Role of Forkhead Box O Transcription Factors in Oxidative Stress-Induced Chondrocyte Dysfunction: Possible Therapeutic Target for Osteoarthritis?


Abstract
:1. Introduction
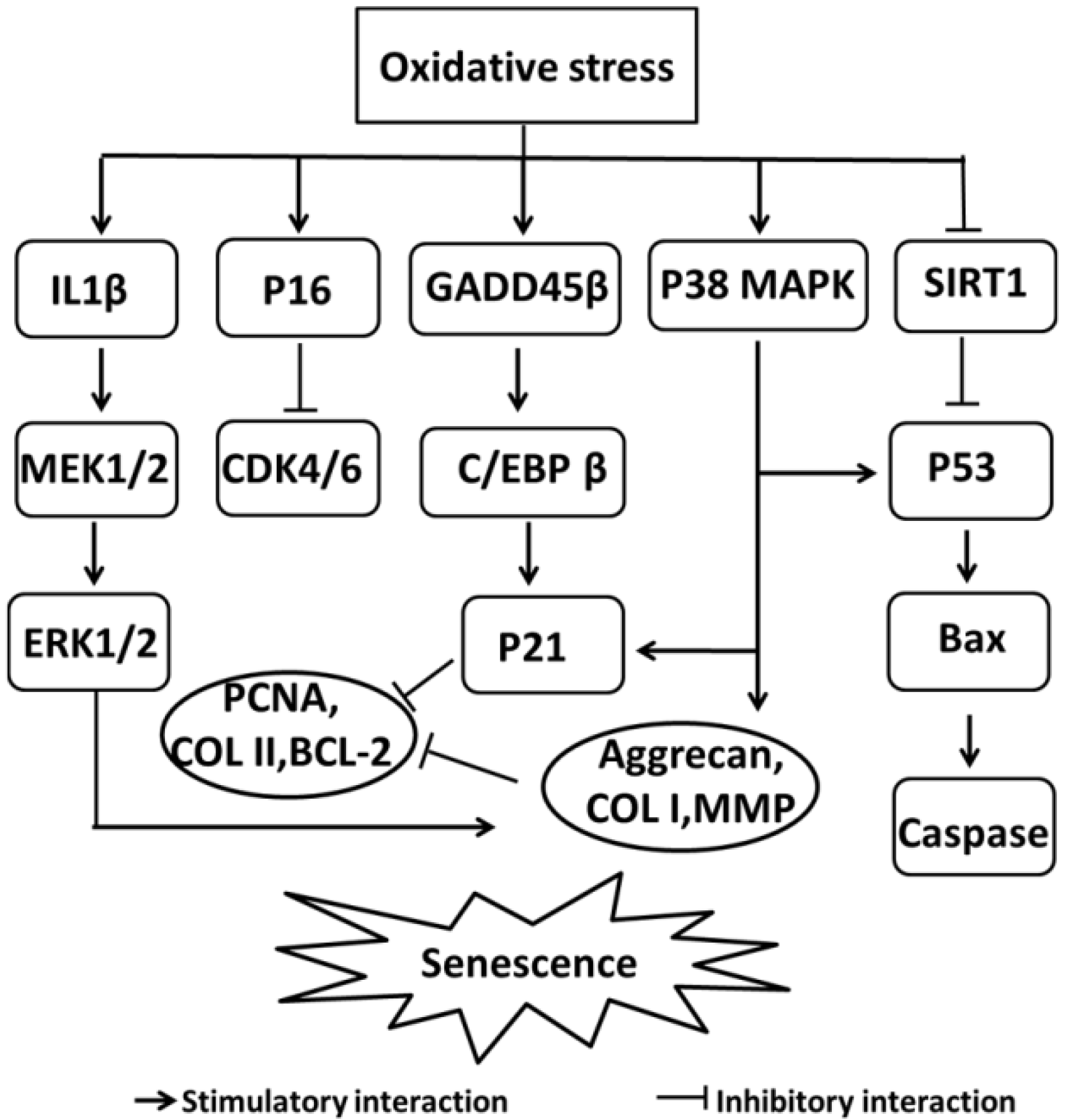
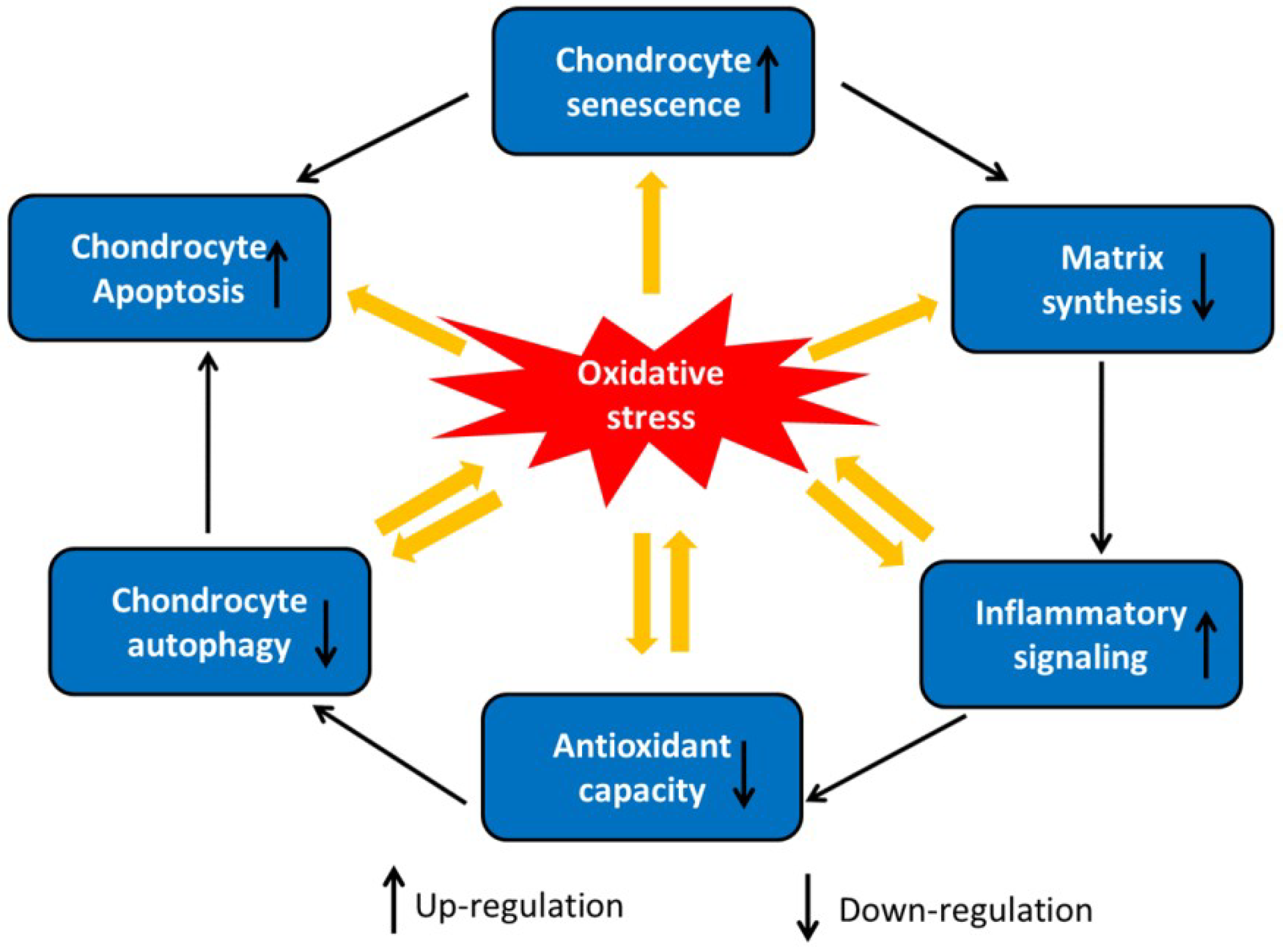
2. Role of Chondrocyte Dysfunction in OA
3. Oxidative Stress-Induced Chondrocyte Dysfunction
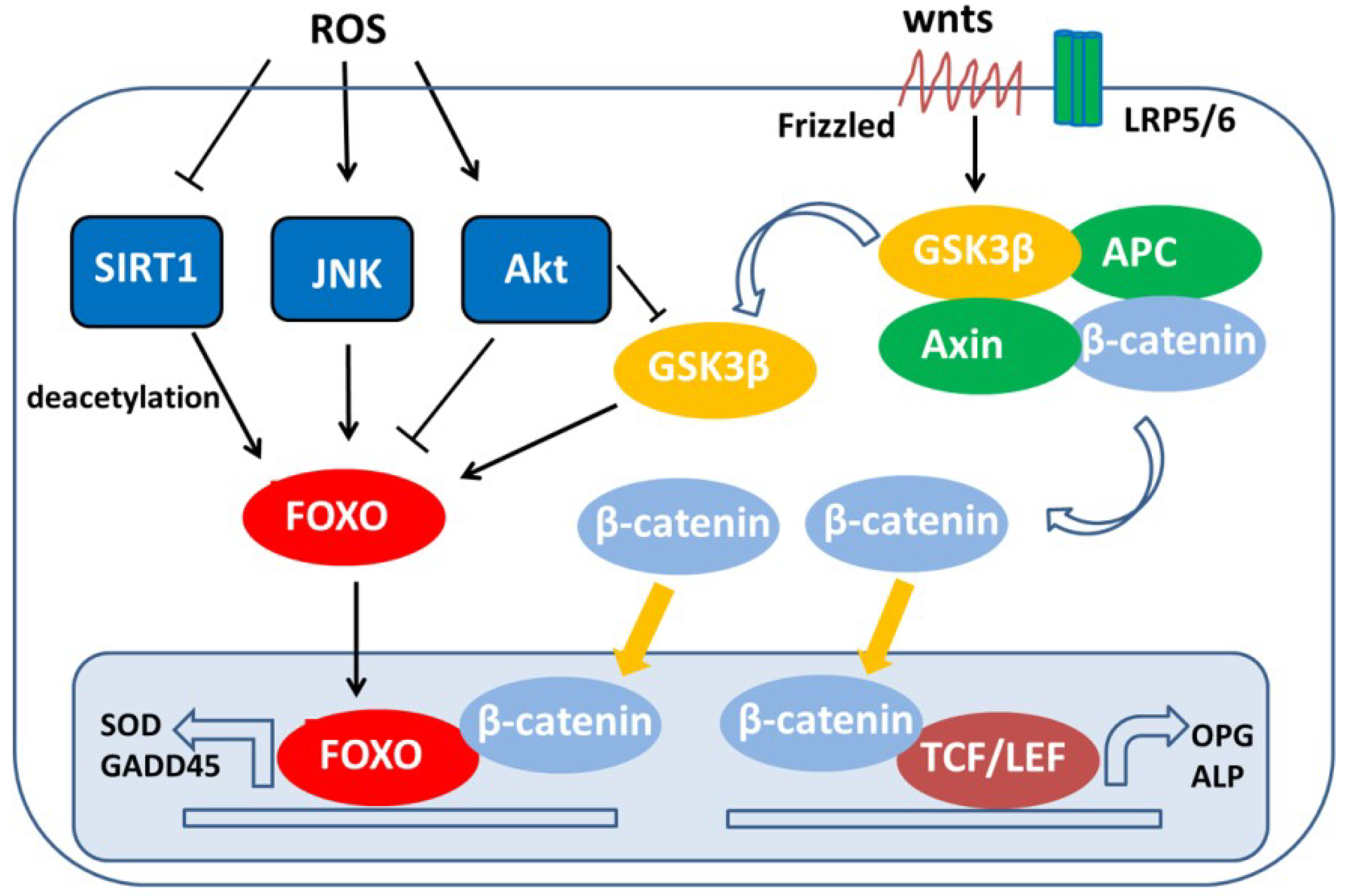
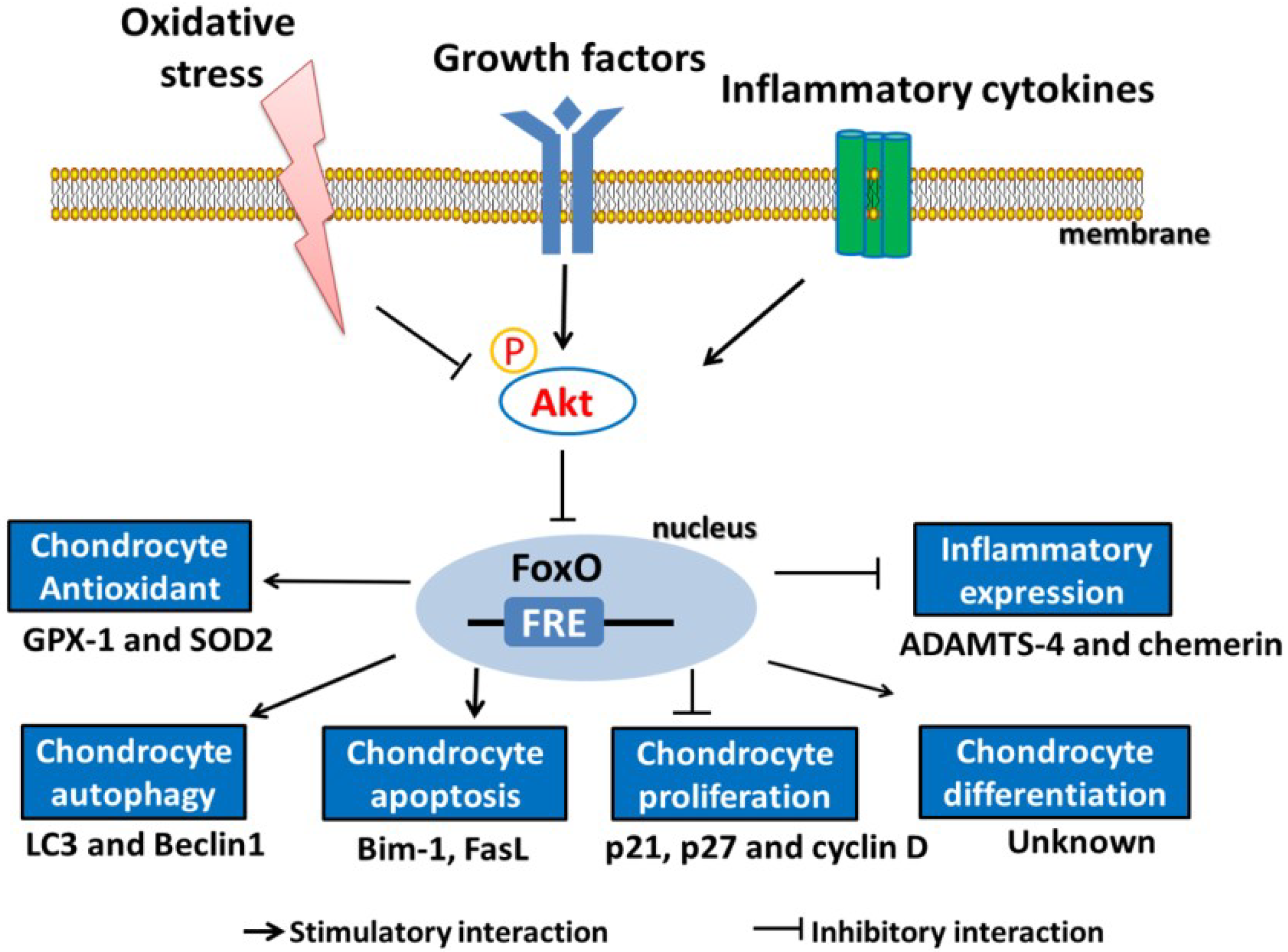
4. Role of FoxOs in Oxidative Stress-Induced Chondrocyte Dysfunction
4.1. Regulation of FoxOs by Oxidative Stress
4.2. Role of FoxOs in Defending against Oxidative Stress
4.3. Expression Patterns of FoxO in Articular Cartilage under Normal and OA Conditions
4.4. Role of FoxOs in the Regulation of Inflammation in Chondrocytes
4.5. FoxOs Regulate the Proliferation, Maturation, and Matrix Production of Chondrocytes
4.6. FoxOs Regulate Chondrocyte Autophagy
4.7. Role of FoxOs in Aging and Longevity
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Taruc-Uy, R.L.; Lynch, S.A. Diagnosis and treatment of osteoarthritis. Primary Care 2013, 40, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.A.; Wood, S.T.; Nelson, K.J.; Rowe, M.A.; Carlson, C.S.; Chubinskaya, S.; Poole, L.B.; Furdui, C.M.; Loeser, R.F. Oxidative Stress Promotes Peroxiredoxin Hyperoxidation and Attenuates Pro-survival Signaling in Aging Chondrocytes. J. Biol. Chem. 2016, 291, 6641–6654. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.M.; Kim, S.J. The thymoquinone-induced production of reactive oxygen species promotes dedifferentiation through the ERK pathway and inflammation through the p38 and PI3K pathways in rabbit articular chondrocytes. Int. J. Mol. Med. 2015, 35, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Ntoumou, E.; Tzetis, M.; Braoudaki, M.; Lambrou, G.; Poulou, M.; Malizos, K.; Stefanou, N.; Anastasopoulou, L.; Tsezou, A. Serum microRNA array analysis identifies miR-140-3p, miR-33b-3p and miR-671-3p as potential osteoarthritis biomarkers involved in metabolic processes. Clin. Epigenet. 2017, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef]
- Akasaki, Y.; Hasegawa, A.; Saito, M.; Asahara, H.; Iwamoto, Y.; Lotz, M.K. Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthr. Cartil. 2014, 22, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Moussaif, M.; Kuan, C.J.; Gargus, J.J.; Sze, J.Y. Serotonin targets the DAF-16/FOXO signaling pathway to modulate stress responses. Cell Metab. 2006, 4, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Tia, N.; Singh, A.K.; Pandey, P.; Azad, C.S.; Chaudhary, P.; Gambhir, I.S. Role of Forkhead Box O (FOXO) transcription factor in aging and diseases. Gene 2018, 648, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Zheng, W.H.; Kar, S.; Quirion, R. Insulin-like growth factor-1-induced phosphorylation of the forkhead family transcription factor FKHRL1 is mediated by Akt kinase in PC12 cells. J. Biol. Chem. 2000, 275, 39152–39158. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biggs, W.H., 3rd; Meisenhelder, J.; Hunter, T.; Cavenee, W.K.; Arden, K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 1999, 96, 7421–7426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J., Jr.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Low, P. The role of ubiquitin-proteasome system in ageing. Gen. Comp. Endocrinol. 2011, 172, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Goldberg, A.L. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy 2008, 4, 378–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rached, M.T.; Kode, A.; Xu, L.; Yoshikawa, Y.; Paik, J.H.; Depinho, R.A.; Kousteni, S. FoxO1 is a positive regulator of bone formation by favoring protein synthesis and resistance to oxidative stress in osteoblasts. Cell Metab. 