Combining PARP Inhibition, Radiation, and Immunotherapy: A Possible Strategy to Improve the Treatment of Cancer?

 and
and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Immune System in Tumor Control
1.2. Combination of Treatments with Immunotherapies
2. Interactions and Synergy between PARPis and Immunotherapies in Tumor Control
2.1. PARPis as a Cytotoxic Treatment
2.2. PARPis and the Antitumor Immune Response
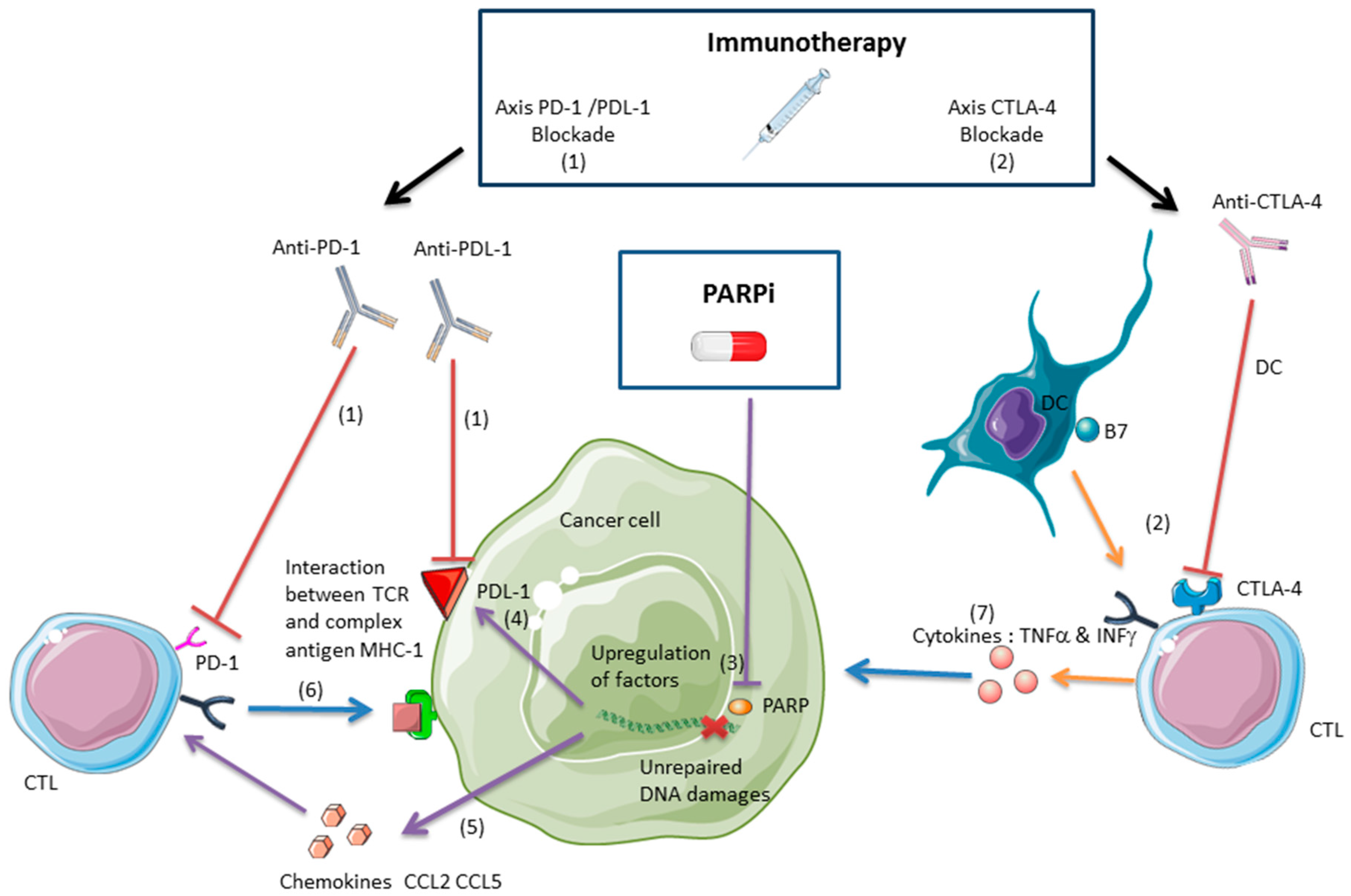
2.2.1. Anti-CTLA-4 and PARPis
2.2.2. Anti-PDL-1/PD-1 and PARPis
3. Immunotherapy and Radiation: A Synergistic Effect Mediated by Cytotoxic Lymphocytes
3.1. Ionizing Radiation Induces an Antitumor Immune Response Mediated by Cytotoxic Lymphocytes
3.2. Radiation and Immunotherapies Can Synergize to Control the Tumor
3.3. Radiation-Induced Immunosuppressive Effects Can Be Reversed with Immunotherapy
3.4. Inducing an Antitumor Immune Response with Proton or Carbon Ion Radiation
4. PARPis and Ionizing Radiation: A Promising Combination Therapy
5. Conclusions and Perspectives: A Rationale for Combining PARPis, Ionizing Radiation, and Immunotherapy
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | Antigen-presenting cell |
| BRCA | BReast CAncer |
| CCL2 | Chemokine (C-C motif) ligand 2 |
| CCL5 | Chemokine (C-C motif) ligand 5 |
| CTLs | specific activated CD8+ lymphocytes against tumor antigens |
| CTLA-4 | Cytotoxic T lymphocyte-associated antigen 4 |
| CXCL-16 | Chemokine (C-X-C motif) ligand 16 |
| CXCL-10 | Chemokine (C-X-C motif) ligand 10 |
| DC | Dendritic cell |
| DSB | Double-strand break |
| GBM | Glioblastoma |
| Gy | Gray |
| HMGB1 | High-mobility group box 1 |
| MDSC | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| NSCLC | Non-small-cell lung cancer |
| PARP | Poly(ADP-ribose) polymerase |
| PARPi | PARP inhibitor |
| PD-1 | Programmed cell death protein 1 |
| PDL-1 | Programmed cell death ligand 1 |
| TAA | Tumor-associated antigen |
| TCR | T cell receptor |
| TIL | Tumor-infiltrating lymphocyte |
| TNBC | Triple-negative breast cancer |
| TMZ | Temozolomide |
References
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Badalamenti, G.; Fanale, D.; Incorvaia, L.; Barraco, N.; Listì, A.; Maragliano, R.; Vincenzi, B.; Calò, V.; Iovanna, J.L.; Bazan, V.; et al. Role of tumor-infiltrating lymphocytes in patients with solid tumors: Can a drop dig a stone? Cell. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Geukes Foppen, M.H.; Donia, M.; Svane, I.M.; Haanen, J.B.A.G. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol. Oncol. 2015, 9, 1918–1935. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Candeias, S.M.; Gaipl, U.S. The Immune System in Cancer Prevention, Development and Therapy. Anticancer Agents Med. Chem. 2016, 16, 101–107. [Google Scholar] [CrossRef]
- Angell, H.; Galon, J. From the immune contexture to the Immunoscore: The role of prognostic and predictive immune markers in cancer. Curr. Opin. Immunol. 2013, 25, 261–267. [Google Scholar] [CrossRef]
- Taube, J.M.; Galon, J.; Sholl, L.M.; Rodig, S.J.; Cottrell, T.R.; Giraldo, N.A.; Baras, A.S.; Patel, S.S.; Anders, R.A.; Rimm, D.L.; et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod. Pathol. 2018, 31, 214–234. [Google Scholar] [CrossRef]
