.A.; Gray, H.B.; Winkler, J.R. α-synuclein structures from fluorescence energy-transfer kinetics: Implications for the role of the protein in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 16466–16471. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, E.R.; Ramlall, T.F.; Borbat, P.P.; Freed, J.H.; Eliezer, D. The lipid-binding domain of wild type and mutant α-synuclein: Compactness and interconversion between the broken and extended helix forms. J. Biol. Chem. 2010, 285, 28261–28274. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.C.; Rochet, J.C.; Lansbury, P.T., Jr. The N-terminal repeat domain of α-synuclein inhibits β-sheet and amyloid fibril formation. Biochemistry 2003, 42, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Vilar, M.; Chou, H.T.; Luhrs, T.; Maji, S.K.; Riek-Loher, D.; Verel, R.; Manning, G.; Stahlberg, H.; Riek, R. The fold of α-synuclein fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 8637–8642. [Google Scholar] [CrossRef] [PubMed]
- Rospigliosi, C.C.; McClendon, S.; Schmid, A.W.; Ramlall, T.F.; Barre, P.; Lashuel, H.A.; Eliezer, D. E46k Parkinson’s-linked mutation enhances C-terminal-to-N-terminal contacts in α-synuclein. J. Mol. Biol. 2009, 388, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Sanjeev, A.; Mattaparthi, V.S.K. Computational investigation on the effects of h50q and g51d mutations on the α-synuclein aggregation propensity. J. Biomol. Struct. Dyn. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Sharikov, Y.; Kouznetsova, V.L.; Greenberg, J.P.; Wrasidlo, W.; Overk, C.; Gonzalez, T.; Trejo, M.; Spencer, B.; Kosberg, K.; et al. Molecular determinants of α-synuclein mutants’ oligomerization and membrane interactions. ACS Chem. Neurosci. 2015, 6, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Shvadchak, V.V.; Yushchenko, D.A.; Pievo, R.; Jovin, T.M. The mode of α-synuclein binding to membranes depends on lipid composition and lipid to protein ratio. FEBS Lett. 2011, 585, 3513–3519. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N. Molecular basis for the glycosphingolipid-binding specificity of α-synuclein: Key role of tyrosine 39 in membrane insertion. J. Mol. Biol. 2011, 408, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Mahfoud, R.; Garmy, N.; Maresca, M.; Yahi, N.; Puigserver, A.; Fantini, J. Identification of a common sphingolipid-binding domain in Alzheimer, prion, and hiv-1 proteins. J. Biol. Chem. 2002, 277, 11292–11296. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, E.; Fantini, J.; Chahinian, H.; Maresca, M.; Taieb, N.; Yahi, N. Altered ion channel formation by the Parkinson’s-disease-linked e46k mutant of α-synuclein is corrected by gm3 but not by gm1 gangliosides. J. Mol. Biol. 2010, 397, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Mammi, S.; Bubacco, L. Structural insights on physiological functions and pathological effects of α-synuclein. FASEB J. 2009, 23, 329–340. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. α-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef] [PubMed]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length α-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. α-synuclein in central nervous system and from erythrocytes, mammalian cells, and escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef] [PubMed]
- Waudby, C.A.; Camilloni, C.; Fitzpatrick, A.W.; Cabrita, L.D.; Dobson, C.M.; Vendruscolo, M.; Christodoulou, J. In-cell NMR characterization of the secondary structure populations of a disordered conformation of α-synuclein within E. Coli cells. PLoS ONE 2013, 8, e72286. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; De Simone, A.; Gopinath, T.; Vostrikov, V.; Vendruscolo, M.; Dobson, C.M.; Veglia, G. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 2014, 5, 3827. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.P.; Soragni, A.; Rabe, M.; Verdes, D.; Liverani, E.; Handschin, S.; Riek, R.; Seeger, S. Mechanism of membrane interaction and disruption by α-synuclein. J. Am. Chem. Soc. 2011, 133, 19366–19375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtain, C.C.; Kirby, N.M.; Mertens, H.D.; Barnham, K.J.; Knott, R.B.; Masters, C.L.; Cappai, R.; Rekas, A.; Kenche, V.B.; Ryan, T. α-synuclein oligomers and fibrils originate in two distinct conformer pools: A small angle X-ray scattering and ensemble optimisation modelling study. Mol. Biosyst. 2015, 11, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Munder, T.; Pfeffer, A.; Schreyer, S.; Guo, J.; Braun, J.; Sack, I.; Steiner, B.; Klein, C. Mr elastography detection of early viscoelastic response of the murine hippocampus to amyloid β accumulation and neuronal cell loss due to Alzheimer’s disease. J. Magn. Reson. Imag. 2018, 47, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castano, E.M.; Frangione, B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat. Med. 1998, 4, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent β-sheet conformation. Biochem. Biophys. Res. Commun. 1996, 226, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Barrow, C.J.; Yasuda, A.; Kenny, P.T.; Zagorski, M.G. Solution conformations and aggregational properties of synthetic amyloid β-peptides of Alzheimer’s disease. Analysis of circular dichroism spectra. J. Mol. Biol. 1992, 225, 1075–1093. [Google Scholar] [CrossRef]