2010, 11, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Manolopoulos, K.N.; Klotz, L.O.; Korsten, P.; Bornstein, S.R.; Barthel, A. Linking Alzheimer’s disease to insulin resistance: The FoxO response to oxidative stress. Mol. Psychiatry 2010, 15, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Cai, G.Q.; Peng, J.P.; Chen, X.D. Autophagy protects chondrocytes from glucocorticoids-induced apoptosis via ROS/Akt/FOXO3 signaling. Osteoarthr. Cartil. 2015, 23, 2279–2287. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Alvarez-Garcia, O.; Saito, M.; Carames, B.; Iwamoto, Y.; Lotz, M.K. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthr. Rheumatol. 2014, 66, 3349–3358. [Google Scholar] [CrossRef] [PubMed]
- Akeson, G.; Malemud, C.J. A Role for Soluble IL-6 Receptor in Osteoarthritis. J. Funct. Morphol. Kinesiol. 2017, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.; Putavet, D.A.; Klein, J.D.; Derks, K.W.; Bourgeois, B.R.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Verlinden, L.; Maes, C.; Beullens, I.; Gysemans, C.; Paik, J.H.; DePinho, R.A.; Bouillon, R.; Carmeliet, G.; Verstuyf, A. Forkhead box O transcription factors in chondrocytes regulate endochondral bone formation. J. Steroid Biochem. Mol. Biol. 2016, 164, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Zhang, F.J.; Zeng, C.; Luo, W.; Xiao, W.F.; Gao, S.G.; Lei, G.H. Autophagy in osteoarthritis. Jt. Bone Spine 2016, 83, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Toh, W.S.; Brittberg, M.; Farr, J.; Foldager, C.B.; Gomoll, A.H.; Hui, J.H.; Richardson, J.B.; Roberts, S.; Spector, M. Cellular senescence in aging and osteoarthritis. Acta Orthop. 2016, 87, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Giunta, S.; Castorina, A.; Marzagalli, R.; Szychlinska, M.A.; Pichler, K.; Mobasheri, A.; Musumeci, G. Ameliorative effects of PACAP against cartilage degeneration. Morphological, immunohistochemical and biochemical evidence from in vivo and in vitro models of rat osteoarthritis. Int. J. Mol. Sci. 2015, 16, 5922–5944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musumeci, G.; Szychlinska, M.A.; Mobasheri, A. Age-related degeneration of articular cartilage in the pathogenesis of osteoarthritis: Molecular markers of senescent chondrocytes. Histol. Histopathol. 2015, 30, 1–12. [Google Scholar] [PubMed]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Lee, G.; Won, Y.; Lee, M.; Kwak, J.S.; Chun, C.H.; Chun, J.S. Matrix cross-linking-mediated mechanotransduction promotes posttraumatic osteoarthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 9424–9429. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, W.B. Osteoarthritis year 2010 in review: Pathomechanisms. Osteoarthr. Cartil. 2011, 19, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Kawakami, Y.; Kobayashi, M.; Greco, N.; Cummins, J.H.; Matsushita, T.; Kuroda, R.; Kurosaka, M.; Fu, F.H.; Huard, J. Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res. Ther. 2014, 16, 482. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.E.; Bruckner, P.; Pujol, J.P. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthr. Cartil. 2003, 11, 747–755. [Google Scholar] [CrossRef]
- Henrotin, Y.; Kurz, B.; Aigner, T. Oxygen and reactive oxygen species in cartilage degradation: Friends or foes? Osteoarthr. Cartil. 2005, 13, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.Y.; Wong, J.M.; Cruz, T.F. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J. Biol. Chem. 1996, 271, 15703–15707. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.; Attur, M.; Patel, R.N.; Thakker, G.D.; Marshall, P.J.; Rediske, J.; Stuchin, S.A.; Patel, I.R.; Abramson, S.B. Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J. Clin. Investig. 