- Sharabi, A.B.; Lim, M.; DeWeese, T.L.; Drake, C.G. Radiation and checkpoint blockade immunotherapy: Radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015, 16, e498–e509. [Google Scholar] [CrossRef]
- Walle, T.; Martinez Monge, R.; Cerwenka, A.; Ajona, D.; Melero, I.; Lecanda, F. Radiation effects on antitumor immune responses: Current perspectives and challenges. Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Caruso, D.; Strudel, M.; Tomao, S.; Tomao, F. Update on Poly-ADP-ribose polymerase inhibition for ovarian cancer treatment. J. Transl. Med. 2016, 14, 267. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudley, J.C.; Lin, M.-T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Egen, J.G.; Kuhns, M.S.; Allison, J.P. CTLA-4: New insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 2002, 3, 611–618. [Google Scholar] [CrossRef]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Herzberg, B.; Campo, M.J.; Gainor, J.F. Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer. Oncologist 2017, 22, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gotwals, P.; Cameron, S.; Cipolletta, D.; Cremasco, V.; Crystal, A.; Hewes, B.; Mueller, B.; Quaratino, S.; Sabatos-Peyton, C.; Petruzzelli, L.; et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 2017, 17, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, X. The role of poly(ADP-ribosyl)ation in DNA damage response and cancer chemotherapy. Oncogene 2015, 34, 3349–3356. [Google Scholar] [CrossRef]
- Scarpa, E.S.; Fabrizio, G.; Di Girolamo, M. A role of intracellular mono-ADP-ribosylation in cancer biology. FEBS J. 2013, 280, 3551–3562. [Google Scholar] [CrossRef] [Green Version]
- Dulaney, C.; Marcrom, S.; Stanley, J.; Yang, E.S. Poly(ADP-ribose) polymerase activity and inhibition in cancer. Semin. Cell Dev. Biol. 2017, 63, 144–153. [Google Scholar] [CrossRef]
- Morgan, R.D.; Clamp, A.R.; Evans, D.G.R.; Edmondson, R.J.; Jayson, G.C. PARP inhibitors in platinum-sensitive high-grade serous ovarian cancer. Cancer Chemother. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Okuma, H.S.; Yonemori, K. BRCA Gene Mutations and Poly(ADP-Ribose) Polymerase Inhibitors in Triple-Negative Breast Cancer. Adv. Exp. Med. Biol. 2017, 1026, 271–286. [Google Scholar] [CrossRef]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Genta, S.; Aglietta, M.; Sapino, A.; Valabrega, G. PARP Inhibitors in Ovarian Cancer. Recent Patents Anticancer Drug Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Haddad, M.; Rhinn, H.; Bloquel, C.; Coqueran, B.; Szabó, C.; Plotkine, M.; Scherman, D.; Margaill, I. Anti-inflammatory effects of PJ34, a poly(ADP-ribose) polymerase inhibitor, in transient focal cerebral ischemia in mice. Br. J. Pharmacol. 2006, 149, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jijon, H.B.; Churchill, T.; Malfair, D.; Wessler, A.; Jewell, L.D.; Parsons, H.G.; Madsen, K.L. Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G641–G651. [Google Scholar] [CrossRef] [PubMed]
- Laudisi, F.; Sambucci, M.; Pioli, C. Poly (ADP-ribose) polymerase-1 (PARP-1) as immune regulator. Endocr. Metab. Immune Disord. Drug Targets 2011, 11, 326–333. [Google Scholar] [CrossRef]