- Hilbich, C.; Kisters-Woike, B.; Reed, J.; Masters, C.L.; Beyreuther, K. Aggregation and secondary structure of synthetic amyloid β a4 peptides of Alzheimer’s disease. J. Mol. Biol. 1991, 218, 149–163. [Google Scholar] [CrossRef]
- Sipe, J.D.; Cohen, A.S. Review: History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Balbirnie, M.; Grothe, R.; Eisenberg, D.S. An amyloid-forming peptide from the yeast prion sup35 reveals a dehydrated β-sheet structure for amyloid. Proc. Natl. Acad. Sci. USA 2001, 98, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Cobb, N.J.; Sonnichsen, F.D.; McHaourab, H.; Surewicz, W.K. Molecular architecture of human prion protein amyloid: A parallel, in-register β-structure. Proc. Natl. Acad. Sci. USA 2007, 104, 18946–18951. [Google Scholar] [CrossRef] [PubMed]
- Fandrich, M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell. Mol. Life Sci. 2007, 64, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Shivaprasad, S.; Wetzel, R. Analysis of amyloid fibril structure by scanning cysteine mutagenesis. Methods Enzymol. 2006, 413, 182–198. [Google Scholar] [PubMed]
- Kheterpal, I.; Chen, M.; Cook, K.D.; Wetzel, R. Structural differences in Aβ amyloid protofibrils and fibrils mapped by hydrogen exchange--mass spectrometry with on-line proteolytic fragmentation. J. Mol. Biol. 2006, 361, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.D.; Shivaprasad, S.; Wetzel, R. Alanine scanning mutagenesis of Aβ(1–40) amyloid fibril stability. J. Mol. Biol. 2006, 357, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Shivaprasad, S.; Wetzel, R. Scanning cysteine mutagenesis analysis of Aβ-(1–40) amyloid fibrils. J. Biol. Chem. 2006, 281, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, N.A.; Mishra, R.; Kheterpal, I.; Williams, A.D.; Wetzel, R.; Serpersu, E.H. Hydrogen-deuterium (h/d) exchange mapping of Aβ 1–40 amyloid fibril secondary structure using nuclear magnetic resonance spectroscopy. Biochemistry 2005, 44, 4434–4441. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.D.; Portelius, E.; Kheterpal, I.; Guo, J.T.; Cook, K.D.; Xu, Y.; Wetzel, R. Mapping Aβ amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol. 2004, 335, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Almeida, B.; Fraga, J.S.; Taboada, P.; Martins, P.M.; Macedo-Ribeiro, S. Distribution of amyloid-like and oligomeric species from protein aggregation kinetics. Angew. Chem. Int. Ed. Engl. 2017, 56, 14042–14045. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.; Zhao, R.; So, M.; Adachi, M.; Rivas, G.; Carver, J.A.; Goto, Y. Recognizing and analyzing variability in amyloid formation kinetics: Simulation and statistical methods. Anal. Biochem. 2016, 510, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Girvan, P.; Miyake, T.; Teng, X.; Branch, T.; Ying, L. Kinetics of the interactions between copper and amyloid-β with fad mutations and phosphorylation at the n terminus. ChemBioChem 2016, 17, 1732–1737. [Google Scholar] [CrossRef] [PubMed]
- Villar-Pique, A.; Espargaro, A.; Ventura, S.; Sabate, R. In vivo amyloid aggregation kinetics tracked by time-lapse confocal microscopy in real-time. Biotechnol. J. 2016, 11, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Crespo, R.; Rocha, F.A.; Damas, A.M.; Martins, P.M. A generic crystallization-like model that describes the kinetics of amyloid fibril formation. J. Biol. Chem. 2012, 287, 30585–30594. [Google Scholar] [CrossRef] [PubMed]
- Price, J.C.; Klunk, W.E.; Lopresti, B.J.; Lu, X.; Hoge, J.A.; Ziolko, S.K.; Holt, D.P.; Meltzer, C.C.; DeKosky, S.T.; Mathis, C.A. Kinetic modeling of amyloid binding in humans using pet imaging and pittsburgh compound-b. J. Cereb. Blood Flow Metab. 2005, 25, 1528–1547. [Google Scholar] [CrossRef] [PubMed]
- Naiki, H.; Gejyo, F. Kinetic analysis of amyloid fibril formation. Methods Enzymol. 1999, 309, 305–318. [Google Scholar] [PubMed]
- Lomakin, A.; Teplow, D.B.; Kirschner, D.A.; Benedek, G.B. Kinetic theory of fibrillogenesis of amyloid β-protein. Proc. Natl. Acad. Sci. USA 1997, 94, 7942–7947. [Google Scholar] [CrossRef] [PubMed]
- Naiki, H.; Nakakuki, K. First-order kinetic model of Alzheimer’s β-amyloid fibril extension in vitro. Lab. Investig. 1996, 74, 374–383. [Google Scholar] [PubMed]
- Come, J.H.; Fraser, P.E.; Lansbury, P.T., Jr. A kinetic model for amyloid formation in the prion diseases: Importance of seeding. Proc. Natl. Acad. Sci. USA 1993, 90, 5959–5963. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Nguyen, M.T.; Nguyen, N.T.; Vu, V.V. The effects of a21g mutation on transmembrane amyloid β (11–40) trimer: An in silico study. J. Phys. Chem. B 2017, 121, 8467–8474. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Benilova, I.; Gallardo, R.; Ungureanu, A.A.; Castillo Cano, V.; Snellinx, A.; Ramakers, M.; Bartic, C.; Rousseau, F.; Schymkowitz, J.; De Strooper, B. The Alzheimer disease protective mutation a2t modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J. Biol. Chem. 2014, 289, 30977–30989. [Google Scholar] [CrossRef] [PubMed]