1997, 99, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Boileau, C.; Martel-Pelletier, J.; Moldovan, F.; Jouzeau, J.Y.; Netter, P.; Manning, P.T.; Pelletier, J.P. The in situ up-regulation of chondrocyte interleukin-1-converting enzyme and interleukin-18 levels in experimental osteoarthritis is mediated by nitric oxide. Arthritis Rheum. 2002, 46, 2637–2647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Zheng, J.; Yao, X.; Shan, H.; Li, Y.; Xu, P.; Guo, X. Defective autophagy in chondrocytes with Kashin-Beck disease but higher than osteoarthritis. Osteoarthr. Cartil. 2014, 22, 1936–1946. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.M.; Kim, S.J. Withaferin A-caused production of intracellular reactive oxygen species modulates apoptosis via PI3K/Akt and JNKinase in rabbit articular chondrocytes. J. Korean Med. Sci. 2014, 29, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Fernandes, J.C.; Jovanovic, D.V.; Reboul, P.; Martel-Pelletier, J. Chondrocyte death in experimental osteoarthritis is mediated by MEK 1/2 and p38 pathways: Role of cyclooxygenase-2 and inducible nitric oxide synthase. J. Rheum. 2001, 28, 2509–2519. [Google Scholar] [PubMed]
- Brandl, A.; Hartmann, A.; Bechmann, V.; Graf, B.; Nerlich, M.; Angele, P. Oxidative stress induces senescence in chondrocytes. J. Orthop. Res. 2011, 29, 1114–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, F.; Fan, C.; Wang, C.; Ruan, H. Effects and relationship of ERK1 and ERK2 in interleukin-1beta-induced alterations in MMP3, MMP13, type II collagen and aggrecan expression in human chondrocytes. Int. J. Mol. Med. 2011, 27, 583–589. [Google Scholar] [PubMed]
- Sondergaard, B.C.; Schultz, N.; Madsen, S.H.; Bay-Jensen, A.C.; Kassem, M.; Karsdal, M.A. MAPKs are essential upstream signaling pathways in proteolytic cartilage degradation--divergence in pathways leading to aggrecanase and MMP-mediated articular cartilage degradation. Osteoarthr. Cartil. 2010, 18, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Cha, B.H.; Kim, J.S.; Ahn, J.; Han, I.; Park, H.; Lee, S.H. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthr. Cartil. 2016, 24, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Sakakima, H.; Tsuchimochi, K.; Matsuda, F.; Komiya, S.; Goldring, M.B.; Ijiri, K. Senescence of chondrocytes in aging articular cartilage: GADD45beta mediates p21 expression in association with C/EBPbeta in senescence-accelerated mice. Pathol. Res. Pract. 2011, 207, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Park, S.R.; Park, H.J.; Choi, B.H.; Min, B.H. Potential predictive markers for proliferative capacity of cultured human articular chondrocytes: PCNA and p21. Artif. Organs 2005, 29, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Diverse and dynamic functions of the Sir silencing complex. Nat. Genet. 1999, 23, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Philipot, D.; Guerit, D.; Platano, D.; Chuchana, P.; Olivotto, E.; Espinoza, F.; Dorandeu, A.; Pers, Y.M.; Piette, J.; Borzi, R.M.; et al. p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodeling in osteoarthritis. Arthritis Res. Ther. 2014, 16, R58. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, S.; Nishizawa, H.; Yamada, M.; Higashi, H.; Kondo, T.; Ozawa, H.; Kakita, A.; Hatakeyama, M. Collective inhibition of pRB family proteins by phosphorylation in cells with p16INK4a loss or cyclin E overexpression. J. Biol. Chem. 2001, 276, 11362–11370. [Google Scholar] [CrossRef] [PubMed]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys. Acta 2016, 1862, 576–591. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liu, S.; Huang, J.; Guo, W.; Chen, J.; Zhang, L.; Zhao, B.; Peng, J.; Wang, A.; Wang, Y.; et al. The ECM-cell interaction of cartilage extracellular matrix on chondrocytes. BioMed Res. Int. 2014, 2014, 648459. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.S.; Feigan, J.; Lowther, D.A. The mechanism of chondrocyte hydrogen peroxide damage. Depletion of intracellular ATP due to suppression of glycolysis caused by oxidation of glyceraldehyde-3-phosphate dehydrogenase. J. Rheum. 1989, 16, 7–14. [Google Scholar] [PubMed]