- Aldinucci, A.; Gerlini, G.; Fossati, S.; Cipriani, G.; Ballerini, C.; Biagioli, T.; Pimpinelli, N.; Borgognoni, L.; Massacesi, L.; Moroni, F.; et al. A key role for poly(ADP-ribose) polymerase-1 activity during human dendritic cell maturation. J. Immunol. 2007, 179, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Schreiber, V.; Saenz, L.; Martínez, T.; Muñoz-Suano, A.; Dominguez-Villar, M.; Ramírez, P.; Parrilla, P.; Aguado, E.; García-Cózar, F.; et al. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol. Immunol. 2008, 45, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Thorén, F.B.; Romero, A.I.; Hellstrand, K. Oxygen radicals induce poly(ADP-ribose) polymerase-dependent cell death in cytotoxic lymphocytes. J. Immunol. 2006, 176, 7301–7307. [Google Scholar] [CrossRef]
- Davalli, P.; Marverti, G.; Lauriola, A.; D’Arca, D. Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies. Oxid. Med. Cell. Longev. 2018. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1(-/-) murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Lança, T.; Costa, M.F.; Gonçalves-Sousa, N.; Rei, M.; Grosso, A.R.; Penido, C.; Silva-Santos, B. Protective role of the inflammatory CCR2/CCL2 chemokine pathway through recruitment of type 1 cytotoxic γδ T lymphocytes to tumor beds. J. Immunol. 2013, 190, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nebot-Bral, L.; Brandao, D.; Verlingue, L.; Rouleau, E.; Caron, O.; Despras, E.; El-Dakdouki, Y.; Champiat, S.; Aoufouchi, S.; Leary, A.; et al. Hypermutated tumours in the era of immunotherapy: The paradigm of personalised medicine. Eur. J. Cancer 2017, 84, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, H.; Hosoi, A.; Ueha, S.; Abe, J.; Fujieda, N.; Tomura, M.; Maekawa, R.; Matsushima, K.; Ohara, O.; Kakimi, K. Cytotoxic T lymphocytes block tumor growth both by lytic activity and IFNγ-dependent cell-cycle arrest. Cancer Immunol. Res. 2015, 3, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Wall, L.; Burke, F.; Barton, C.; Smyth, J.; Balkwill, F. IFN-gamma induces apoptosis in ovarian cancer cells in vivo and in vitro. Clin. Cancer Res. 2003, 9, 2487–2496. [Google Scholar]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Du, X.; Tang, F.; Liu, M.; Su, J.; Zhang, Y.; Wu, W.; Devenport, M.; Lazarski, C.A.; Zhang, P.; Wang, X.; et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. 2018, 28, 416. [Google Scholar] [CrossRef]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.A.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Paradis, T.J.; Floyd, E.; Burkwit, J.; Cole, S.H.; Brunson, B.; Elliott, E.; Gilman, S.; Gladue, R.P. The anti-tumor activity of anti-CTLA-4 is mediated through its induction of IFN gamma. Cancer Immunol. Immunother. 2001, 50, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Cimino-Mathews, A.; Peer, C.J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Cao, L.; Harrell, M.I.; Swisher, E.M.; Houston, N.; et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination with Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1-3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, Phase I Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 2193–2202. [Google Scholar] [CrossRef]
