Hepatocellular Carcinoma in β-Thalassemia Patients: Review of the Literature with Molecular Insight into Liver Carcinogenesis


Abstract
:1. Introduction
2. HCC in Β-Thalassemia—Incidence and Characteristics
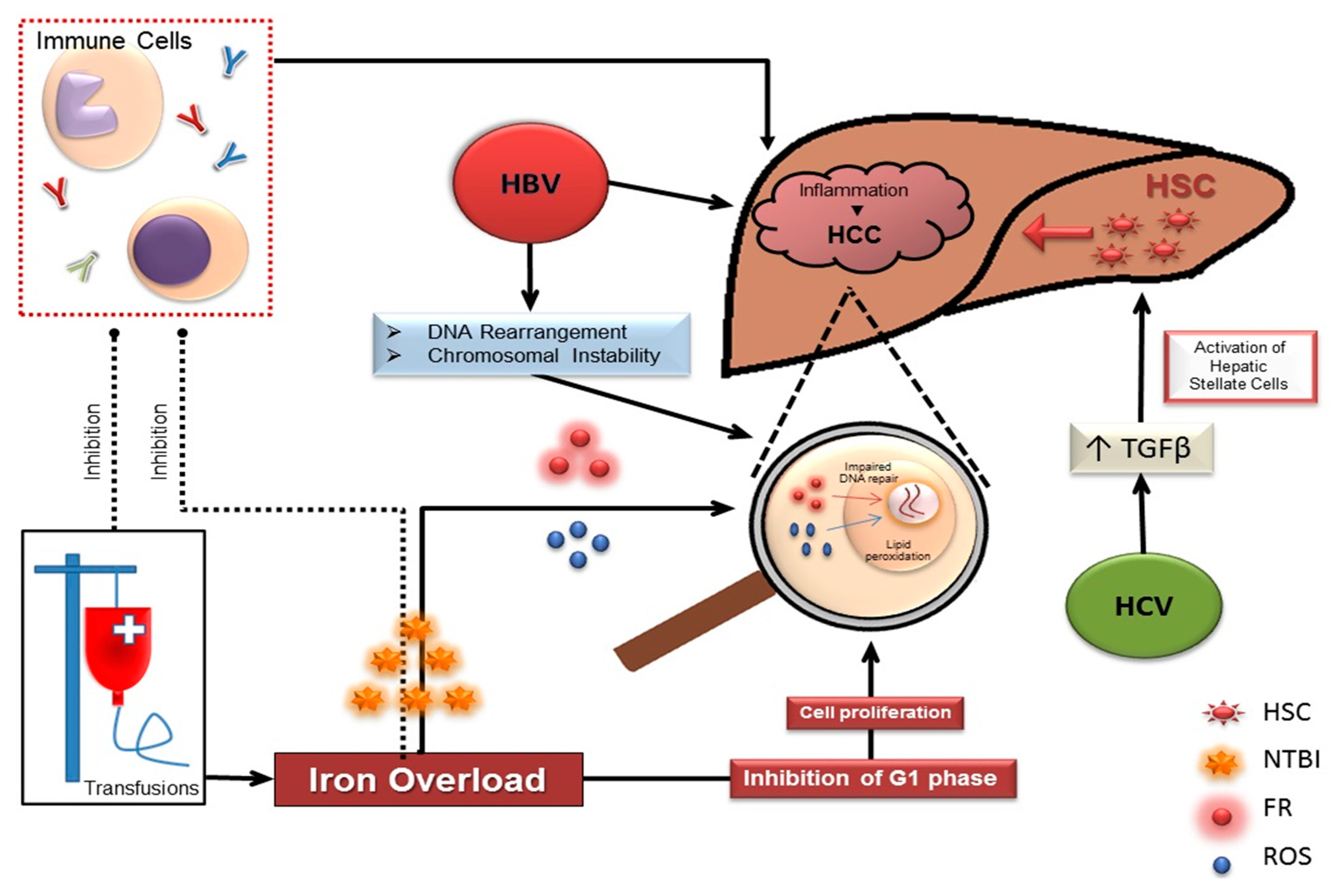
3. HCC in Thalassemia Risk Factors: Hepatitis B/Cirrhosis
4. HCC in Thalassemia Risk Factors: Hepatitis C
5. HCC in Thalassemia Risk Factors: Iron Overload
6. Intracellular Effects of Iron on Proliferation
7. Intranuclear Effects of Iron
8. Effect of Iron on the Immune System
9. Management of HCC in Thalassemia Patients—Screening
10. Management of HCC in Thalassemia Patients—Prevention
11. Management of HCC in Thalassemia Patients—Modalities of Treatment
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 2016, 172, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, A. Hepatocellular carcinoma in thalassemia: A critical review. World J. Hepatol. 2010, 2, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; Rugolotto, S.; De Stefano, P.; Zhao, H.; Cappellini, M.D.; Del Vecchio, G.C.; Romeo, M.A.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.O.; et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004, 89, 1187–1193. [Google Scholar] [PubMed]
- Moukhadder, H.M.; Halawi, R.; Cappellini, M.D.; Taher, A.T. Hepatocellular carcinoma as an emerging morbidity in the thalassemia syndromes: A comprehensive review. Cancer 2017, 123, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Shiratori, Y.; Yoshida, H.; Omata, M. Different clinicopathological features of hepatocellular carcinoma in relation to causative agents. J. Gastroenterol. 2001, 36, 73–78. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Mason, A.C. Rising incidence of hepatocellular carcinoma in the United States. N. Engl. J. Med. 1999, 340, 745–750. [Google Scholar] [CrossRef]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar] [CrossRef] [Green Version]
- Prati, D. Benefits and complications of regular blood transfusion in patients with β-thalassaemia major. Vox Sang. 2000, 79, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; De Stefano, P.; Sessa, F.; Avato, F. Hepatocellular carcinoma in thalassemia major. Med. Pediatr. Oncol. 1986, 14, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Zurlo, M.; De Stefano, P.; Borgna-Pignatti, C.; Di Palma, A.; Melevendi, C.; Piga, A.; Di Gregorio, F.; Burattini, M.; Terzoli, S. Survival and causes of death in thalassaemia major. Lancet 1989, 334, 27–30. [Google Scholar] [CrossRef]
- Ladis, V.; Chouliaras, G.; Berdoukas, V.; Chatziliami, A.; Fragodimitri, C.; Karabatsos, F.; Youssef, J.; Kattamis, A.; Karagiorga-Lagana, M. Survival in a large cohort of Greek patients with transfusion-dependent β thalassaemia and mortality ratios compared to the general population. Eur. J. Haematol. 2011, 86, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; Garani, M.C.; Forni, G.L.; Cappellini, M.D.; Cassinerio, E.; Fidone, C.; Spadola, V.; Maggio, A.; Restivo Pantalone, G.; Piga, A.; et al. Hepatocellular carcinoma in thalassaemia: An update of the Italian Registry. Br. J. Haematol. 2014, 167, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Azarkivan, A.; Halagi, F. Incidence of hepatocellular carcinoma in patients with thalassemia who had hepatitis C. Acta Med. Iran. 2013, 51, 404–407. [Google Scholar] [PubMed]