- Hauselmann, H.J.; Stefanovic-Racic, M.; Michel, B.A.; Evans, C.H. Differences in nitric oxide production by superficial and deep human articular chondrocytes: Implications for proteoglycan turnover in inflammatory joint diseases. J. Immunol. 1998, 160, 1444–1448. [Google Scholar] [PubMed]
- Studer, R.K.; Levicoff, E.; Georgescu, H.; Miller, L.; Jaffurs, D.; Evans, C.H. Nitric oxide inhibits chondrocyte response to IGF-I: Inhibition of IGF-IRbeta tyrosine phosphorylation. Am. J. Physiol. Cell Physiol. 2000, 279, C961–C969. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Park, J.I.; Loeser, R.F. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK signaling pathways. J. Biol. Chem. 2009, 284, 31972–31981. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.M.; Kim, S.J. Production of reactive oxygen species by withaferin A causes loss of type collagen expression and COX-2 expression through the PI3K/Akt, p38, and JNK pathways in rabbit articular chondrocytes. Exp. Cell Res. 2013, 319, 2822–2834. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.; Weijzen, S.; de Vries-Smits, A.M.; Saarloos, I.; de Ruiter, N.D.; Bos, J.L.; Burgering, B.M. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23, 4802–4812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccitto, M.; Kalb, R.G. Regulation of Foxo-dependent transcription by post-translational modifications. Curr. Drug Targets 2011, 12, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Jang, Y.; You-Ten, A.I.; Okada, H.; Liepa, J.; Wakeham, A.; Zaugg, K.; Mak, T.W. p53-dependent inhibition of FKHRL1 in response to DNA damage through protein kinase SGK1. Proc. Natl. Acad. Sci. USA 2004, 101, 14057–14062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloet, D.E.; Burgering, B.M. The PKB/FOXO switch in aging and cancer. Biochim. Biophys. Acta 2011, 1813, 1926–1937. [Google Scholar] [CrossRef] [PubMed]
- Liu-Bryan, R.; Terkeltaub, R. Emerging regulators of the inflammatory process in osteoarthritis. Nat. Rev. Rheum. 2015, 11, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Petursson, F.; Viollet, B.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Peroxisome proliferator-activated receptor gamma coactivator 1alpha and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheum. 2014, 66, 3073–3082. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Han, L.; Iyer, S.; de Cabo, R.; Zhao, H.; O’Brien, C.A.; Manolagas, S.C.; Almeida, M. Sirtuin1 Suppresses Osteoclastogenesis by Deacetylating FoxOs. Mol. Endocrinol. 2015, 29, 1498–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, K.; Ishida, K.; Matsushita, T.; Fujita, N.; Hayashi, S.; Sasaki, K.; Tei, K.; Kubo, S.; Matsumoto, T.; Fujioka, H.; et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum. 2009, 60, 2731–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, M.; Wang, J.G.; Xiao, D.M.; Fan, M.; Wang, D.P.; Xiong, J.Y.; Chen, Y.; Ding, Y.; Liu, S.L. Resveratrol inhibits interleukin 1beta-mediated inducible nitric oxide synthase expression in articular chondrocytes by activating SIRT1 and thereby suppressing nuclear factor-kappaB activity. Eur. J. Pharmacol. 2012, 674, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Furukawa-Hibi, Y.; Chen, C.; Horio, Y.; Isobe, K.; Ikeda, K.; Motoyama, N. SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. Int. J. Mol. Med. 2005, 16, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Hoogeboom, D.; Essers, M.A.; Polderman, P.E.; Voets, E.; Smits, L.M.; Burgering, B.M. Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J. Biol. Chem. 2008, 283, 9224–9230. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Martin-Millan, M.; O’Brien, C.A.; Manolagas, S.C. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J. Biol. Chem. 2007, 282, 27298–27305. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.G.; de Vries-Smits, L.M.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.T.; Korswagen, H.C. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Huang, J.H.; Sampson, E.R.; Kim, K.O.; Zuscik, M.J.; O’Keefe, R.J.; Chen, D.; Rosier, R.N. Smurf2 induces degradation of GSK-3beta and upregulates beta-catenin in chondrocytes: A potential mechanism for Smurf2-induced degeneration of articular cartilage. Exp. Cell Res. 2009, 315, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Litherland, G.J.; Hui, W.; Elias, M.S.; Wilkinson, D.J.; Watson, S.; Huesa, C.; Young, D.A.; Rowan, A.D. Glycogen synthase kinase 3 inhibition stimulates human cartilage destruction and exacerbates murine osteoarthritis. Arthritis Rheum. 2014, 66, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Miclea, R.L.; Siebelt, M.; Finos, L.; Goeman, J.J.; Lowik, C.W.; Oostdijk, W.; Weinans, H.; Wit, J.M.; Robanus-Maandag, E.C.; Karperien, M. Inhibition of Gsk3beta in cartilage induces osteoarthritic features through activation of the canonical Wnt signaling pathway. Osteoarthr. Cartil. 2011, 19, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Mattila, J.; Kallijarvi, J.; Puig, O. RNAi screening for kinases and phosphatases identifies FoxO regulators. Proc. Natl. Acad. Sci. USA 2008, 105, 14873–14878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Romero, C.; Calamia, V.; Mateos, J.; Carreira, V.; Martinez-Gomariz, M.; Fernandez, M.; Blanco, F.J. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: A decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol. Cell. Proteom. 2009, 8, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Carlo, M.D., Jr.; Loeser, R.F. Increased oxidative stress with aging reduces chondrocyte survival: Correlation with intracellular glutathione levels. Arthritis Rheum. 2003, 48, 3419–3430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, E.A.; Bowler, R.P.; Crapo, J.D. Joint fluid antioxidants are decreased in osteoarthritic joints compared to joints with macroscopically intact cartilage and subacute injury. Osteoarthr. Cartil. 2008, 16, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.M.; Guilak, F.; Weinberg, J.B.; Fermor, B. Reactive nitrogen and oxygen species in interleukin-1-mediated DNA damage associated with osteoarthritis. Osteoarthr. Cartil. 2008, 16, 624–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid. Redox Signal. 2011, 14, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Alvarez-Garcia, O.; Mokuda, S.; Nagira, K.; Olmer, M.; Gamini, R.; Miyata, K.; Akasaki, Y.; Su, A.I.; Asahara, H.; et al. FoxO transcription factors modulate autophagy and proteoglycan 4 in cartilage homeostasis and osteoarthritis. Sci. Transl. Med. 2018, 10, eaan0746. [Google Scholar] [CrossRef] [PubMed]
- De Isla, N.; Charif, N.; Stoltz, J.F. Are FoxO transcription factors implicated in osteoarthritis? Influence of Diacerhein. Bio-Med. Mat. Eng. 2010, 20, 227–233. [Google Scholar]
- Scott, J.L.; Gabrielides, C.; Davidson, R.K.; Swingler, T.E.; Clark, I.M.; Wallis, G.A.; Boot-Handford, R.P.; Kirkwood, T.B.; Taylor, R.W.; Young, D.A. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis. 2010, 69, 1502–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, J.F.; Tyler, J.A. Upregulation of insulin-like growth factor I gene expression in the lesions of osteoarthritic human articular cartilage. Ann. Rheum. Dis. 1992, 51, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Dore, S.; Pelletier, J.P.; DiBattista, J.A.; Tardif, G.; Brazeau, P.; Martel-Pelletier, J. Human osteoarthritic chondrocytes possess an increased number of insulin-like growth factor 1 binding sites but are unresponsive to its stimulation. Possible role of IGF-1-binding proteins. Arthritis Rheum. 1994, 37, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Verschure, P.J.; Marle, J.V.; Joosten, L.A.; Helsen, M.M.; Lafeber, F.P.; Berg, W.B. Localization of insulin-like growth factor-1 receptor in human normal and osteoarthritic cartilage in relation to proteoglycan synthesis and content. Brit. J. Rheum. 1996, 35, 1044–1055. [Google Scholar] [CrossRef]