- Ko, E.C.; Formenti, S.C. Radiotherapy and checkpoint inhibitors: A winning new combination? Ther. Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rückert, M.; Deloch, L.; Rühle, P.F.; Derer, A.; Fietkau, R.; Gaipl, U.S. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol. Rev. 2017, 280, 231–248. [Google Scholar] [CrossRef]
- Schaue, D.; Kachikwu, E.L.; McBride, W.H. Cytokines in radiobiological responses: A review. Radiat. Res. 2012, 178, 505–523. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, A.B.; Nirschl, C.J.; Kochel, C.M.; Nirschl, T.R.; Francica, B.J.; Velarde, E.; Deweese, T.L.; Drake, C.G. Stereotactic Radiation Therapy Augments Antigen-Specific PD-1-Mediated Antitumor Immune Responses via Cross-Presentation of Tumor Antigen. Cancer Immunol. Res. 2015, 3, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, S.R.; Jammeh, M.L.; Wattenberg, M.M.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 2014, 5, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Vandenabeele, P.; Vandecasteele, K.; Bachert, C.; Krysko, O.; Krysko, D.V. Immunogenic Apoptotic Cell Death and Anticancer Immunity. Adv. Exp. Med. Biol. 2016, 930, 133–149. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Parker, J.J.; Jones, J.C.; Strober, S.; Knox, S.J. Characterization of direct radiation-induced immune function and molecular signaling changes in an antigen presenting cell line. Clin. Immunol. 2013, 148, 44–55. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Probst, H.C.; Vuong, V.; Landshammer, A.; Muth, S.; Yagita, H.; Schwendener, R.; Pruschy, M.; Knuth, A.; van den Broek, M. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J. Immunol. 2012, 189, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Hallahan, D.; Kuchibhotla, J.; Wyble, C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996, 56, 5150–5155. [Google Scholar] [PubMed]
- Chow, M.T.; Luster, A.D. Chemokines in cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Demaria, S. Up-regulation of the pro-inflammatory chemokine CXCL16 is a common response of tumor cells to ionizing radiation. Radiat. Res. 2010, 173, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Sorensen, E.W.; Gerber, S.A.; Moran, J.P.; Frelinger, J.G.; Lord, E.M. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J. Immunol. 2008, 180, 3132–3139. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Cushman, T.R.; Tang, C.; Welsh, J.W. Toxicity of radiation and immunotherapy combinations. Adv. Radiat. Oncol. 2018, 3, 506–511. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef]
- Gong, X.; Li, X.; Jiang, T.; Xie, H.; Zhu, Z.; Zhou, F.; Zhou, C. Combined Radiotherapy and Anti-PD-L1 Antibody Synergistically Enhances Antitumor Effect in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1085–1097. [Google Scholar] [CrossRef]
- Derer, A.; Spiljar, M.; Bäumler, M.; Hecht, M.; Fietkau, R.; Frey, B.; Gaipl, U.S. Chemoradiation Increases PD-L1 Expression in Certain Melanoma and Glioblastoma Cells. Front. Immunol. 2016, 7, 610. [Google Scholar] [CrossRef]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef]