- Borgna-Pignatti, C.; Vergine, G.; Lombardo, T.; Cappellini, M.D.; Cianciulli, P.; Maggio, A.; Renda, D.; Lai, M.E.; Mandas, A.; Forni, G.; et al. Hepatocellular carcinoma in the thalassaemia syndromes. Br. J. Haematol. 2004, 124, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Fragatou, S.; Tsourveloudis, I.; Manesis, G. Incidence of hepatocellular carcinoma in a thalassemia unit. Hemoglobin 2010, 34, 221–226. [Google Scholar] [CrossRef]
- Maakaron, J.E.; Cappellini, M.D.; Graziadei, G.; Ayache, J.B.; Taher, A.T. Hepatocellular carcinoma in hepatitis-negative patients with thalassemia intermedia: A closer look at the role of siderosis. Ann. Hepatol. 2013, 12, 142–146. [Google Scholar]
- Maakaron, J.E.; Musallam, K.M.; Ayache, J.B.; Jabbour, M.; Tawil, A.N.; Taher, A.T. A liver mass in an iron-overloaded thalassaemia intermedia patient. Br. J. Haematol. 2013, 161. [Google Scholar] [CrossRef]
- Mancuso, A.; Rigano, P.; Renda, D.; Di Salvo, V.; Pignatti, C.B.; Guddo, F.; Buccellato, A.; Nicoli, N.; Maggio, A. Hepatocellular carcinoma on cirrhosis-free liver in a HCV-infected thalassemic. Am. J. Hematol. 2005, 78, 158–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancuso, A.; Sciarrino, E.; Renda, M.C.; Maggio, A. A prospective study of hepatocellular carcinoma incidence in thalassemia. Hemoglobin 2006, 30, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Moukhadder, H.M.; Roumi, J.E.; Bou-Fakhredin, R.; Taher, A.T. Hepatocellular Carcinoma in a β-Thalassemia Intermedia Patient: Yet Another Case in the Expanding Epidemic. Hemoglobin 2018, 42, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Restivo Pantalone, G.; Renda, D.; Valenza, F.; D’Amato, F.; Vitrano, A.; Cassara, F.; Rigano, P.; Salvo, V.D.; Giangreco, A.; Bevacqua, E.; et al. Hepatocellular carcinoma in patients with thalassaemia syndromes: Clinical characteristics and outcome in a long term single centre experience. Br. J. Haematol. 2010, 150, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Venook, A.P.; Papandreou, C.; Furuse, J.; de Guevara, L.L. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist 2010, 15 (Suppl. 4), 5–13. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M. Risk of hepatocellular carcinoma in hepatitis B and prevention through treatment. Clevel. Clin. J. Med. 2009, 76 (Suppl. 3), S6–S9. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Yang, H.I.; Su, J.; Jen, C.L.; You, S.L.; Lu, S.N.; Huang, G.T.; Iloeje, U.H.; Reveal-HBV Study Group. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006, 295, 65–73. [Google Scholar] [CrossRef]
- Zamor, P.J.; deLemos, A.S.; Russo, M.W. Viral hepatitis and hepatocellular carcinoma: Etiology and management. J. Gastrointest. Oncol. 2017, 8, 229–242. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Lee, J.M.; Wong, C.M.; Ng, I.O. Hepatitis B virus-associated multistep hepatocarcinogenesis: A stepwise increase in allelic alterations. Cancer Res. 2008, 68, 5988–5996. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y. Genome-wide DNA methylation profiles in precancerous conditions and cancers. Cancer Sci. 2010, 101, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, T.H.; Kim, H.; Oh, B.K.; Kim, M.S.; Kim, K.S.; Jung, G.; Park, Y.N. Aberrant CpG island hypermethylation in dysplastic nodules and early HCC of hepatitis B virus-related human multistep hepatocarcinogenesis. J. Hepatol. 