- Fawzi, N.L.; Kohlstedt, K.L.; Okabe, Y.; Head-Gordon, T. Protofibril assemblies of the Arctic, Dutch, and Flemish mutants of the Alzheimer’s Aβ1–40 peptide. Biophys. J. 2008, 94, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Hasegawa, K.; Matsuzaki, K.; Naiki, H.; Yanagisawa, K. Environment- and mutation-dependent aggregation behavior of Alzheimer amyloid β-protein. J. Neurochem. 2004, 90, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘arctic’ app mutation (e693g) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Zagorski, M.G.; Barrow, C.J. NMR studies of amyloid β-peptides: Proton assignments, secondary structure, and mechanism of an α-helix-β-sheet conversion for a homologous, 28-residue, N-terminal fragment. Biochemistry 1992, 31, 5621–5631. [Google Scholar] [CrossRef] [PubMed]
- Burdick, D.; Soreghan, B.; Kwon, M.; Kosmoski, J.; Knauer, M.; Henschen, A.; Yates, J.; Cotman, C.; Glabe, C. Assembly and aggregation properties of synthetic Alzheimer’s a4/β amyloid peptide analogs. J. Biol. Chem. 1992, 267, 546–554. [Google Scholar] [PubMed]
- Velez-Vega, C.; Escobedo, F.A. Characterizing the structural behavior of selected Aβ-42 monomers with different solubilities. J. Phys. Chem. B 2011, 115, 4900–4910. [Google Scholar] [CrossRef] [PubMed]
- Sgourakis, N.G.; Merced-Serrano, M.; Boutsidis, C.; Drineas, P.; Du, Z.; Wang, C.; Garcia, A.E. Atomic-level characterization of the ensemble of the Aβ(1–42) monomer in water using unbiased molecular dynamics simulations and spectral algorithms. J. Mol. Biol. 2011, 405, 570–583. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, N.F.; Wu, C.; Shea, J.E.; Bowers, M.T. Human islet amyloid polypeptide monomers form ordered β-hairpins: A possible direct amyloidogenic precursor. J. Am. Chem. Soc. 2009, 131, 18283–18292. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Teplow, D.B. Amyloid β-protein monomer folding: Free-energy surfaces reveal alloform-specific differences. J. Mol. Biol. 2008, 384, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Sgourakis, N.G.; Yan, Y.; McCallum, S.A.; Wang, C.; Garcia, A.E. The Alzheimer’s peptides Aβ40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J. Mol. Biol. 2007, 368, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Tarus, B.; Straub, J.E.; Thirumalai, D. Dynamics of asp23-lys28 salt-bridge formation in Aβ10–35 monomers. J. Am. Chem. Soc. 2006, 128, 16159–16168. [Google Scholar] [CrossRef] [PubMed]
- Luttmann, E.; Fels, G. All-atom molecular dynamics studies of the full-length β-amyloid peptides. Chem. Phys. 2006, 323, 138–147. [Google Scholar] [CrossRef]
- Yoshiike, Y.; Akagi, T.; Takashima, A. Surface structure of amyloid-β fibrils contributes to cytotoxicity. Biochemistry 2007, 46, 9805–9812. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, S.; Lee, E.; Manna, M.; Steinert, S.; Kumar, G.S.; Wenk, M.; Wohland, T.; Kraut, R. A fluorescent sphingolipid binding domain peptide probe interacts with sphingolipids and cholesterol-dependent raft domains. J. Lipid Res. 2008, 49, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B. Immunological approach for the treatment of Alzheimer’s disease. J. Mol. Neurosci. 2003, 20, 283–286. [Google Scholar] [CrossRef]
- Frenkel, D.; Balass, M.; Katchalski-Katzir, E.; Solomon, B. High affinity binding of monoclonal antibodies to the sequential epitope efrh of β-amyloid peptide is essential for modulation of fibrillar aggregation. J. Neuroimmunol. 1999, 95, 136–142. [Google Scholar] [CrossRef]
- Frenkel, D.; Balass, M.; Solomon, B. N-terminal efrh sequence of Alzheimer’s β-amyloid peptide represents the epitope of its anti-aggregating antibodies. J. Neuroimmunol. 1998, 88, 85–90. [Google Scholar] [CrossRef]
- Coskuner, O.; Wise-Scira, O. Arginine and disordered amyloid-β peptide structures: Molecular level insights into the toxicity in Alzheimer’s disease. ACS Chem. Neurosci. 2013, 4, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Barone, E.; Di Domenico, F.; Butterfield, D.A. Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Biophys. Acta 2016, 1862, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Coutadeur, S.; Benyamine, H.; Delalonde, L.; de Oliveira, C.; Leblond, B.; Foucourt, A.; Besson, T.; Casagrande, A.S.; Taverne, T.; Girard, A.; et al. A novel dyrk1a (dual specificity tyrosine phosphorylation-regulated kinase 1a) inhibitor for the treatment of Alzheimer’s disease: Effect on tau and amyloid pathologies in vitro. J. Neurochem. 2015, 133, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Yeo, W.S.; Kim, Y.J.; Kabir, M.H.; Kang, J.W.; Ahsan-Ul-Bari, M.; Kim, K.P. Mass spectrometric analysis of protein tyrosine nitration in aging and neurodegenerative diseases. Mass Spectrom. Rev. 2015, 34, 166–183. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Miura, T.; Takeuchi, H. Inhibitory effect of copper(ii) on zinc(ii)-induced aggregation of amyloid β-peptide. Biochem. Biophys. Res. Commun. 2001, 285, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Suzuki, K.; Takeuchi, H. Binding of iron(iii) to the single tyrosine residue of amyloid β-peptide probed by raman spectroscopy. J. Mol. Struct. 2001, 598, 79–84. [Google Scholar] [CrossRef]