- Watroba, M.; Maslinska, D.; Maslinski, S. Current overview of functions of FoxO proteins, with special regards to cellular homeostasis, cell response to stress, as well as inflammation and aging. Adv. Med. Sci. 2012, 57, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Ludikhuize, J.; de Launay, D.; Groot, D.; Smeets, T.J.; Vinkenoog, M.; Sanders, M.E.; Tas, S.W.; Tak, P.P.; Reedquist, K.A. Inhibition of forkhead box class O family member transcription factors in rheumatoid synovial tissue. Arthritis Rheum. 2007, 56, 2180–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Sun, C.; Zhang, S.; Xu, X.; Zhai, L.; Wang, Y.; Wang, S.; Liu, Z.; Cheng, H.; Xiao, M.; et al. Sam68 Promotes NF-kappaB Activation and Apoptosis Signaling in Articular Chondrocytes during Osteoarthritis. Inflamm. Res. 2015, 64, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Kayal, R.A.; Siqueira, M.; Alblowi, J.; McLean, J.; Krothapalli, N.; Faibish, D.; Einhorn, T.A.; Gerstenfeld, L.C.; Graves, D.T. TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. J. Bone Miner. Res. 2010, 25, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Mobasheri, A.; Trovato, F.M.; Szychlinska, M.A.; Graziano, A.C.; Lo Furno, D.; Avola, R.; Mangano, S.; Giuffrida, R.; Cardile, V. Biosynthesis of collagen I, II, RUNX2 and lubricin at different time points of chondrogenic differentiation in a 3D in vitro model of human mesenchymal stem cells derived from adipose tissue. Acta Histochem. 2014, 116, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Ford-Hutchinson, A.F.; Ali, Z.; Lines, S.E.; Hallgrimsson, B.; Boyd, S.K.; Jirik, F.R. Inactivation of Pten in osteo-chondroprogenitor cells leads to epiphyseal growth plate abnormalities and skeletal overgrowth. J. Bone Miner. Res. 2007, 22, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.C.; Chen, N.T.; Lo, S.H. Conditional loss of PTEN leads to skeletal abnormalities and lipoma formation. Mol. Carcinog. 2009, 48, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, K.; Miyamoto, T.; Fujita, N.; Kubota, Y.; Ito, K.; Takubo, K.; Miyamoto, K.; Ninomiya, K.; Suzuki, T.; Iwasaki, R.; et al. Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J. Exp. Med. 2007, 204, 1613–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidotti, S.; Minguzzi, M.; Platano, D.; Santi, S.; Trisolino, G.; Filardo, G.; Mariani, E.; Borzi, R.M. Glycogen Synthase Kinase-3beta Inhibition Links Mitochondrial Dysfunction, Extracellular Matrix Remodelling and Terminal Differentiation in Chondrocytes. Sci. Rep. 2017, 7, 12059. [Google Scholar] [CrossRef] [PubMed]
- Rokutanda, S.; Fujita, T.; Kanatani, N.; Yoshida, C.A.; Komori, H.; Liu, W.; Mizuno, A.; Komori, T. Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev. Biol. 2009, 328, 78–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.; Chen, Y.; Zhang, R.; Dai, H.; Zeng, C.; Zeng, H.; Feng, H.; Du, G.; Fang, H.; Cai, D. c-Jun N-terminal kinase - c-Jun pathway transactivates Bim to promote osteoarthritis. Can. J. Physiol. Pharmacol. 2014, 92, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Takayama, K.