2011, 54, 939–947. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Kanwal, F. Epidemiology of hepatocellular carcinoma in the United States: Where are we? Where do we go? Hepatology 2014, 60, 1767–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze-Krebs, A.; Preimel, D.; Popov, Y.; Bartenschlager, R.; Lohmann, V.; Pinzani, M.; Schuppan, D. Hepatitis C virus-replicating hepatocytes induce fibrogenic activation of hepatic stellate cells. Gastroenterology 2005, 129, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Wonke, B.; Hoffbrand, A.V.; Brown, D.; Dusheiko, G. Antibody to hepatitis C virus in multiply transfused patients with thalassaemia major. J. Clin. Pathol. 1990, 43, 638–640. [Google Scholar] [CrossRef]
- Izumi, N.; Asahina, Y.; Kurosaki, M.; Yamada, G.; Kawai, T.; Kajiwara, E.; Okamura, Y.; Takeuchi, T.; Yokosuka, O.; Kariyama, K.; et al. Inhibition of hepatocellular carcinoma by PegIFNα-2a in patients with chronic hepatitis C: A nationwide multicenter cooperative study. J. Gastroenterol. 2013, 48, 382–390. [Google Scholar] [CrossRef]
- Shiratori, Y.; Ito, Y.; Yokosuka, O.; Imazeki, F.; Nakata, R.; Tanaka, N.; Arakawa, Y.; Hashimoto, E.; Hirota, K.; Yoshida, H.; et al. Antiviral therapy for cirrhotic hepatitis C: Association with reduced hepatocellular carcinoma development and improved survival. Ann. Intern. Med. 2005, 142, 105–114. [Google Scholar] [CrossRef]
- Cabibbo, G.; Maida, M.; Genco, C.; Antonucci, M.; Camma, C. Causes of and prevention strategies for hepatocellular carcinoma. Semin. Oncol. 2012, 39, 374–383. [Google Scholar] [CrossRef]
- Makiyama, A.; Itoh, Y.; Kasahara, A.; Imai, Y.; Kawata, S.; Yoshioka, K.; Tsubouchi, H.; Kiyosawa, K.; Kakumu, S.; Okita, K.; et al. Characteristics of patients with chronic hepatitis C who develop hepatocellular carcinoma after a sustained response to interferon therapy. Cancer 2004, 101, 1616–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Kumada, T.; Nakano, S.; Takeda, I.; Kiriyama, S.; Sone, Y.; Toyoda, H.; Shimizu, H.; Honda, T. Incidence of hepatocellular carcinoma in chronic hepatitis C after interferon therapy. Hepatogastroenterology 2002, 49, 508–512. [Google Scholar] [PubMed]
- Puoti, C.; Bellis, L.; Martellino, F.; Durola, L.; Spilabotti, L.; Dell’Unto, O.; Galossi, A.; Guarisco, R. Occurrence of hepatocellular carcinoma in an apparently ‘healthy’ HCV patient. Eur. J. Gastroenterol. Hepatol. 2005, 17, 1263–1264. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Lu, S.N.; Wang, J.H.; Hu, T.H.; Chen, C.H.; Huang, C.M.; Lee, C.M. Sustained HCV clearance by interferon-based therapy reduces hepatocellular carcinoma in hepatitis B and C dually-infected patients. Antivir. Ther. 2011, 16, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Schietroma, I.; Scheri, G.C.; Pinacchio, C.; Statzu, M.; Petruzziello, A.; Vullo, V. Hepatitis C Virus and Hepatocellular Carcinoma: Pathogenetic Mechanisms and Impact of Direct-Acting Antivirals. Open Virol. J. 2018, 12, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004, 127 (Suppl. 1), S79–S86. [Google Scholar] [CrossRef]