- Coskuner, O.; Murray, I.V.J. Adenosine triphosphate (atp) reduces amyloid-β protein misfolding in vitro. J. Alzheimer’s Dis. 2014, 41, 561–574. [Google Scholar]
- Barnham, K.J.; Haeffner, F.; Ciccotosto, G.D.; Curtain, C.C.; Tew, D.; Mavros, C.; Beyreuther, K.; Carrington, D.; Masters, C.L.; Cherny, R.A.; et al. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease β-amyloid. Faseb J. 2004, 18, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Coskuner, O.; Uversky, V.N. Tyrosine regulates β-sheet structure formation in amyloid-β42: A new clustering algorithm for disordered proteins. J. Chem. Inf. Model. 2017, 57, 1342–1358. [Google Scholar] [CrossRef] [PubMed]
- Aran Terol, P.; Kumita, J.R.; Hook, S.C.; Dobson, C.M.; Esbjorner, E.K. Solvent exposure of tyr10 as a probe of structural differences between monomeric and aggregated forms of the amyloid-β peptide. Biochem. Biophys. Res. Commun. 2015, 468, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.H.; Raleigh, D.P. Role of aromatic interactions in amyloid formation by islet amyloid polypeptide. Biochemistry 2013, 52, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, F.; Taddei, N.; Stefani, M.; Chiti, F. Assessing the role of aromatic residues in the amyloid aggregation of human muscle acylphosphatase. Protein Sci. 2006, 15, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Luu, X.C.; Nguyen, M.T.; Le, C.N.; Vu, V.V. In silico studies of solvated f19w amyloid β (11–40) trimer. RSC Adv. 2017, 7, 42379–42386. [Google Scholar] [CrossRef]
- Demuro, A.; Smith, M.; Parker, I. Single-channel Ca(2+) imaging implicates Aβ1–42 amyloid pores in Alzheimer’s disease pathology. J. Cell Biol. 2011, 195, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Glabe, C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [Google Scholar] [CrossRef] [PubMed]
- Shirwany, N.A.; Payette, D.; Xie, J.; Guo, Q. The amyloid β ion channel hypothesis of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 597–612. [Google Scholar] [PubMed]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Bhatia, R.; Lal, R. Amyloid β protein forms ion channels: Implications for Alzheimer’s disease pathophysiology. FASEB J. 2001, 15, 2433–2444. [Google Scholar] [CrossRef] [PubMed]
- De Groot, N.S.; Aviles, F.X.; Vendrell, J.; Ventura, S. Mutagenesis of the central hydrophobic cluster in Aβ42 Alzheimer’s peptide. Side-chain properties correlate with aggregation propensities. FEBS J. 2006, 273, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Iversen, L.L.; Mortishire-Smith, R.J.; Pollack, S.J.; Shearman, M.S. The toxicity in vitro of β-amyloid protein. Biochem. J. 1995, 311, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, S.L.; Wyttenbach, T.; Baumketner, A.; Shea, J.E.; Bitan, G.; Teplow, D.B.; Bowers, M.T. Amyloid β-protein: Monomer structure and early aggregation states of Aβ42 and its pro19 alloform. J. Am. Chem. Soc. 2005, 127, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Lomakin, A.; Benedek, G.B.; Condron, M.M.; Teplow, D.B. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J. Biol. Chem. 1997, 272, 22364–22372. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.J.; Wetzel, R.; Martin, J.D.; Hurle, M.R. Prolines and amyloidogenicity in fragments of the Alzheimer’s peptide β/a4. Biochemistry 1995, 34, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Zheng, J.; Lal, R.; Nussinov, R. New structures help the modeling of toxic amyloid β ion channels. Trends Biochem. Sci. 2008, 33, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Arispe, N. Architecture of the Alzheimer’s Aβ p ion channel pore. J. Membr. Biol. 2004, 197, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Durell, S.R.; Guy, H.R.; Arispe, N.; Rojas, E.; Pollard, H.B. Theoretical models of the ion channel structure of amyloid β-protein. Biophys. J. 1994, 67, 2137–2145. [Google Scholar] [CrossRef]
- Connelly, L.; Jang, H.; Arce, F.T.; Ramachandran, S.; Kagan, B.L.; Nussinov, R.; Lal, R. Effects of point substitutions on the structure of toxic Alzheimer’s β-amyloid channels: Atomic force microscopy and molecular dynamics simulations. Biochemistry 2012, 51, 3031–3038. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Lal, R.; Nussinov, R. Β-barrel topology of Alzheimer’s β-amyloid ion channels. J. Mol. Biol. 2010, 404, 917–934. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Azimova, R.; Kagan, B.L.; Nussinov, R.; Lal, R. Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer’s disease and down syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 6538–6543. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Zheng, J.; Nussinov, R. Models of β-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys. J. 2007, 93, 1938–1949. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Jayakumar, R. Structural analysis of amyloid β peptide fragment (25–35) in different microenvironments. Biopolymers 2004, 76, 421–434. [Google Scholar] [CrossRef] [PubMed]