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Fujita, N.; Oka, S.; Kurosaka, M.; Kuroda, R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012, 64, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Yan, J.; Erkocak, O.F.; Zheng, X.F.; Chen, X.D. Nitric oxide inhibits autophagy via suppression of JNK in meniscal cells. Rheumatology 2014, 53, 1022–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferdous, A.; Battiprolu, P.K.; Ni, Y.G.; Rothermel, B.A.; Hill, J.A. FoxO, autophagy, and cardiac remodeling. J. Cardiovasc. Transl. Res. 2010, 3, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. SIRT1: Regulation of longevity via autophagy. Cell. Signal. 2009, 21, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Tang, B.L. Sirtuins’ modulation of autophagy. J. Cell. Physiol. 2013, 228, 2262–2270. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, W.; Zhang, H.; Hu, Y.; Wang, M.; Yin, Z. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int. J. Mol. Med. 2013, 32, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Seroude, L.; Benzer, S. Extended life-span and stress resistance in the Drosophila mutant methuselah. Science 1998, 282, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Orr, W.C.; Sohal, R.S. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science 1994, 263, 1128–1130. [Google Scholar] [CrossRef] [PubMed]
- Kampkotter, A.; Nkwonkam, C.G.; Zurawski, R.F.; Timpel, C.; Chovolou, Y.; Watjen, W.; Kahl, R. Effects of the flavonoids kaempferol and fisetin on thermotolerance, oxidative stress and FoxO transcription factor DAF-16 in the model organism Caenorhabditis elegans. Arch. Toxicol. 2007, 81, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Heinegård, D. I-6 BASIC PERSPECTIVE ON THE ROLE OF BIOMARKERS IN THE DIAGNOSIS AND MONITORING OF OSTEOARTHRITIS. Osteoarthr. Cartil. 2009, 17, S2–S3. [Google Scholar] [CrossRef]
- Alvarez-Garcia, O.; Matsuzaki, T.; Olmer, M.; Masuda, K.; Lotz, M.K. Age-related reduction in the expression of FOXO transcription factors and correlations with intervertebral disc degeneration. J. Orthop. Res. 2017, 35, 2682–2691. [Google Scholar] [CrossRef] [PubMed]
- Ambrogini, E.; Almeida, M.; Martin-Millan, M.; Paik, J.H.; Depinho, R.A.; Han, L.; Goellner, J.; Weinstein, R.S.; Jilka, R.L.; O’Brien, C.A.; et al. FoxO-mediated defense against oxidative stress in osteoblasts is indispensable for skeletal homeostasis in mice. Cell Metab. 2010, 11, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, T.C.; Macdougald, O.A. Wnt/beta-catenin signaling in adipogenesis and metabolism. Curr. Opin. Cell Biol. 2007, 19, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Ambrogini, E.; Bartell, S.M.; Han, L.; Roberson, P.K.; de Cabo, R.; Jilka, R.L.; Weinstein, R.S.; O’Brien, C.A.; Manolagas, S.C.; et al. FOXOs attenuate bone formation by suppressing Wnt signaling. J. Clin. Investig. 2013, 123, 3409–3419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.; Han, L.; Bartell, S.M.; Kim, H.N.; Gubrij, I.; de Cabo, R.; O’Brien, C.A.; Manolagas, S.C.; Almeida, M. Sirtuin1 (Sirt1) promotes cortical bone formation by preventing beta-catenin sequestration by FoxO transcription factors in osteoblast progenitors. J. Biol. Chem. 2014, 289, 24069–24078. [Google Scholar] [CrossRef] [PubMed]
- Sakata, S.; Hayashi, S.; Fujishiro, T.; Kawakita, K.; Kanzaki, N.; Hashimoto, S.; Iwasa, K.; Chinzei, N.; Kihara, S.; Haneda, M.; et al. Oxidative stress-induced apoptosis and matrix loss of chondrocytes is inhibited by eicosapentaenoic acid. J. Orthop. Res. 2015, 33, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Jallali, N.; Ridha, H.; Thrasivoulou, C.; Underwood, C.; Butler, P.E.; Cowen, T. Vulnerability to ROS-induced cell death in ageing articular cartilage: The role of antioxidant enzyme activity. Osteoarthr. Cartil. 