- Adams, P.C. Hepatocellular carcinoma in hereditary hemochromatosis. Can. J. Gastroenterol. Hepatol. 1993, 7, 37–41. [Google Scholar] [CrossRef]
- Haddow, J.E.; Palomaki, G.E.; McClain, M.; Craig, W. Hereditary haemochromatosis and hepatocellular carcinoma in males: A strategy for estimating the potential for primary prevention. J. Med. Screen. 2003, 10, 11–13. [Google Scholar] [CrossRef]
- Mandishona, E.; MacPhail, A.P.; Gordeuk, V.R.; Kedda, M.A.; Paterson, A.C.; Rouault, T.A.; Kew, M.C. Dietary iron overload as a risk factor for hepatocellular carcinoma in Black Africans. Hepatology 1998, 27, 1563–1566. [Google Scholar] [CrossRef]
- Blanc, J.F.; De Ledinghen, V.; Bernard, P.H.; de Verneuil, H.; Winnock, M.; Le Bail, B.; Carles, J.; Saric, J.; Balabaud, C.; Bioulac-Sage, P. Increased incidence of HFE C282Y mutations in patients with iron overload and hepatocellular carcinoma developed in non-cirrhotic liver. J. Hepatol. 2000, 32, 805–811. [Google Scholar] [CrossRef]
- Bralet, M.P.; Regimbeau, J.M.; Pineau, P.; Dubois, S.; Loas, G.; Degos, F.; Valla, D.; Belghiti, J.; Degott, C.; Terris, B. Hepatocellular carcinoma occurring in nonfibrotic liver: Epidemiologic and histopathologic analysis of 80 French cases. Hepatology 2000, 32, 200–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turlin, B.; Juguet, F.; Moirand, R.; Le Quilleuc, D.; Loreal, O.; Campion, J.P.; Launois, B.; Ramée, M.P.; Brissot, P.; Deugnier, Y. Increased liver iron stores in patients with hepatocellular carcinoma developed on a noncirrhotic liver. Hepatology 1995, 22, 446–450. [Google Scholar] [PubMed]
- Nemeth, E. Hepcidin in β-thalassemia. Ann. N. Y. Acad. Sci. 2010, 1202, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.; Musallam, K.M.; El Rassi, F.; Duca, L.; Inati, A.; Koussa, S.; Cappellini, M.D. Levels of non-transferrin-bound iron as an index of iron overload in patients with thalassaemia intermedia. Br. J. Haematol. 2009, 146, 569–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, G.N.; Kowdley, K.V. Iron, HFE mutations, and hepatocellular carcinoma: Is hepatic iron a carcinogen? Clin. Gastroenterol. Hepatol. 2003, 1, 246–248. [Google Scholar] [CrossRef]
- Toyokuni, S. Iron-induced carcinogenesis: The role of redox regulation. Free Radic. Biol. Med. 1996, 20, 553–566. [Google Scholar] [CrossRef]
- Djeha, A.; Brock, J.H. Effect of transferrin, lactoferrin and chelated iron on human T-lymphocytes. Br. J. Haematol. 1992, 80, 235–241. [Google Scholar] [CrossRef]
- Deugnier, Y.; Turlin, B. Iron and hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2001, 16, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Hann, H.W.; Stahlhut, M.W.; Hann, C.L. Effect of iron and desferoxamine on cell growth and in vitro ferritin synthesis in human hepatoma cell lines. Hepatology 1990, 11, 566–569. [Google Scholar] [CrossRef]
- Brodie, C.; Siriwardana, G.; Lucas, J.; Schleicher, R.; Terada, N.; Szepesi, A.; Gelfand, E.; Seligman, P. Neuroblastoma sensitivity to growth inhibition by deferrioxamine: Evidence for a block in G1 phase of the cell cycle. Cancer Res. 1993, 53, 3968–3975. [Google Scholar]
- Fabbri, E.; Guerrini, G.; Borgna, C. The risk of Hepatocellular carcinoma in patients with thalassemia. A review. Acta Pediatr Mediterr. 2009, 25, 47–52. [Google Scholar]