- D’Ursi, A.M.; Armenante, M.R.; Guerrini, R.; Salvadori, S.; Sorrentino, G.; Picone, D. Solution structure of amyloid β-peptide (25–35) in different media. J. Med. Chem. 2004, 47, 4231–4238. [Google Scholar] [CrossRef] [PubMed]
- Ippel, J.H.; Olofsson, A.; Schleucher, J.; Lundgren, E.; Wijmenga, S.S. Probing solvent accessibility of amyloid fibrils by solution nmr spectroscopy. Proc. Natl. Acad. Sci. USA 2002, 99, 8648–8653. [Google Scholar] [CrossRef] [PubMed]
- Konno, T. Amyloid-induced aggregation and precipitation of soluble proteins: An electrostatic contribution of the Alzheimer’s β(25–35) amyloid fibril. Biochemistry 2001, 40, 2148–2154. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Nussinov, R. The stability of monomeric intermediates controls amyloid formation: Aβ25-35 and its n27q mutant. Biophys. J. 2006, 90, 3365–3374. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Kiefhaber, T. Intermediates can accelerate protein folding. Proc. Natl. Acad. Sci. USA 1999, 96, 6716–6721. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fukuda, H.; Murayama, S.; Izumiyama, N.; Shirasawa, T. Isoaspartate formation at position 23 of amyloid β peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Klimov, D.K. Side chain interactions can impede amyloid fibril growth: Replica exchange simulations of Aβ peptide mutant. J. Phys. Chem. B 2009, 113, 11848–11857. [Google Scholar] [CrossRef] [PubMed]
- Harmeier, A.; Wozny, C.; Rost, B.R.; Munter, L.M.; Hua, H.; Georgiev, O.; Beyermann, M.; Hildebrand, P.W.; Weise, C.; Schaffner, W.; et al. Role of amyloid-β glycine 33 in oligomerization, toxicity, and neuronal plasticity. J. Neurosci. 2009, 29, 7582–7590. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.W.; Ciccotosto, G.D.; Giannakis, E.; Tew, D.J.; Perez, K.; Masters, C.L.; Cappai, R.; Wade, J.D.; Barnham, K.J. Amyloid-β peptide (Aβ) neurotoxicity is modulated by the rate of peptide aggregation: Aβ dimers and trimers correlate with neurotoxicity. J. Neurosci. 2008, 28, 11950–11958. [Google Scholar] [CrossRef] [PubMed]
- Munter, L.M.; Voigt, P.; Harmeier, A.; Kaden, D.; Gottschalk, K.E.; Weise, C.; Pipkorn, R.; Schaefer, M.; Langosch, D.; Multhaup, G. Gxxxg motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Aβ42. EMBO J. 2007, 26, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wei, G.; Derreumaux, P. Effects of g33a and g33i mutations on the structures of monomer and dimer of the amyloid-β fragment 29-42 by replica exchange molecular dynamics simulations. J. Phys. Chem. B 2011, 115, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Lansbury, P.T., Jr.; Costa, P.R.; Griffiths, J.M.; Simon, E.J.; Auger, M.; Halverson, K.J.; Kocisko, D.A.; Hendsch, Z.S.; Ashburn, T.T.; Spencer, R.G.; et al. Structural model for the β-amyloid fibril based on interstrand alignment of an antiparallel-sheet comprising a c-terminal peptide. Nat. Struct. Biol. 1995, 2, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.G.; Okamoto, Y. Amyloid-β(29-42) dimer formations studied by a multicanonical-multioverlap molecular dynamics simulation. J. Phys. Chem. B 2008, 112, 2767–2770. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Sterpone, F.; Campanera, J.M.; Nasica-Labouze, J.; Derreumaux, P. Impact of the a2v mutation on the heterozygous and homozygous Aβ1–40 dimer structures from atomistic simulations. ACS Chem. Neurosci. 2016, 7, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Maloney, J.A.; Bainbridge, T.; Gustafson, A.; Zhang, S.; Kyauk, R.; Steiner, P.; van der Brug, M.; Liu, Y.; Ernst, J.A.; Watts, R.J.; et al. Molecular mechanisms of Alzheimer disease protection by the a673t allele of amyloid precursor protein. J. Biol. Chem. 2014, 289, 30990–31000. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Murray, B.; Belfort, G. Alzheimer’s protective a2t mutation changes the conformational landscape of the Aβ(1)(-)(4)(2) monomer differently than does the a2v mutation. Biophys. J. 2015, 108, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Huet, A.; Derreumaux, P. Impact of the mutation a21g (Flemish variant) on Alzheimer’s β-amyloid dimers by molecular dynamics simulations. Biophys. J. 2006, 91, 3829–3840. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.R.; Teplow, D.B.; Stanley, H.E.; Urbanc, B. Effects of the arctic (e22→g) mutation on amyloid β-protein folding: Discrete molecular dynamics study. J. Am. Chem. Soc. 2008, 130, 17413–17422. [Google Scholar] [CrossRef] [PubMed]
- Gursky, O.; Aleshkov, S. Temperature-dependent β-sheet formation in β-amyloid Aβ(1–40) peptide in water: Uncoupling β-structure folding from aggregation. Biochim. Biophys. Acta 2000, 1476, 93–102. [Google Scholar] [CrossRef]