- Park, S.S.; Dong, H.; Liu, X.; Harrington, S.M.; Krco, C.J.; Grams, M.P.; Mansfield, A.S.; Furutani, K.M.; Olivier, K.R.; Kwon, E.D. PD-1 Restrains Radiotherapy-Induced Abscopal Effect. Cancer Immunol. Res. 2015, 3, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed]
- Rückert, M.; Deloch, L.; Fietkau, R.; Frey, B.; Hecht, M.; Gaipl, U.S. Immune modulatory effects of radiotherapy as basis for well-reasoned radioimmunotherapies. Strahlenther. Onkol. 2018, 194, 509–519. [Google Scholar] [CrossRef]
- Schmidt, E.V. Developing combination strategies using PD-1 checkpoint inhibitors to treat cancer. Semin. Immunopathol. 2018. [Google Scholar] [CrossRef]
- Samstein, R.M.; Riaz, N. The DNA damage response in immunotherapy and radiation. Adv. Radiat. Oncol. 2018, 3, 527–533. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.-X. Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Pilones, K.A.; Wennerberg, E.; Formenti, S.C.; Demaria, S. In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine 2015, 33, 7415–7422. [Google Scholar] [CrossRef] [Green Version]
- Burnette, B.C.; Liang, H.; Lee, Y.; Chlewicki, L.; Khodarev, N.N.; Weichselbaum, R.R.; Fu, Y.-X.; Auh, S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011, 71, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.T.; Herman, J.M.; Dholakia, A.S.; Moningi, S.; Lu, Y.; Rosati, L.M.; Hacker-Prietz, A.; Assadi, R.K.; Saeed, A.M.; Pawlik, T.M.; et al. Lymphocyte-Sparing Effect of Stereotactic Body Radiation Therapy in Patients with Unresectable Pancreatic Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Demaria, S.; Schiff, P.B.; Chachoua, A.; Formenti, S.C. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol. Res. 2013, 1, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, A.M.; Simeone, E.; Giannarelli, D.; Muto, P.; Falivene, S.; Borzillo, V.; Giugliano, F.M.; Sandomenico, F.; Petrillo, A.; Curvietto, M.; et al. Abscopal effects of radiotherapy on advanced melanoma patients who progressed after ipilimumab immunotherapy. Oncoimmunology 2014, 3, e28780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, E.C.; Benjamin, K.T.; Formenti, S.C. Generating antitumor immunity by targeted radiation therapy: Role of dose and fractionation. Adv. Radiat. Oncol. 2018, 3, 486–493. [Google Scholar] [CrossRef]
- Tormoen, G.W.; Crittenden, M.R.; Gough, M.J. Role of the immunosuppressive microenvironment in immunotherapy. Adv. Radiat. Oncol. 2018, 3, 520–526. [Google Scholar] [CrossRef]
- Azad, A.; Yin Lim, S.; D’Costa, Z.; Jones, K.; Diana, A.; Sansom, O.J.; Kruger, P.; Liu, S.; McKenna, W.G.; Dushek, O.; et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol. Med. 2017, 9, 167–180. [Google Scholar] [CrossRef]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachikwu, E.L.; Iwamoto, K.S.; Liao, Y.-P.; DeMarco, J.J.; Agazaryan, N.; Economou, J.S.; McBride, W.H.; Schaue, D. Radiation enhances regulatory T cell representation. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Limkin, E.J.; Vakalopoulou, M.; Dercle, L.; Champiat, S.; Han, S.R.; Verlingue, L.; Brandao, D.; Lancia, A.; Ammari, S.; et al. A radiomics approach to assess tumour-infiltrating CD8 cells and response to anti-PD-1 or anti-PD-L1 immunotherapy: An imaging biomarker, retrospective multicohort study. Lancet Oncol. 2018. [Google Scholar] [CrossRef]