- Loeb, L.A.; James, E.A.; Waltersdorph, A.M.; Klebanoff, S.J. Mutagenesis by the autoxidation of iron with isolated DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 3918–3922. [Google Scholar] [CrossRef]
- Meneghini, R. Iron homeostasis, oxidative stress, and DNA damage. Free Radic. Biol. Med. 1997, 23, 783–792. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Vautier, G.; Bomford, A.B.; Portmann, B.C.; Metivier, E.; Williams, R.; Ryder, S.D. p53 mutations in british patients with hepatocellular carcinoma: Clustering in genetic hemochromatosis. Gastroenterology 1999, 117, 154–160. [Google Scholar] [CrossRef]
- Hussain, S.P.; Raja, K.; Amstad, P.A.; Sawyer, M.; Trudel, L.J.; Wogan, G.N.; Hofseth, L.J.; Shields, P.G.; Billiar, T.R.; Trautwein, C.; et al. Increased p53 mutation load in nontumorous human liver of wilson disease and hemochromatosis: Oxyradical overload diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 12770–12775. [Google Scholar] [CrossRef] [PubMed]
- Marrogi, A.J.; Khan, M.A.; van Gijssel, H.E.; Welsh, J.A.; Rahim, H.; Demetris, A.J.; Kowdley, K.V.; Hussain, S.P.; Nair, J.; Bartsch, H.; et al. Oxidative stress and p53 mutations in the carcinogenesis of iron overload-associated hepatocellular carcinoma. J. Natl. Cancer Inst. 2001, 93, 1652–1655. [Google Scholar] [CrossRef]
- Alagol, H.; Erdem, E.; Sancak, B.; Turkmen, G.; Camlibel, M.; Bugdayci, G. Nitric oxide biosynthesis and malondialdehyde levels in advanced breast cancer. Aust. N. Z. J. Surg. 1999, 69, 647–650. [Google Scholar] [CrossRef]
- Gal, A.; Wogan, G.N. Mutagenesis associated with nitric oxide production in transgenic SJL mice. Proc. Natl. Acad. Sci. USA 1996, 93, 15102–15107. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, J.C.; Wright, T.L.; deRojas-Walker, T.; Tannenbaum, S.R.; Wogan, G.N. Nitric oxide-induced mutations in the HPRT gene of human lymphoblastoid TK6 cells and in Salmonella typhimurium. Environ. Mol. Mutagen. 2000, 35, 39–47. [Google Scholar] [CrossRef]
- Wink, D.A.; Vodovotz, Y.; Laval, J.; Laval, F.; Dewhirst, M.W.; Mitchell, J.B. The multifaceted roles of nitric oxide in cancer. Carcinogenesis 1998, 19, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, J.; Carmichael, P.L.; Fernando, R.C.; Phillips, D.H.; Strain, A.J.; Bartsch, H. Lipid peroxidation-induced etheno-DNA adducts in the liver of patients with the genetic metal storage disorders Wilson’s disease and primary hemochromatosis. Cancer Epidemiol. Biomark. Prev. 1998, 7, 435–440. [Google Scholar]
- Niemela, O.; Parkkila, S.; Britton, R.S.; Brunt, E.; Janney, C.; Bacon, B. Hepatic lipid peroxidation in hereditary hemochromatosis and alcoholic liver injury. J. Lab. Clin. Med. 1999, 133, 451–460. [Google Scholar] [CrossRef]
- Green, R.; Esparza, I.; Schreiber, R. Iron inhibits the nonspecific tumoricidal activity of macrophages. A possible contributory mechanism for neoplasia in hemochromatosis. Ann. N. Y. Acad. Sci. 1988, 526, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Seligman, P.A.; Kovar, J.; Gelfand, E.W. Lymphocyte proliferation is controlled by both iron availability and regulation of iron uptake pathways. Pathobiology 1992, 60, 19–26. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, M.; Porto, G.; Arosa, F.A.; Cardoso, C.; Cabeda, J.M.; LaCerda, R.; Fraga, J. T-lymphocyte expression and function in hemochromatosis. In Hemochromatosis: Genetics, Pathophysiology, Diagnosis and Treatment; Cambridge University Press: Cambridge, UK, 2000; pp. 396–407. [Google Scholar]