- Lim, K.H.; Collver, H.H.; Le, Y.T.; Nagchowdhuri, P.; Kenney, J.M. Characterizations of distinct amyloidogenic conformations of the Aβ(1–40) and (1–42) peptides. Biochem. Biophys. Res. Commun. 2007, 353, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Baumketner, A.; Shea, J.E. The structure of the Alzheimer amyloid β 10–35 peptide probed through replica-exchange molecular dynamics simulations in explicit solvent. J. Mol. Biol. 2007, 366, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Wu, Y.D. A strand-loop-strand structure is a possible intermediate in fibril elongation: Long time simulations of amyloid-β peptide (10–35). J. Am. Chem. Soc. 2005, 127, 15408–15416. [Google Scholar] [CrossRef] [PubMed]
- Massi, F.; Peng, J.W.; Lee, J.P.; Straub, J.E. Simulation study of the structure and dynamics of the Alzheimer’s amyloid peptide congener in solution. Biophys. J. 2001, 80, 31–44. [Google Scholar] [CrossRef]
- Zhang, S.; Iwata, K.; Lachenmann, M.J.; Peng, J.W.; Li, S.; Stimson, E.R.; Lu, Y.; Felix, A.M.; Maggio, J.E.; Lee, J.P. The Alzheimer’s peptide Aβ adopts a collapsed coil structure in water. J. Struct. Biol. 2000, 130, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid β -protein (Aβ) assembly: Aβ 40 and Aβ 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, K.N.; Manelli, A.M.; Stine, W.B., Jr.; Baker, L.K.; Krafft, G.A.; LaDu, M.J. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 2002, 277, 32046–32053. [Google Scholar] [CrossRef] [PubMed]
- Whalen, B.M.; Selkoe, D.J.; Hartley, D.M. Small non-fibrillar assemblies of amyloid β-protein bearing the arctic mutation induce rapid neuritic degeneration. Neurobiol. Dis. 2005, 20, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Krone, M.G.; Baumketner, A.; Bernstein, S.L.; Wyttenbach, T.; Lazo, N.D.; Teplow, D.B.; Bowers, M.T.; Shea, J.E. Effects of familial Alzheimer’s disease mutations on the folding nucleation of the amyloid β-protein. J. Mol. Biol. 2008, 381, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Mousseau, N.; Derreumaux, P. The conformations of the amyloid-β (21–30) fragment can be described by three families in solution. J. Chem. Phys. 2006, 125, 084911. [Google Scholar] [CrossRef] [PubMed]
- Baumketner, A.; Bernstein, S.L.; Wyttenbach, T.; Lazo, N.D.; Teplow, D.B.; Bowers, M.T.; Shea, J.E. Structure of the 21–30 fragment of amyloid β-protein. Protein Sci. 2006, 15, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.; Urbanc, B.; Borreguero, J.M.; Lazo, N.D.; Teplow, D.B.; Stanley, H.E. Solvent and mutation effects on the nucleation of amyloid β-protein folding. Proc. Natl. Acad. Sci. USA 2005, 102, 18258–18263. [Google Scholar] [CrossRef] [PubMed]
- Lazo, N.D.; Grant, M.A.; Condron, M.C.; Rigby, A.C.; Teplow, D.B. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005, 14, 1581–1596. [Google Scholar] [CrossRef] [PubMed]
- Borreguero, J.M.; Urbanc, B.; Lazo, N.D.; Buldyrev, S.V.; Teplow, D.B.; Stanley, H.E. Folding events in the 21-30 region of amyloid β-protein (Aβ) studied in silico. Proc. Natl. Acad. Sci. USA 2005, 102, 6015–6020. [Google Scholar] [CrossRef] [PubMed]
- Kassler, K.; Horn, A.H.; Sticht, H. Effect of pathogenic mutations on the structure and dynamics of Alzheimer’s Aβ 42-amyloid oligomers. J. Mol. Model. 2010, 16, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Pifer, P.M.; Yates, E.A.; Legleiter, J. Point mutations in Aβ result in the formation of distinct polymorphic aggregates in the presence of lipid bilayers. PLoS ONE 2011, 6, e16248. [Google Scholar] [CrossRef] [PubMed]
- Poojari, C.; Strodel, B. Stability of transmembrane amyloid β-peptide and membrane integrity tested by molecular modeling of site-specific Aβ42 mutations. PLoS ONE 2013, 8, e78399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, M.A.; Lazo, N.D.; Lomakin, A.; Condron, M.M.; Arai, H.; Yamin, G.; Rigby, A.C.; Teplow, D.B. Familial Alzheimer’s disease mutations alter the stability of the amyloid β-protein monomer folding nucleus. Proc. Natl. Acad. Sci. USA 2007, 104, 16522–16527. [Google Scholar] [CrossRef] [PubMed]
- Luheshi, L.M.; Tartaglia, G.G.; Brorsson, A.C.; Pawar, A.P.; Watson, I.E.; Chiti, F.; Vendruscolo, M.; Lomas, D.A.; Dobson, C.M.; Crowther, D.C. Systematic in vivo analysis of the intrinsic determinants of amyloid β pathogenicity. PLoS Biol. 2007, 5, e290. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; Freeman: New York, NY, USA, 1999. [Google Scholar]
- Lin, Y.S.; Bowman, G.R.; Beauchamp, K.A.; Pande, V.S. Investigating how peptide length and a pathogenic mutation modify the structural ensemble of amyloid β monomer. Biophys. J. 2012, 102, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Prakash, M.K.; Barducci, A.; Parrinello, M. Replica temperatures for uniform exchange and efficient roundtrip times in explicit solvent parallel tempering simulations. J. Chem. Theory Comput. 2011, 7, 2025–2027. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Kagan, B.L.; Lal, R.; Nussinov, R. Familial Alzheimer’s disease osaka mutant (Δe22) β-barrels suggest an explanation for the different Aβ1-40/42 preferred conformational states observed by experiment. J. Phys. Chem. B 2013, 117, 11518–11529. [Google Scholar] [CrossRef] [PubMed]