- Trowell, O.A. The sensitivity of lymphocytes to ionising radiation. J. Pathol. Bacteriol. 1952, 64, 687–704. [Google Scholar] [CrossRef] [PubMed]
- Song, W.Y.; Huh, S.N.; Liang, Y.; White, G.; Nichols, R.C.; Watkins, W.T.; Mundt, A.J.; Mell, L.K. Dosimetric comparison study between intensity modulated radiation therapy and three-dimensional conformal proton therapy for pelvic bone marrow sparing in the treatment of cervical cancer. J. Appl. Clin. Med. Phys. 2010, 11, 3255. [Google Scholar] [CrossRef] [PubMed]
- Gueulette, J.; Slabbert, J.P.; Böhm, L.; De Coster, B.M.; Rosier, J.F.; Octave-Prignot, M.; Ruifrok, A.; Schreuder, A.N.; Wambersie, A.; Scalliet, P.; et al. Proton RBE for early intestinal tolerance in mice after fractionated irradiation. Radiother. Oncol. 2001, 61, 177–184. [Google Scholar] [CrossRef]
- Uzawa, A.; Ando, K.; Koike, S.; Furusawa, Y.; Matsumoto, Y.; Takai, N.; Hirayama, R.; Watanabe, M.; Scholz, M.; Elsässer, T.; et al. Comparison of Biological Effectiveness of Carbon-Ion Beams in Japan and Germany. Int. J. Radiat. Oncol. 2009, 73, 1545–1551. [Google Scholar] [CrossRef]
- Durante, M.; Reppingen, N.; Held, K.D. Immunologically augmented cancer treatment using modern radiotherapy. Trends Mol. Med. 2013, 19, 565–582. [Google Scholar] [CrossRef]
- Gameiro, S.R.; Malamas, A.S.; Bernstein, M.B.; Tsang, K.Y.; Vassantachart, A.; Sahoo, N.; Tailor, R.; Pidikiti, R.; Guha, C.P.; Hahn, S.M.; et al. Tumor Cells Surviving Exposure to Proton or Photon Radiation Share a Common Immunogenic Modulation Signature, Rendering Them More Sensitive to T Cell-Mediated Killing. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 120–130. [Google Scholar] [CrossRef]
- Lee, K.-S.; Lee, D.-H.; Chun, S.-Y.; Nam, K.-S. Metastatic potential in MDA-MB-231 human breast cancer cells is inhibited by proton beam irradiation via the Akt/nuclear factor-κB signaling pathway. Mol. Med. Rep. 2014, 10, 1007–1012. [Google Scholar] [CrossRef]
- Ogata, T.; Teshima, T.; Kagawa, K.; Hishikawa, Y.; Takahashi, Y.; Kawaguchi, A.; Suzumoto, Y.; Nojima, K.; Furusawa, Y.; Matsuura, N. Particle irradiation suppresses metastatic potential of cancer cells. Cancer Res. 2005, 65, 113–120. [Google Scholar] [PubMed]
- Tamaki, T.; Iwakawa, M.; Ohno, T.; Imadome, K.; Nakawatari, M.; Sakai, M.; Tsujii, H.; Nakano, T.; Imai, T. Application of carbon-ion beams or gamma-rays on primary tumors does not change the expression profiles of metastatic tumors in an in vivo murine model. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Ebner, D.K.; Kamada, T.; Yamada, S. Abscopal effect in recurrent colorectal cancer treated with carbon-ion radiation therapy: 2 case reports. Adv. Radiat. Oncol. 2017, 2, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Mansour, W.Y.; Rhein, T.; Dahm-Daphi, J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010, 38, 6065–6077. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Yoon, C.; Schmidt, B.; Park, D.J.; Zhang, A.Y.; Erkizan, H.V.; Toretsky, J.A.; Kirsch, D.G.; Yoon, S.S. Combining PARP-1 inhibition and radiation in Ewing sarcoma results in lethal DNA damage. Mol. Cancer Ther. 2013, 12, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- Tuli, R.; Surmak, A.J.; Reyes, J.; Armour, M.; Hacker-Prietz, A.; Wong, J.; DeWeese, T.L.; Herman, J.M. Radiosensitization of Pancreatic Cancer Cells In Vitro and In Vivo through Poly (ADP-ribose) Polymerase Inhibition with ABT-888. Transl. Oncol. 2014. [Google Scholar] [CrossRef]
- Lemasson, B.; Wang, H.; Galbán, S.; Li, Y.; Zhu, Y.; Heist, K.A.; Tsein, C.; Chenevert, T.L.; Rehemtulla, A.; Galbán, C.J.; et al. Evaluation of Concurrent Radiation, Temozolomide and ABT-888 Treatment Followed by Maintenance Therapy with Temozolomide and ABT-888 in a Genetically Engineered Glioblastoma Mouse Model. Neoplasia 2016, 18, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Lesueur, P.; Chevalier, F.; Austry, J.-B.; Waissi, W.; Burckel, H.; Noël, G.; Habrand, J.-L.; Saintigny, Y.; Joly, F. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: A systematic review of pre-clinical and clinical human studies. Oncotarget 2017, 8, 69105–69124. [Google Scholar] [CrossRef]
- McLachlan, J.; George, A.; Banerjee, S. The Current Status of PARP Inhibitors in Ovarian Cancer, The Current Status of PARP Inhibitors in Ovarian Cancer. Tumori J. 2016, 102, 433–440. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef]
- Kostine, M.; Cleven, A.H.; de Miranda, N.F.C.C.; Italiano, A.; Cleton-Jansen, A.-M.; Bovée, J.V.M.G. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod. Pathol. 2016, 29, 1028–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enderlin, M.; Kleinmann, E.V.; Struyf, S.; Buracchi, C.; Vecchi, A.; Kinscherf, R.; Kiessling, F.; Paschek, S.; Sozzani, S.; Rommelaere, J.; et al. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. 2009, 16, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.; Schulte, A.; Schnack, C.; Hundhausen, C.; Reiss, K.; Brodway, N.; Held-Feindt, J.; Mentlein, R. Enhanced expression and shedding of the transmembrane chemokine CXCL16 by reactive astrocytes and glioma cells. J. Neurochem. 2005, 93, 1293–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Césaire, M.; Thariat, J.; Candéias, S.M.; Stefan, D.; Saintigny, Y.; Chevalier, F. Combining PARP Inhibition, Radiation, and Immunotherapy: A Possible Strategy to Improve the Treatment of Cancer? Int. J. Mol. Sci. 2018, 19, 3793. https://doi.org/10.3390/ijms19123793
Césaire M, Thariat J, Candéias SM, Stefan D, Saintigny Y, Chevalier F. Combining PARP Inhibition, Radiation, and Immunotherapy: A Possible Strategy to Improve the Treatment of Cancer? International Journal of Molecular Sciences. 2018; 19(12):3793. https://doi.org/10.3390/ijms19123793
Chicago/Turabian StyleCésaire, Mathieu, Juliette Thariat, Serge M. Candéias, Dinu Stefan, Yannick Saintigny, and François Chevalier. 2018. "Combining PARP Inhibition, Radiation, and Immunotherapy: A Possible Strategy to Improve the Treatment of Cancer?" International Journal of Molecular Sciences 19, no. 12: 3793. https://doi.org/10.3390/ijms19123793