- Harada, T.; Baba, M.; Torii, I.; Morikawa, S. Ferritin selectively suppresses delayed-type hypersensitivity responses at induction or effector phase. Cell. Immunol. 1987, 109, 75–88. [Google Scholar] [CrossRef]
- Matzner, Y.; Hershko, C.; Polliack, A.; Konijn, A.M.; Izak, G. Suppressive effect of ferritin on in vitro lymphocyte function. Br. J. Haematol. 1979, 42, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Carrat, F. Liver fibrosis: Screening is not staging. J. Hepatol. 2009, 50, 1268–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockey, D.C.; Caldwell, S.H.; Goodman, Z.D.; Nelson, R.C.; Smith, A.D.; American Association for the Study of Liver, D. Liver biopsy. Hepatology 2009, 49, 1017–1044. [Google Scholar] [CrossRef] [PubMed]
- Chapman, R.W.; Hussain, M.A.; Gorman, A.; Laulicht, M.; Politis, D.; Flynn, D.M.; Sherlock, S.H.E.; Hoffbrand, A.V. Effect of ascorbic acid deficiency on serum ferritin concentration in patients with β-thalassaemia major and iron overload. J. Clin. Pathol. 1982, 35, 487–491. [Google Scholar] [CrossRef]
- Chirico, V.; Rigoli, L.; Lacquaniti, A.; Salpietro, V.; Piraino, B.; Amorini, M.; Salpietro, C.; Arrigo, T. Endocrinopathies, metabolic disorders, and iron overload in major and intermedia thalassemia: Serum ferritin as diagnostic and predictive marker associated with liver and cardiac T2* MRI assessment. Eur. J. Haematol. 2015, 94, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Pakbaz, Z.; Fischer, R.; Fung, E.; Nielsen, P.; Harmatz, P.; Vichinsky, E. Serum ferritin underestimates liver iron concentration in transfusion independent thalassemia patients as compared to regularly transfused thalassemia and sickle cell patients. Pediatr. Blood Cancer 2007, 49, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghe, C.N.; Kontoghiorghes, G.J. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Des. Dev. Ther. 2016, 10, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Sirlin, C.B.; Reeder, S.B. Magnetic resonance imaging quantification of liver iron. Magn. Reson. Imaging Clin. N. Am. 2010, 18, 359–381. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Sherman, M.; Practice Guidelines Committee AAftSoLD. Management of hepatocellular carcinoma. Hepatology 2005, 42, 1208–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmorris, P.; Singal, A.K. Surveillance and Diagnosis of Hepatocellular Carcinoma. Gastroenterol. Hepatol. 2015, 11, 38–46. [Google Scholar]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with β-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef]
- Cappellini, M.-D.; Cohen, A.; Porter, J.; Taher, A.; Viprakasit, V. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT); Thalassaemia International Federation Nicosia: Nicosia, Cyprus, 2014. [Google Scholar]
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-transfusion-dependent thalassemias. Haematologica 2013, 98, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Kuhns, M.C.; Busch, M.P. New strategies for blood donor screening for hepatitis B virus: Nucleic acid testing versus immunoassay methods. Mol. Diagn. Ther. 2006, 10, 77–91. [Google Scholar] [CrossRef]