2005, 13, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, H.; Akagi, M.; Zushi, S.; Teramura, T.; Onodera, Y.; Sawamura, T.; Hamanishi, C. Induction of hypertrophic chondrocyte-like phenotypes by oxidized LDL in cultured bovine articular chondrocytes through increase in oxidative stress. Osteoarthr. Cartil. 2010, 18, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, W.; McCabe, D.; Sauter, E.; Reese, E.; Walter, M.; Buckwalter, J.A.; Martin, J.A. Rotenone prevents impact-induced chondrocyte death. J. Orthop. Res. 2010, 28, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Matta, C.; Zakany, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2015, 80, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.; Matta, C.; Mobasheri, A. Age-Related Alterations in Signaling Pathways in Articular Chondrocytes: Implications for the Pathogenesis and Progression of Osteoarthritis—A Mini-Review. Gerontology 2017, 63, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Castrogiovanni, P.; Trovato, F.M.; Imbesi, R.; Giunta, S.; Szychlinska, M.A.; Loreto, C.; Castorina, S.; Mobasheri, A. Physical activity ameliorates cartilage degeneration in a rat model of aging: A study on lubricin expression. Scand. J. Med. Sci. Sports 2015, 25, e222–e230. [Google Scholar] [CrossRef] [PubMed]
- Rahmati, M.; Nalesso, G.; Mobasheri, A.; Mozafari, M. Aging and osteoarthritis: Central role of the extracellular matrix. Ageing Res. Rev. 2017, 40, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, L.; Yang, S.; Li, F.; Li, X. Behavioral stress-induced activation of FoxO3a in the cerebral cortex of mice. Biol. Psychiatry 2012, 71, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Legendre, F.; Heuze, A.; Boukerrouche, K.; Leclercq, S.; Boumediene, K.; Galera, P.; Domagala, F.; Pujol, J.P.; Ficheux, H. Rhein, the metabolite of diacerhein, reduces the proliferation of osteoarthritic chondrocytes and synoviocytes without inducing apoptosis. Scand. J. Rheumatol. 2009, 38, 104–111. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcription Factors | Targets | Effects | References |
|---|---|---|---|
| FoxO1, FoxO3 | CAT, MnSOD, GPX-1 | Upregulation, antioxidant | [13,24] |
| FoxO3 | GADD45 | Upregulation, DNA repair | [68] |
| FoxO1 | PRG4 | Upregulation, cartilage homeostasis | [81] |
| FoxO1, FoxO3 | ADAMTS-4, chemerin | Downregulation, inflammation | [21] |
| FoxO1, FoxO3 | p21, p27, cyclin G2 | Upregulation, proliferation | [82] |
| FoxO1 | Bim-1, FasL | Upregulation, apoptosis | [82] |
| FoxO1, FoxO3 | Beclin1, LC3 | Upregulation, autophagy | [21] |
| Protein Factors | Transcription Factors | Expression | Transcriptional Activity | References |
|---|---|---|---|---|
| IL-1β | FoxO1, FoxO3, FoxO4 | Suppress | Downregulation | [6] |
| TNF-α | FoxO1 | Suppress | Upregulation | [6,90] |
| TGF-β | FoxO1 | Increase | No effect | [6] |
| PDGF | FoxO1, FoxO3, FoxO4 | Increase (FoxO3) | Downregulation | [6] |
| bFGF | FoxO1, FoxO3 | No effect | Downregulation | [6] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Zhang, S.; Previn, R.; Chen, D.; Jin, Y.; Zhou, G. Role of Forkhead Box O Transcription Factors in Oxidative Stress-Induced Chondrocyte Dysfunction: Possible Therapeutic Target for Osteoarthritis? Int. J. Mol. Sci. 2018, 19, 3794. https://doi.org/10.3390/ijms19123794
Wang R, Zhang S, Previn R, Chen D, Jin Y, Zhou G. Role of Forkhead Box O Transcription Factors in Oxidative Stress-Induced Chondrocyte Dysfunction: Possible Therapeutic Target for Osteoarthritis? International Journal of Molecular Sciences. 2018; 19(12):3794. https://doi.org/10.3390/ijms19123794
Chicago/Turabian StyleWang, Rikang, Shuai Zhang, Rahul Previn, Di Chen, Yi Jin, and Guangqian Zhou. 2018. "Role of Forkhead Box O Transcription Factors in Oxidative Stress-Induced Chondrocyte Dysfunction: Possible Therapeutic Target for Osteoarthritis?" International Journal of Molecular Sciences 19, no. 12: 3794. https://doi.org/10.3390/ijms19123794