- Roychaudhuri, R.; Yang, M.; Deshpande, A.; Cole, G.M.; Frautschy, S.; Lomakin, A.; Benedek, G.B.; Teplow, D.B. C-terminal turn stability determines assembly differences between Aβ40 and Aβ42. J. Mol. Biol. 2013, 425, 292–308. [Google Scholar] [CrossRef] [PubMed]
- Kamino, K.; Orr, H.T.; Payami, H.; Wijsman, E.M.; Alonso, M.E.; Pulst, S.M.; Anderson, L.; O’Dahl, S.; Nemens, E.; White, J.A.; et al. Linkage and mutational analysis of familial Alzheimer disease kindreds for the app gene region. Am. J. Hum. Genet. 1992, 51, 998–1014. [Google Scholar] [PubMed]
- Levy, E.; Carman, M.D.; Fernandez-Madrid, I.J.; Power, M.D.; Lieberburg, I.; van Duinen, S.G.; Bots, G.T.; Luyendijk, W.; Frangione, B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Podlisny, M.B. Deciphering the genetic basis of Alzheimer’s disease. Annu. Rev. Genom. Hum. Genet. 2002, 3, 67–99. [Google Scholar] [CrossRef] [PubMed]
- Inayathullah, M.; Teplow, D.B. Structural dynamics of the Δe22 (Osaka) familial Alzheimer’s disease-linked amyloid β-protein. Amyloid 2011, 18, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikova, O.Y.; Finder, V.H.; Vodopivec, I.; Nitsch, R.M.; Glockshuber, R. The Osaka fad mutation e22Δ leads to the formation of a previously unknown type of amyloid β fibrils and modulates Aβ neurotoxicity. J. Mol. Biol. 2011, 408, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Cloe, A.L.; Orgel, J.P.; Sachleben, J.R.; Tycko, R.; Meredith, S.C. The japanese mutant Aβ (Δe22-Aβ(1–39)) forms fibrils instantaneously, with low-thioflavin t fluorescence: Seeding of WT Aβ(1–40) into atypical fibrils by Δe22-Aβ(1–39). Biochemistry 2011, 50, 2026–2039. [Google Scholar] [CrossRef] [PubMed]
- Esler, W.P.; Felix, A.M.; Stimson, E.R.; Lachenmann, M.J.; Ghilardi, J.R.; Lu, Y.A.; Vinters, H.V.; Mantyh, P.W.; Lee, J.P.; Maggio, J.E. Activation barriers to structural transition determine deposition rates of Alzheimer’s disease Aβ amyloid. J. Struct. Biol. 2000, 130, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Sian, A.K.; Frears, E.R.; El-Agnaf, O.M.; Patel, B.P.; Manca, M.F.; Siligardi, G.; Hussain, R.; Austen, B.M. Oligomerization of β-amyloid of the Alzheimer’s and the Dutch-cerebral-haemorrhage types. Biochem. J. 2000, 349, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.J.; Lander, A.D.; Selkoe, D.J. Heparin-binding properties of the amyloidogenic peptides Abeta and amylin. Dependence on aggregation state and inhibition by Congo Red. J. Biol. Chem. 1997, 272, 31617–31624. [Google Scholar] [CrossRef] [PubMed]
- Massi, F.; Klimov, D.; Thirumalai, D.; Straub, J.E. Charge states rather than propensity for beta-structure determine enhanced fibrillogenesis in WT Alzheimer’s β-amyloid peptide compared to e22q Dutch mutant. Protein Sci. 2002, 11, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Baumketner, A.; Krone, M.G.; Shea, J.E. Role of the familial Dutch mutation e22q in the folding and aggregation of the 15–28 fragment of the Alzheimer amyloid-β protein. Proc. Natl. Acad. Sci. USA 2008, 105, 6027–6032. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Castano, E.M.; Frangione, B.; Inestrosa, N.C. The α-helical to β-strand transition in the amino-terminal fragment of the amyloid β-peptide modulates amyloid formation. J. Biol. Chem. 1995, 270, 3063–3067. [Google Scholar] [CrossRef] [PubMed]
- Truong, P.M.; Viet, M.H.; Nguyen, P.H.; Hu, C.K.; Li, M.S. Effect of Taiwan mutation (d7h) on structures of amyloid-β peptides: Replica exchange molecular dynamics study. J. Phys. Chem. B 2014, 118, 8972–8981. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, J.; Jarvet, J.; Damberg, P.; Graslund, A. The Alzheimer β-peptide shows temperature-dependent transitions between left-handed 3-helix, β-strand and random coil secondary structures. FEBS J. 2005, 272, 3938–3949. [Google Scholar] [CrossRef] [PubMed]
- Viet, M.H.; Nguyen, P.H.; Ngo, S.T.; Li, M.S.; Derreumaux, P. Effect of the tottori familial disease mutation (d7n) on the monomers and dimers of Aβ40 and Aβ42. ACS Chem. Neurosci. 2013, 4, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Hashimoto, T.; Wakutani, Y.; Urakami, K.; Nakashima, K.; Condron, M.M.; Tsubuki, S.; Saido, T.C.; Teplow, D.B.; Iwatsubo, T. The Tottori (d7n) and English (h6r) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 2007, 282, 4916–4923. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.; Marques, S.F.; Ferreiro, E.; Pereira, C.M. Effect of α-Synuclein on Amyloid β Induced Toxicity: Relevance to Lewy Body Variant of Alzheimer Disease. Neurochem. Res. 2013, 38, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Pettegrew, J.; Masliah, E.; Hamilton, R.; Mandal, R. Interaction between Aβ Peptide and αSynuclein: Molecular Mechanisms in Overlapping Pathology of Alzheimer’s and Parkinson’s in Dementia with Lewy Body Disease. Neurochem. Res. 2006, 31, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Crews, L.; Desplats, P.; Shaked, G.M.; Sharikov, Y.; Mizuno, H.; Spencer, B.; Rockenstein, E.; Trejo, M.; Platoshyn, O.; et al. Mechanisms of Hybrid Oligomer Formation in the Pathogenesis of Combined Alzheimer’s and Parkinson’s Diseases. PLoS ONE 2008, 3, e3135. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.L.; Schneider, B.L.; Brown, D.R. α-Synuclein increases β-amyloid secretion by promoting β-/γ-secretase processing of APP. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.C.; Chatterjee, P.; Sengupta, N. Cross Dimerization of Amyloid-β and αSynuclein Proteins in Aqueous Environment: A Molecular Dynamics Simulations Study. PLoS ONE 2014, 9, e106883. [Google Scholar] [CrossRef] [PubMed]