- Rampone, B.; Schiavone, B.; Martino, A.; Viviano, C.; Confuorto, G. Current management strategy of hepatocellular carcinoma. World J. Gastroenterol. 2009, 15, 3210–3216. [Google Scholar] [CrossRef]
- Mancuso, A.; Perricone, G. Time to define a new strategy for management of hepatocellular carcinoma in thalassaemia? Br. J. Haematol. 2015, 168, 304–305. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, A. Management of hepatocellular carcinoma: Enlightening the gray zones. World J. Hepatol. 2013, 5, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, N.F.; Liu, P.P.; Sher, G.D.; Daly, P.A.; Greig, P.D.; McCusker, P.J.; Collins, A.F.; Francombe, W.H.; Templeton, D.M.; Butany, J. Brief report: Combined liver and heart transplantation for end-stage iron-induced organ failure in an adult with homozygous β-thalassemia. N. Engl. J. Med. 1994, 330, 1125–1127. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Patient | Sex | Age at Diagnosis | Phenotype | HCV Ab | HCV RNA | HBV Status | Serum Ferritin | Survival | Remarks |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 48 | TI | − | − | − | 1520 µg/L | alive at 16 months | Grade 4 hemosiderosis no cirrhosis [17] |
| 2 | F | 61 | TI | + | − | − | 369 µg/L | Five months | Grade 4 hemosiderosis no cirrhosis [17] |
| 3 | M | 22 | TM | − | − | − | 2000 µg/L | Five months | [17] |
| 4 | M | 71 | TI | − | − | − | 600 µg/L | Five months | CPC B, Grade 6 fibrosis LIC 5.2 mg/g DW [18] |
| 5 | M | 53 | TI | − | − | − | 1350 µg/L | Six months | CPC B, Grade 6 fibrosis LIC 4.8 mg/g DW [18] |
| 6 | F | 41 | TI | − | − | − | 1450 µg/L | Three months | CPC B, Grade 5 fibrosis LIC 6.9 mg/g DW [18] |
| 7 | F | 59 | TI | − | − | NA | 990 µg/L | 25 months | no cirrhosis [24] |
| 8 | M | 73 | TI | − | − | NA | 574 µg/L | Seven months | no cirrhosis [24] |
| 9 | M | 54 | TI | − | − | − | 1291 µg/L | 1 month | no cirrhosis, LIC 12.3 mg/g DW [19] |
| 10 | M | 55 | TI | − | − | − | 5602 µg/L | 48 months | no cirrhosis, LIC 23.9 mg/g DW Siderosis [19] |
| 11 | M | 55 | TI | − | − | − | 832 µg/L | In remission | CPC B, LIC 1.19 mg/g DW 3 years ealrier: ferritin 3860 µg/L, LIC 7.3 mg/g DW [23] |
| 4 patients | NA | NA | TI | − | − | − | 1063–5678 µg/L | NA | [15] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finianos, A.; Matar, C.F.; Taher, A. Hepatocellular Carcinoma in β-Thalassemia Patients: Review of the Literature with Molecular Insight into Liver Carcinogenesis. Int. J. Mol. Sci. 2018, 19, 4070. https://doi.org/10.3390/ijms19124070
Finianos A, Matar CF, Taher A. Hepatocellular Carcinoma in β-Thalassemia Patients: Review of the Literature with Molecular Insight into Liver Carcinogenesis. International Journal of Molecular Sciences. 2018; 19(12):4070. https://doi.org/10.3390/ijms19124070
Chicago/Turabian StyleFinianos, Antoine, Charbel F. Matar, and Ali Taher. 2018. "Hepatocellular Carcinoma in β-Thalassemia Patients: Review of the Literature with Molecular Insight into Liver Carcinogenesis" International Journal of Molecular Sciences 19, no. 12: 4070. https://doi.org/10.3390/ijms19124070