- Atsmon-Raz, Y.; Miller, Y. Non-Amyloid-β Component of Human α-Synuclein Oligomers Induces Formation of New Aβ Oligomers: Insight into the Mechanisms That Link Parkinson’s and Alzheimer’s Diseases. ACS Chem. Neurosci. 2016, 7, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Fusco, G.; De Simone, A.; Arosio, P.; Vendruscolo, M.; Veglia, G.; Dobson, C.M. Structural Ensembles of Membrane-bound α-Synuclein Reveal the Molecular Determinants of Synaptic Vesicle Affinity. Sci. Rep. 2016, 6, 27125. [Google Scholar] [CrossRef] [PubMed]
- Baram, M.; Atsmon-Raz, Y.; Ma, B.; Nussinov, R.; Miller, Y. Amylin-Aβ oligomers at atomic resolution using molecular dynamics simulations: A link between Type 2 diabetes and Alzheimer’s disease. Phys. Chem. Chem. Phys. 2016, 18, 2330–2338. [Google Scholar] [CrossRef] [PubMed]

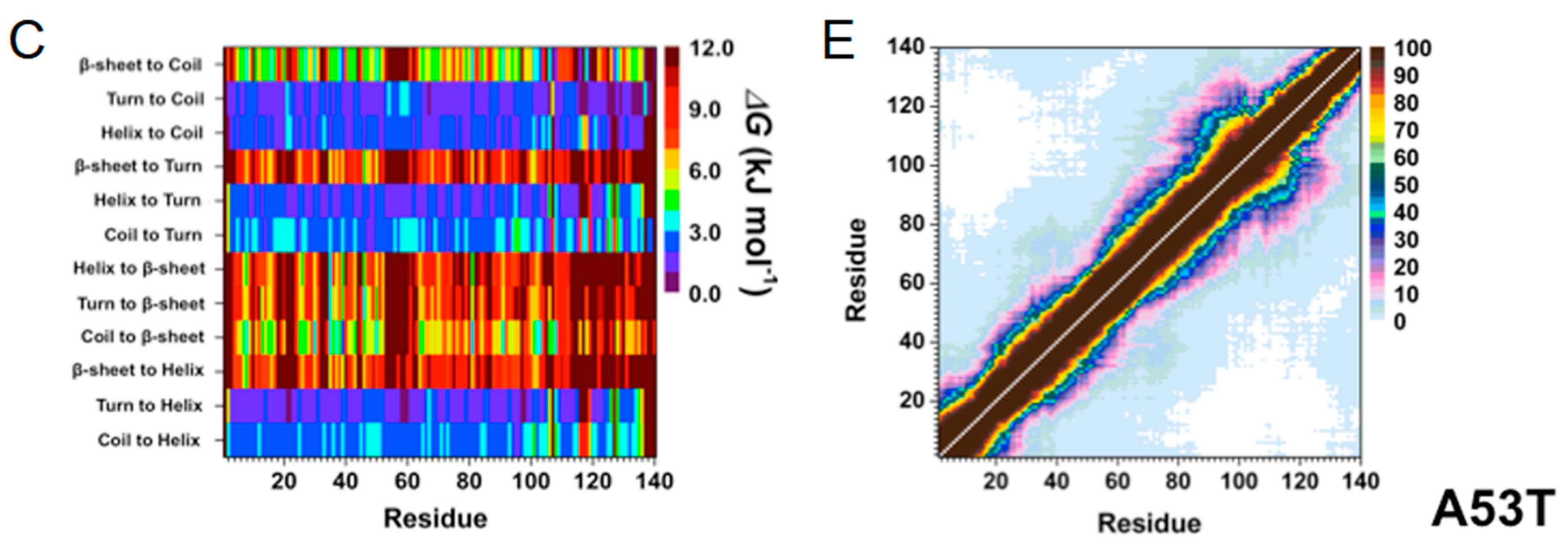

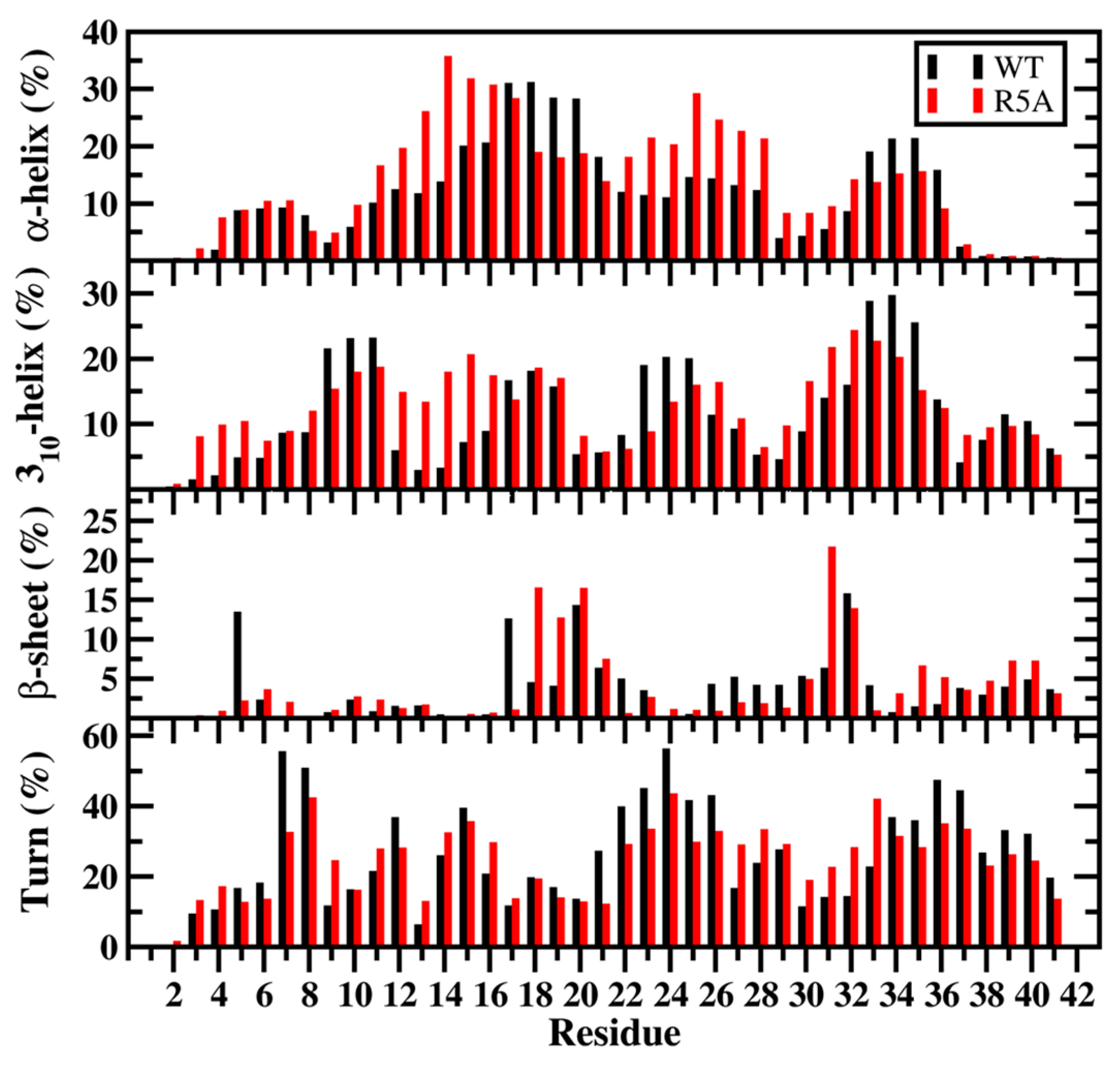

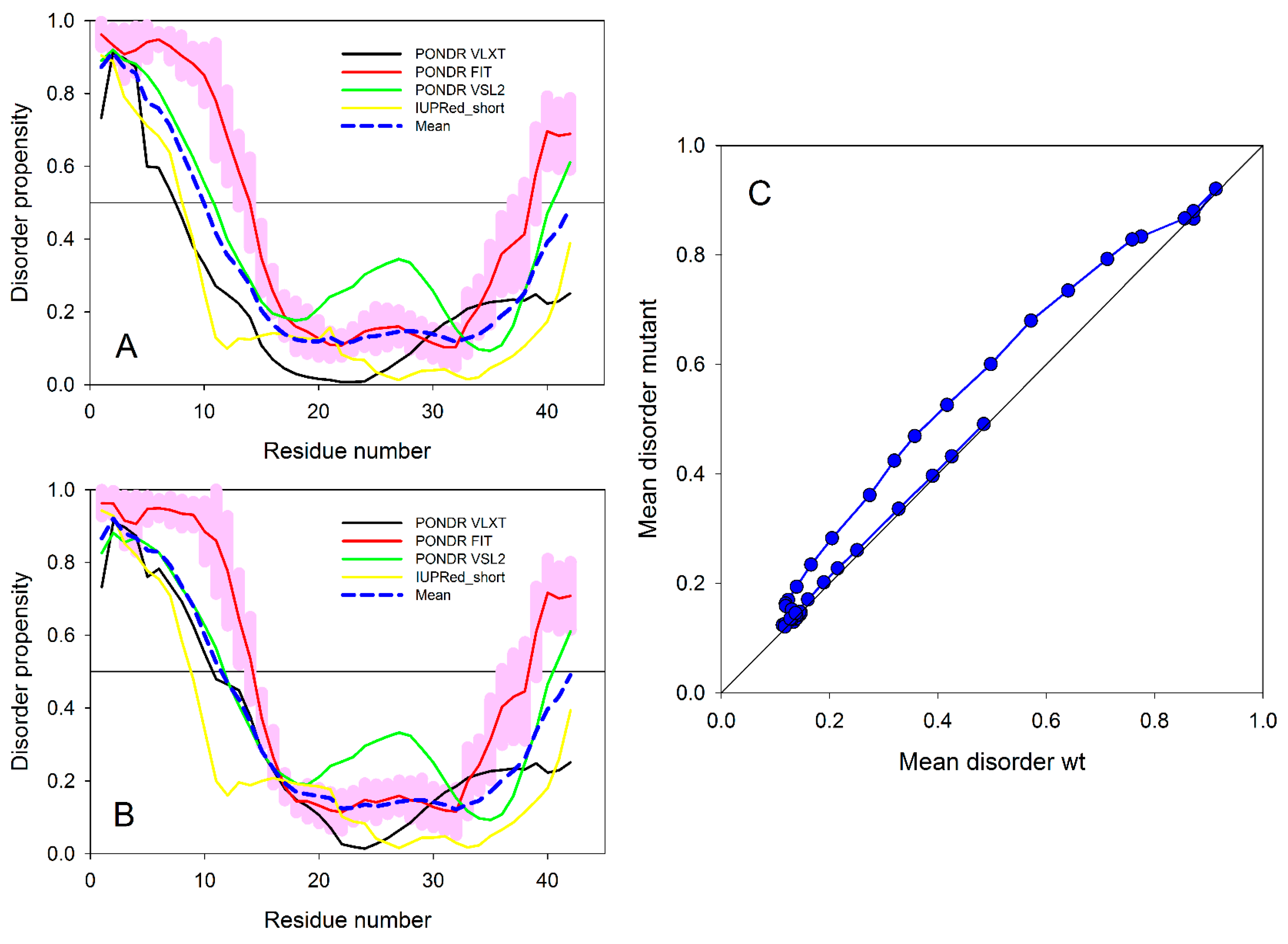
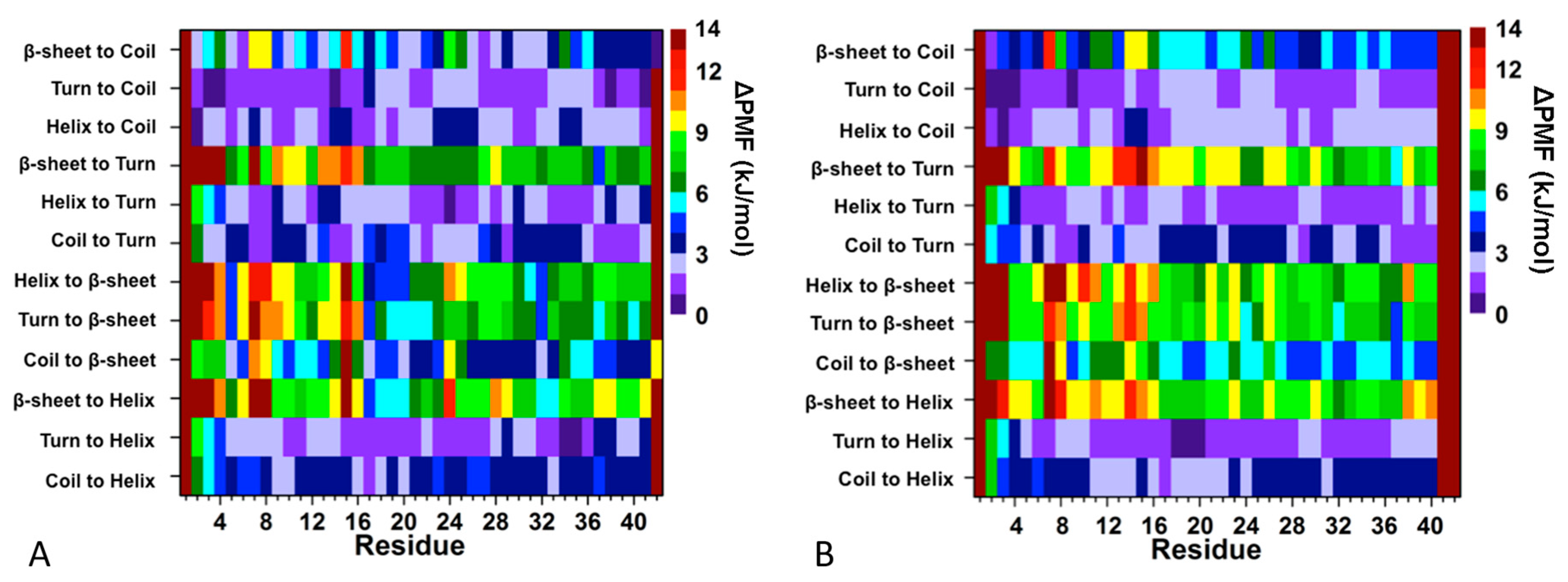
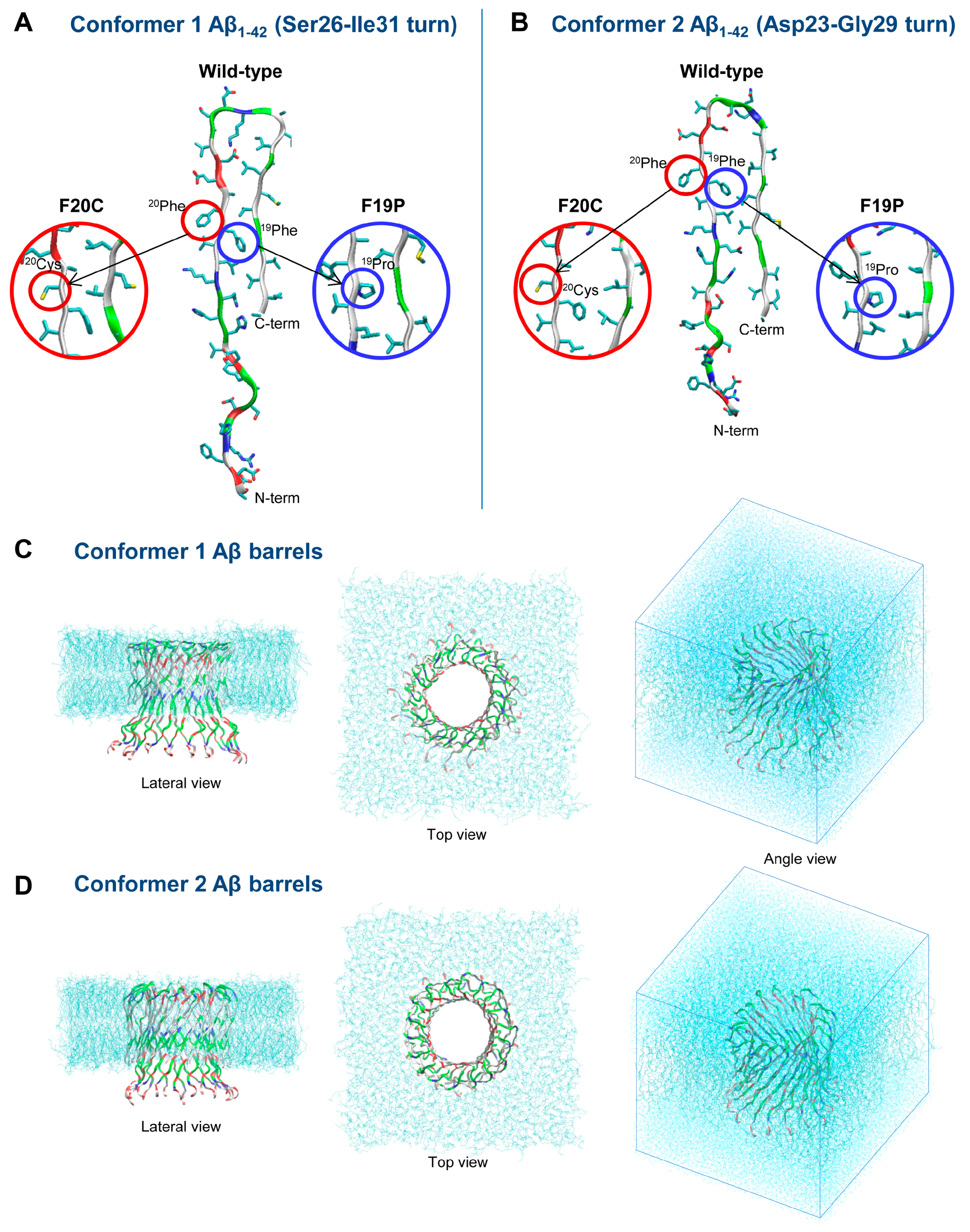
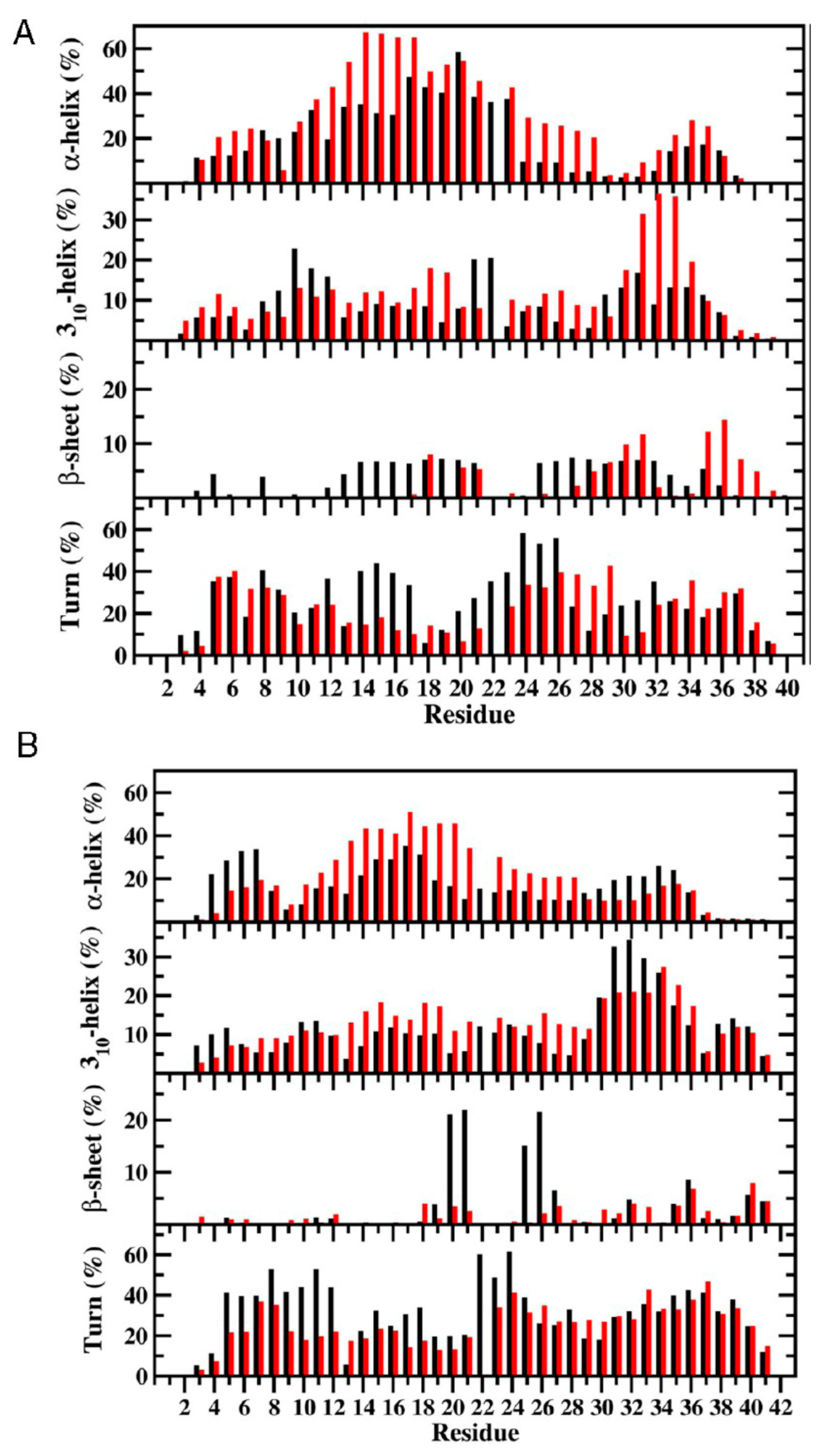

{kind=link}
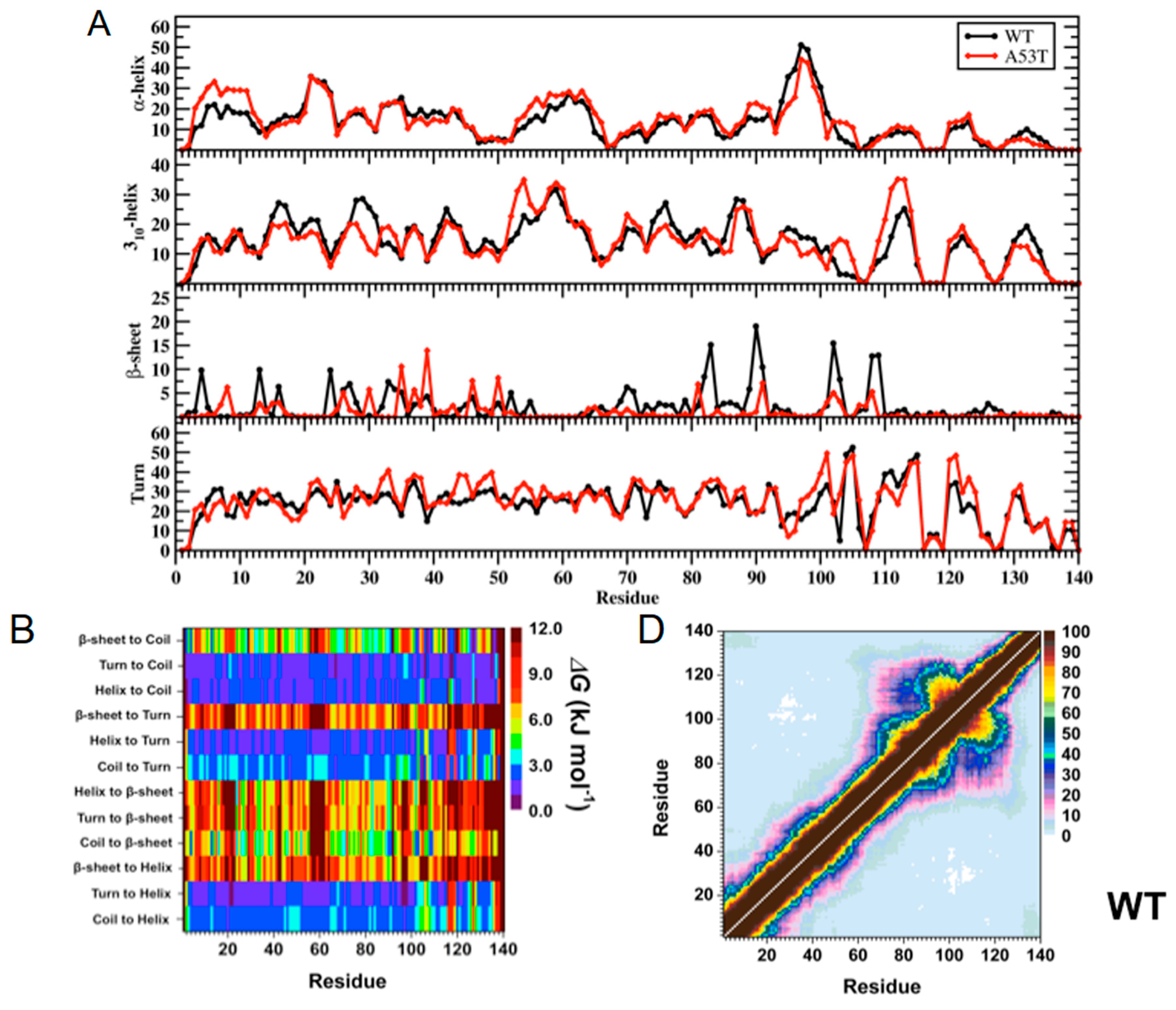{kind=link}
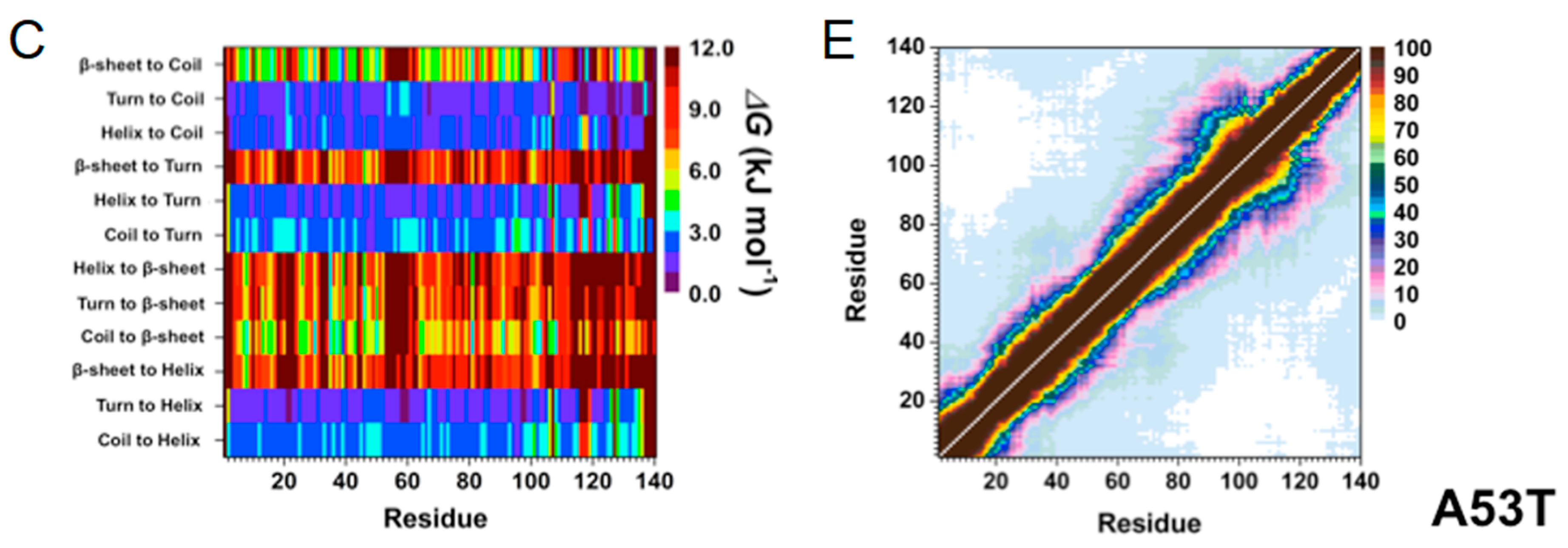{kind=link}
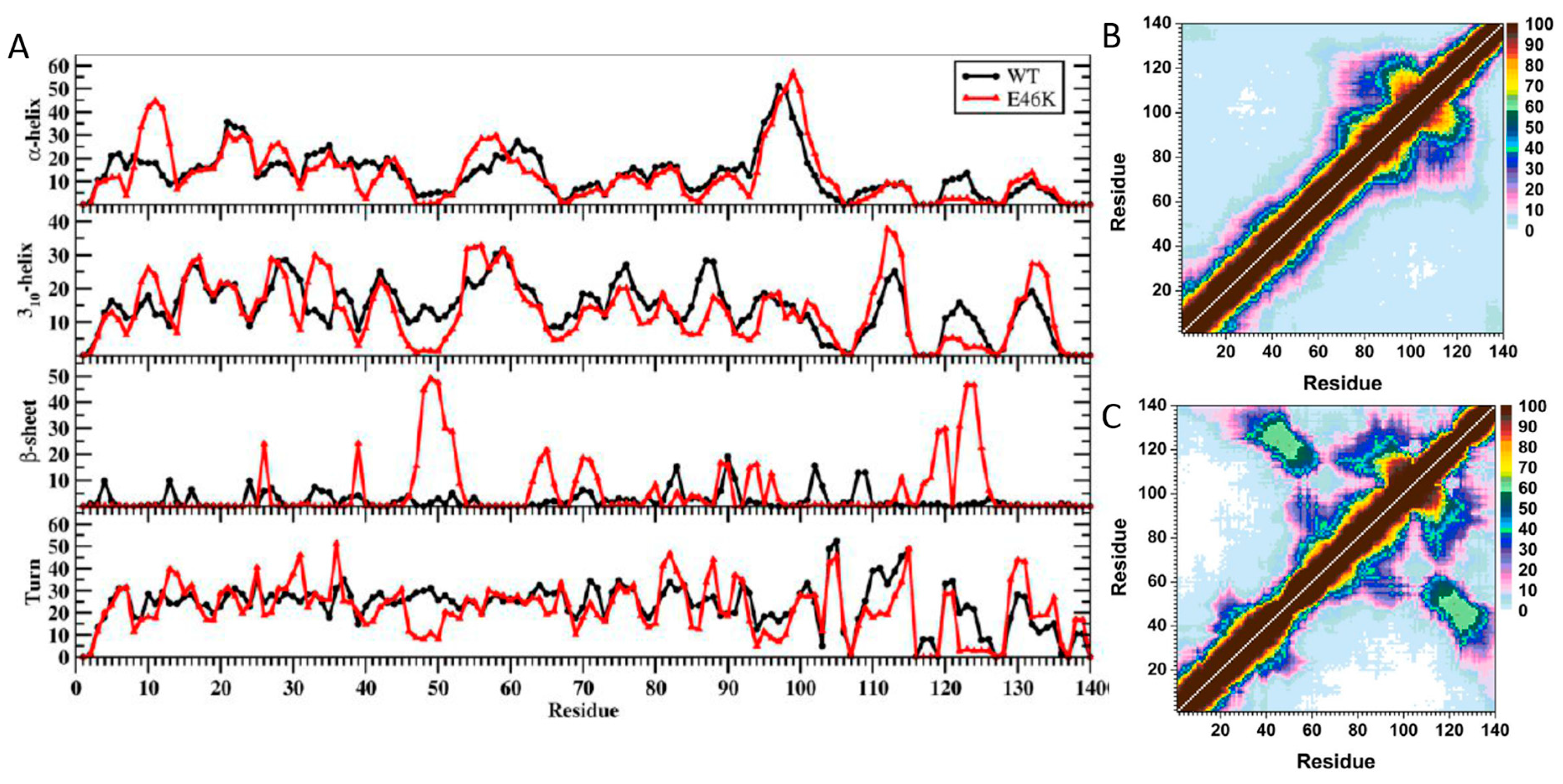{kind=link}
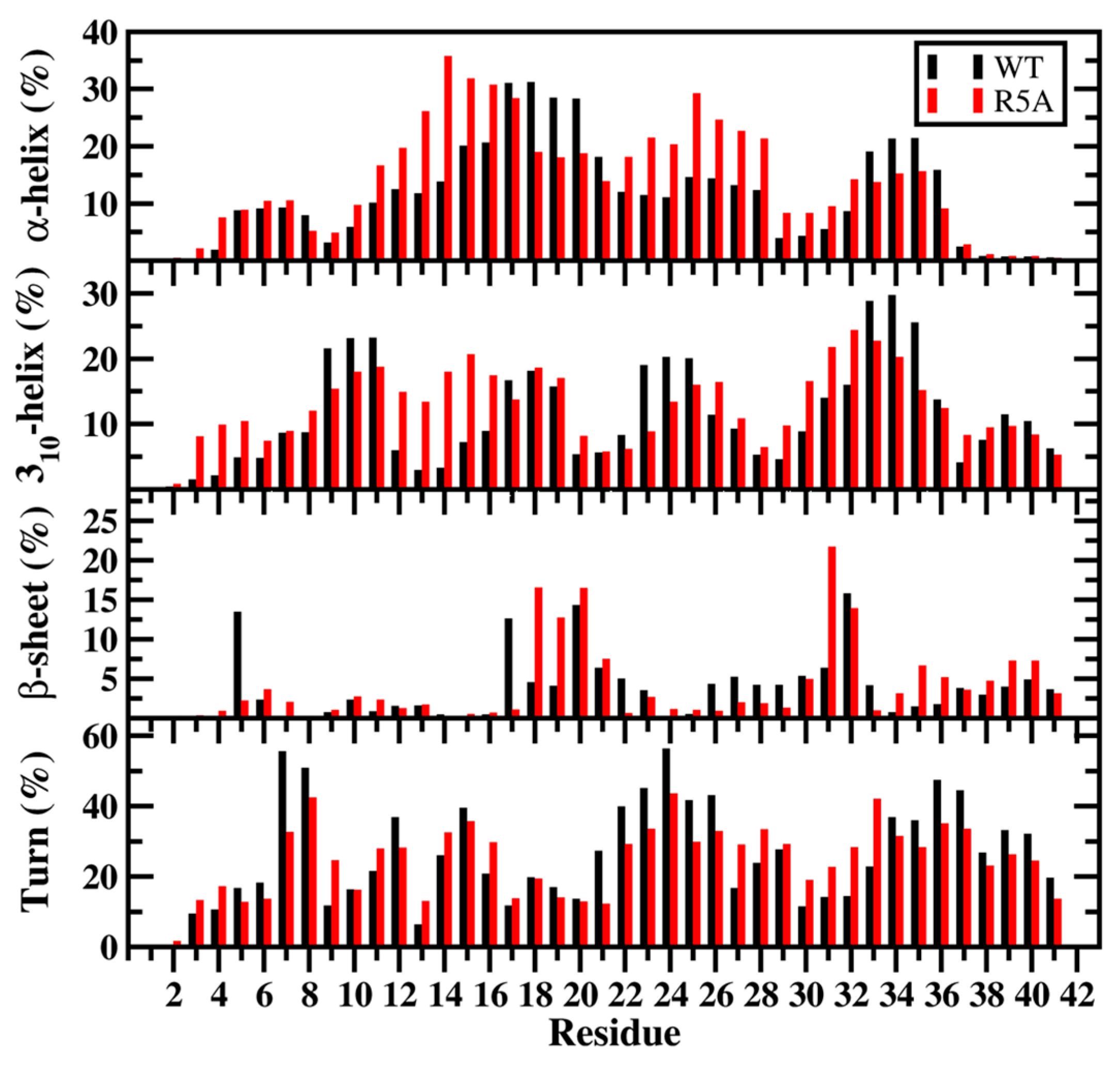{kind=link}
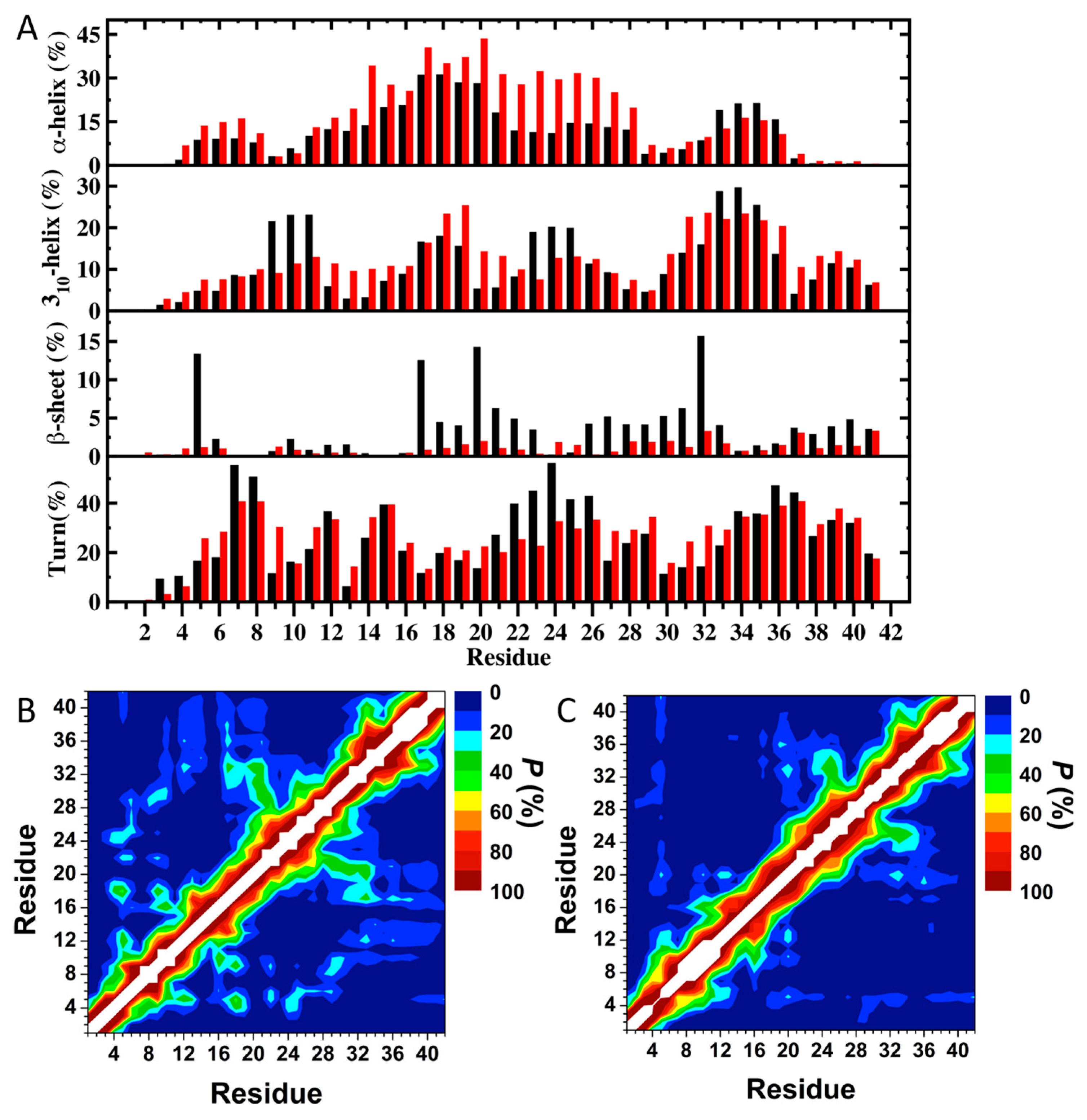{kind=link}
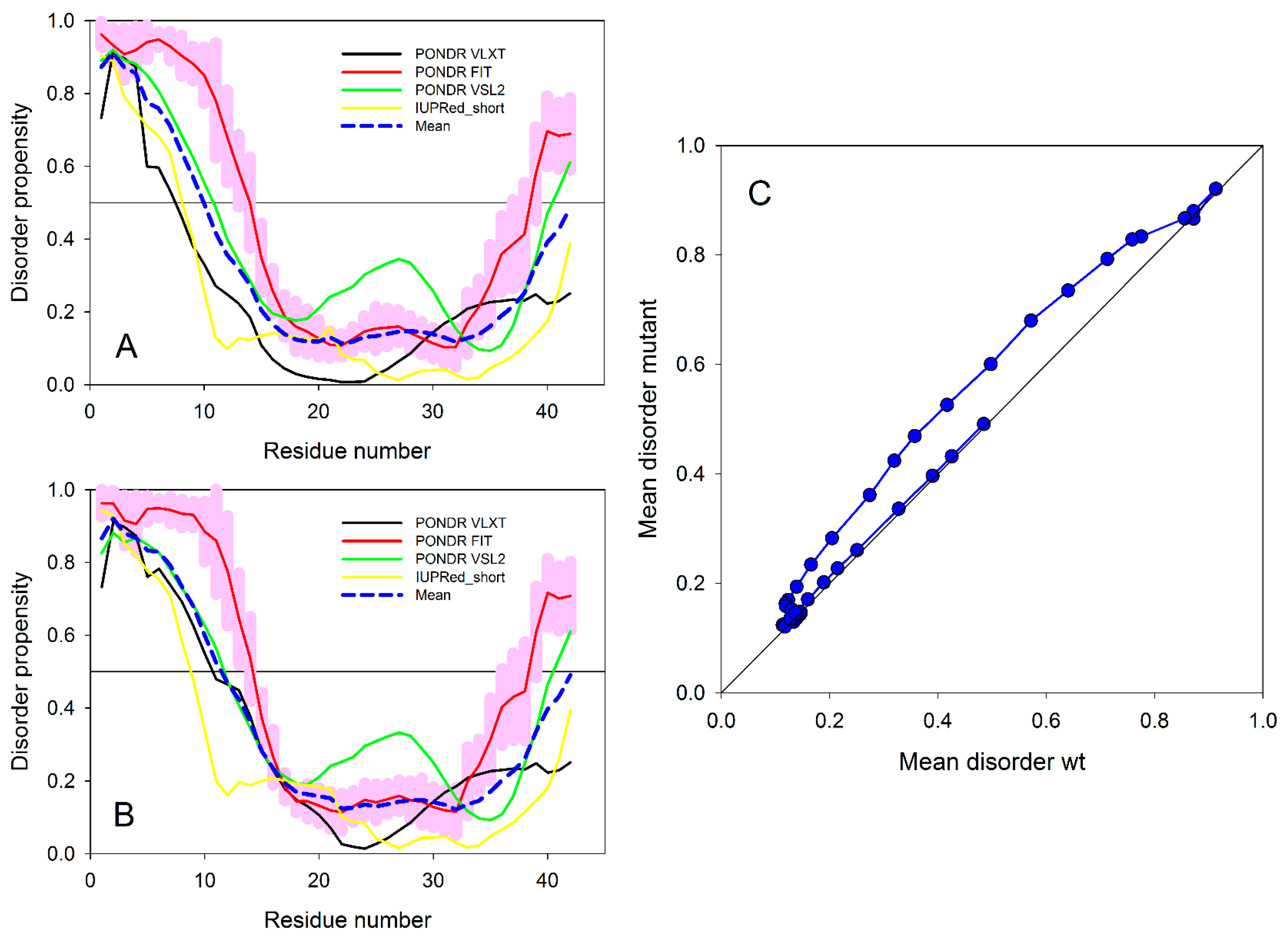{kind=link}
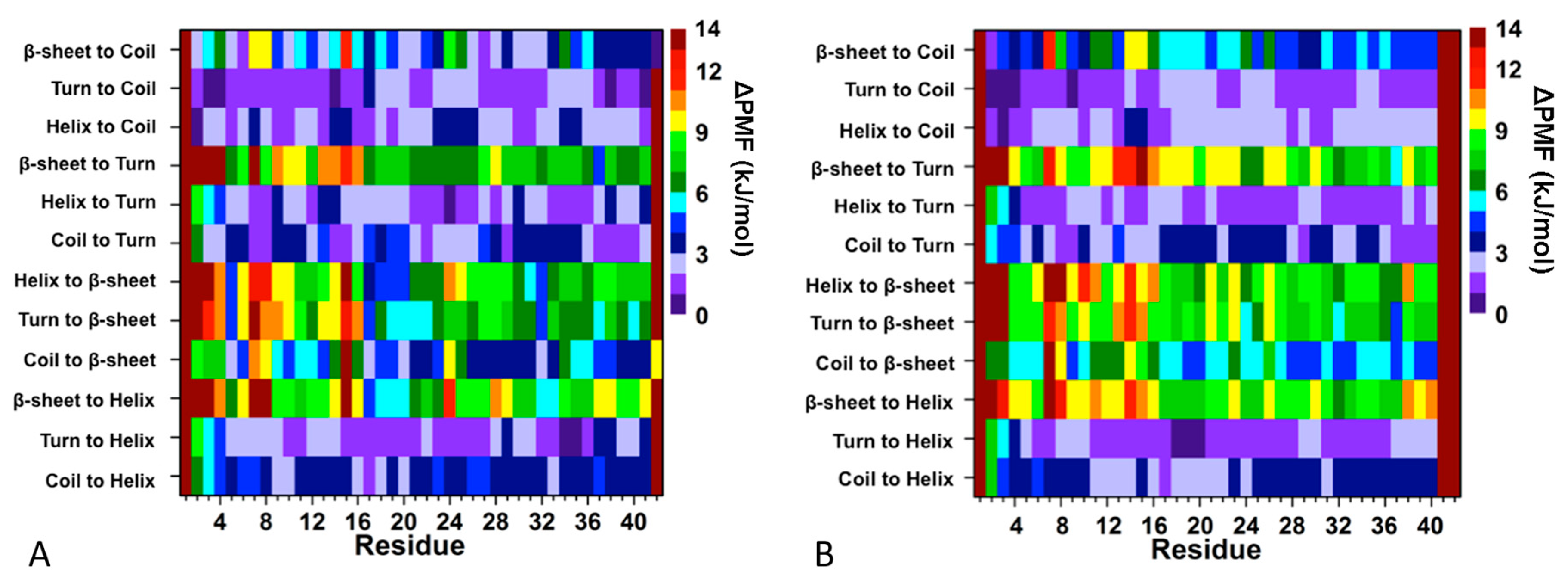{kind=link}
{kind=link}
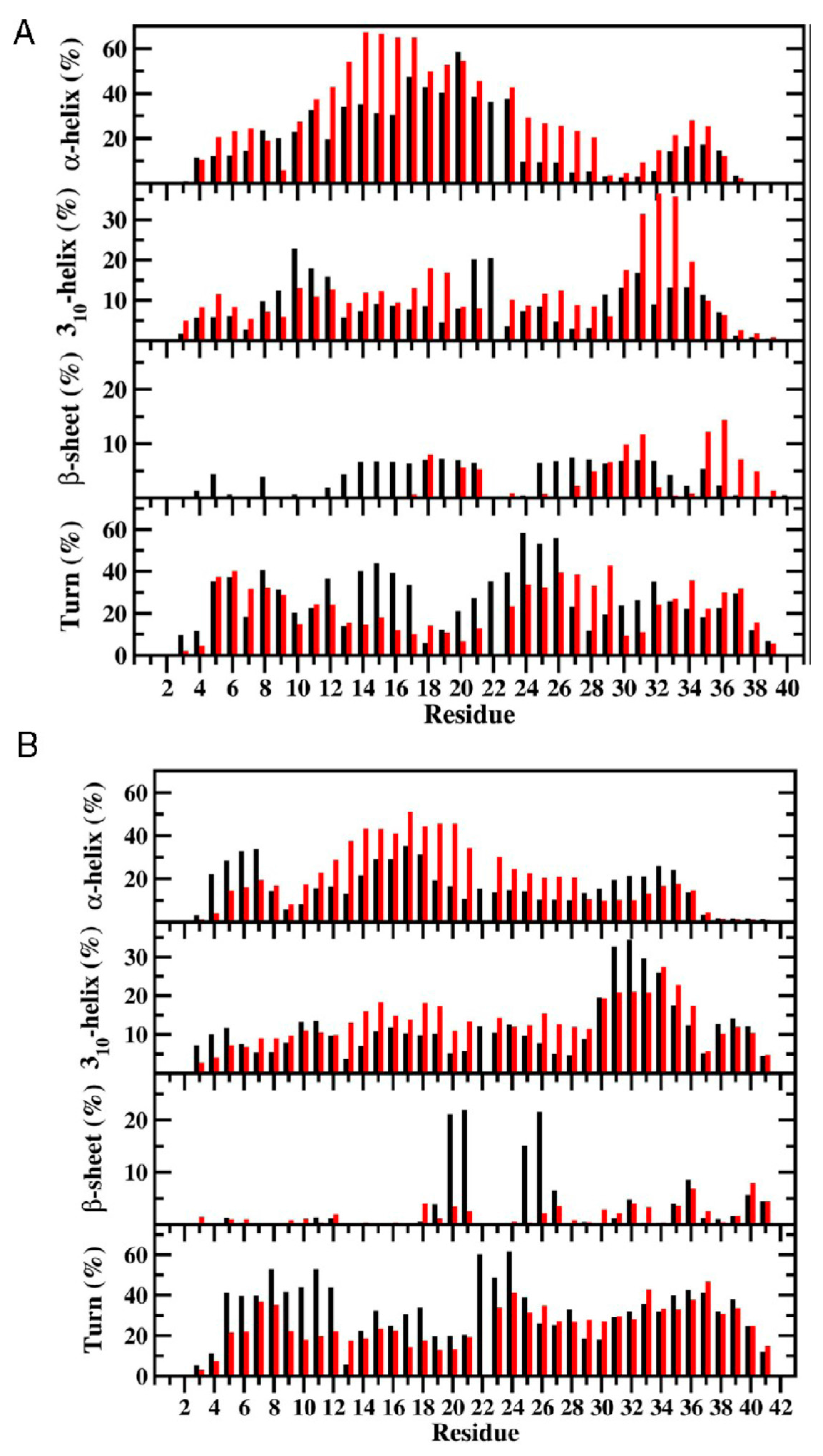{kind=link}
| Protein | <H> (kJ·mol−1) | −T<SNMA> (kJ·mol−1) | <GMM/PBSA> (kJ·mol−1) |
|---|---|---|---|
| WT αS | −9557.4 (±18.4) | −7043.8 (±8.4) | −16,601.4 (±13.0) |
| A53T αS | −9662.3 (±21.0) | −7072.0 (±9.6) | −16,734.3 (±10.2) |
| A30P αS | −9461.4 (±50.8) | −7057.3 (±18.8) | −16,518.7 (±32.1) |
| E46K αS | −9495.6 (±60.9) | −6991.3 (±13.7) | −16,486.9 (±54.2) |
| Protein | <H> (kJ·mol−1) | −T<SNMA> (kJ·mol−1) | −T<SQH> (kJ·mol−1) | <GNMA> | <GQH> |
|---|---|---|---|---|---|
| WT Aβ42 | −2612.9 (±127.4) | −2196.4 (±39.3) | −5895.3 (±18.0) | −4809.3 (±126.3) | −8508.2 (±128.7) |
| R5A Aβ42 | −1736.9 (±127.9) | −2196.8 (±43.5) | −6151.3 (±44.3) | −3933.6 (±125.0) | −7888.2 (±135.4) |
| DONOR | Acceptor | WT Aβ42 (P %) | Tyr10Ala Aβ42 (P %) | ||||
|---|---|---|---|---|---|---|---|
| R(C-N) ≤ 4 Å | R(C-N) ≤ 5 Å | R(C-N) ≤ 6 Å | R(C-N) ≤ 4 Å | R(C-N) ≤ 5 Å | R(C-N) ≤ 6 Å | ||
| R5 | E3 | 57.7 | 59.2 | 59.6 | 61.2 | 64.3 | 65.5 |
| R5 | E22 | 26.4 | 26.7 | 26.7 | 9.3 | 9.8 | 10.0 |
| R5 | A42 (-COO−) | 20.2 | 21.0 | 21.3 | 20.0 | 20.7 | 21.2 |
| R5 | E11 | 14.1 | 15.9 | 16.7 | 16.6 | 19.3 | 20.4 |
| R5 | D1 | 13.9 | 14.8 | 15.0 | 18.3 | 19.5 | 20.9 |
| R5 | D23 | 8.2 | 8.4 | 8.4 | 10.9 | 12.7 | 13.4 |
| K28 | E22 | 7.3 | 11.6 | 13.2 | 3.9 | 6.6 | 7.4 |
| K16 | A42 (-COO−) | 5.6 | 8.1 | 9.4 | 4.2 | 5.7 | 6.3 |
| K28 | D23 | 3.9 | 6.2 | 7.1 | 7.9 | 11.4 | 13.2 |
| K16 | E11 | 3.8 | 6.7 | 8.0 | 10.9 | 19.1 | 22.8 |
| K16 | D7 | 3.8 | 8.3 | 11.1 | 7.0 | 9.3 | 9.9 |
| D1 (-NH3+) | E3 | 2.8 | 4.5 | 5.3 | 2.0 | 2.8 | 3.1 |
| K16 | D23 | 1.9 | 3.6 | 4.1 | 0.3 | 0.6 | 0.7 |
| K28 | A42 (-COO−) | 1.8 | 2.6 | 2.9 | 1.8 | 2.6 | 2.8 |
| K16 | E3 | 0.9 | 1.6 | 1.8 | 1.6 | 2.5 | 2.9 |
| R5 | D7 | 0.7 | 0.9 | 1.2 | 4.2 | 5.0 | 5.9 |
| K16 | D1 | 0.7 | 0.9 | 1.0 | 1.8 | 2.9 | 3.1 |
| K28 | D1 | – | – | – | 4.2 | 5.9 | 6.3 |
| Peptide | <H> (kJ·mol−1) | −T<SNMA> (kJ·mol−1) | <SQH> (kJ·mol−1) | <GNMA> (kJ·mol−1) | <GQH> (kJ·mol−1) |
|---|---|---|---|---|---|
| WT Aβ40 | −2788.2 (±55.6) | −2114.4 (±9.9) | −5334.3 (±78.2) | −4902.5 (±45.9) | −8361.5 (±95.9) |
| WT Aβ42 | −2579.9 (±24.2) | −2206.6 (±4.1) | −5781.3 (±131.3) | −4786.5 (±20.3) | −8122.2 (±93.5) |
| ΔE22 Aβ40 | −2202.7 (±41.0) | −2177.7 (±10.5) | −5810.1 (±123.3) | −4380.4 (±30.7) | −7821.8 (±99.9) |
| ΔE22 Aβ42 | −2454.9 (±38.3) | −2072.9 (±8.7) | −5367.0 (±86.6) | −4526.6 (±30.5) | −8012.7 (±94.7) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coskuner-Weber, O.; Uversky, V.N. Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology. Int. J. Mol. Sci. 2018, 19, 336. https://doi.org/10.3390/ijms19020336
Coskuner-Weber O, Uversky VN. Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology. International Journal of Molecular Sciences. 2018; 19(2):336. https://doi.org/10.3390/ijms19020336
Chicago/Turabian StyleCoskuner-Weber, Orkid, and Vladimir N. Uversky. 2018. "Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology" International Journal of Molecular Sciences 19, no. 2: 336. https://doi.org/10.3390/ijms19020336