PSMA-Oriented Target Delivery of Novel Anticancer Prodrugs: Design, Synthesis, and Biological Evaluations of Oligopeptide-Camptothecin Conjugates

 , and
, and
Abstract
:
1. Introduction
2. Results
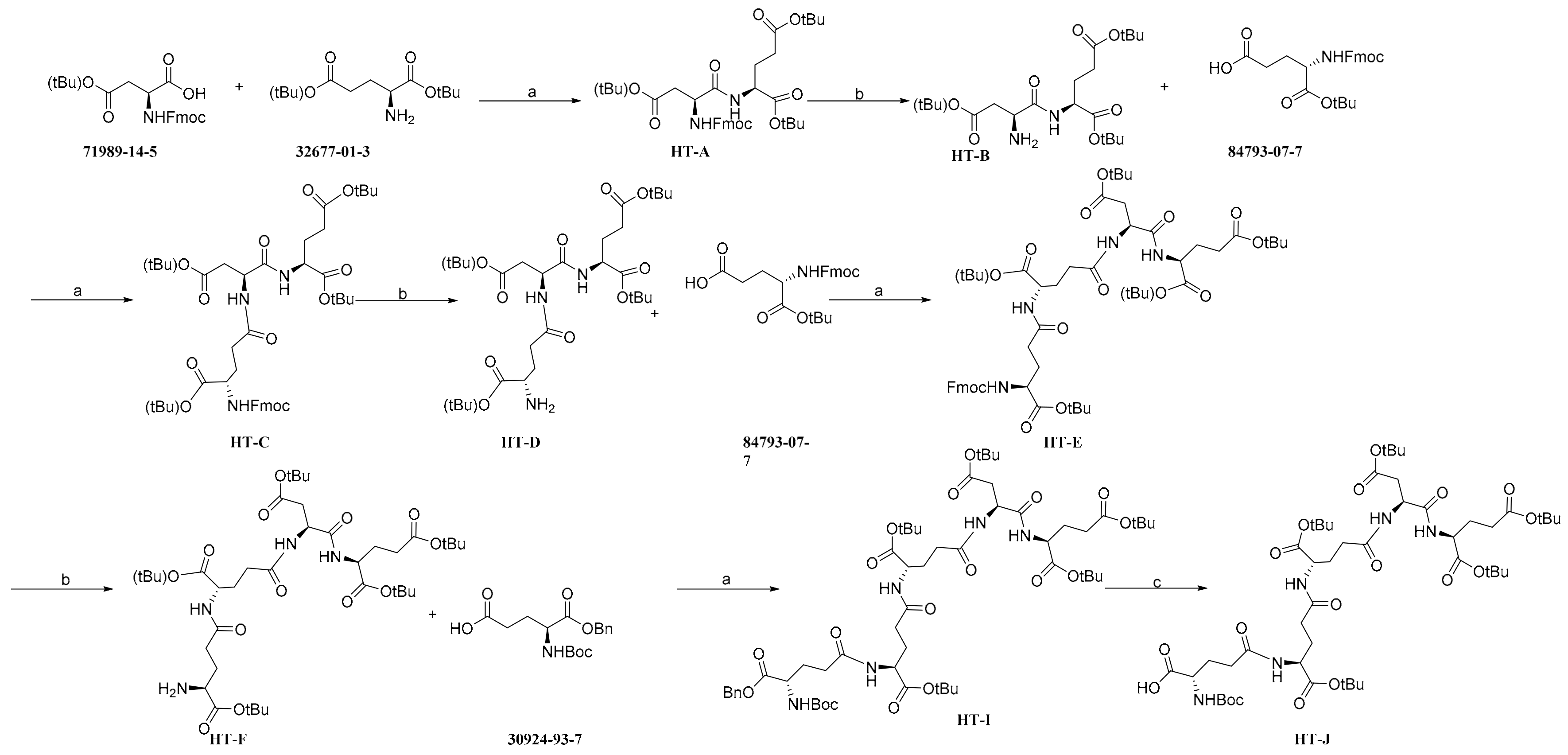
2.1. Chemistry
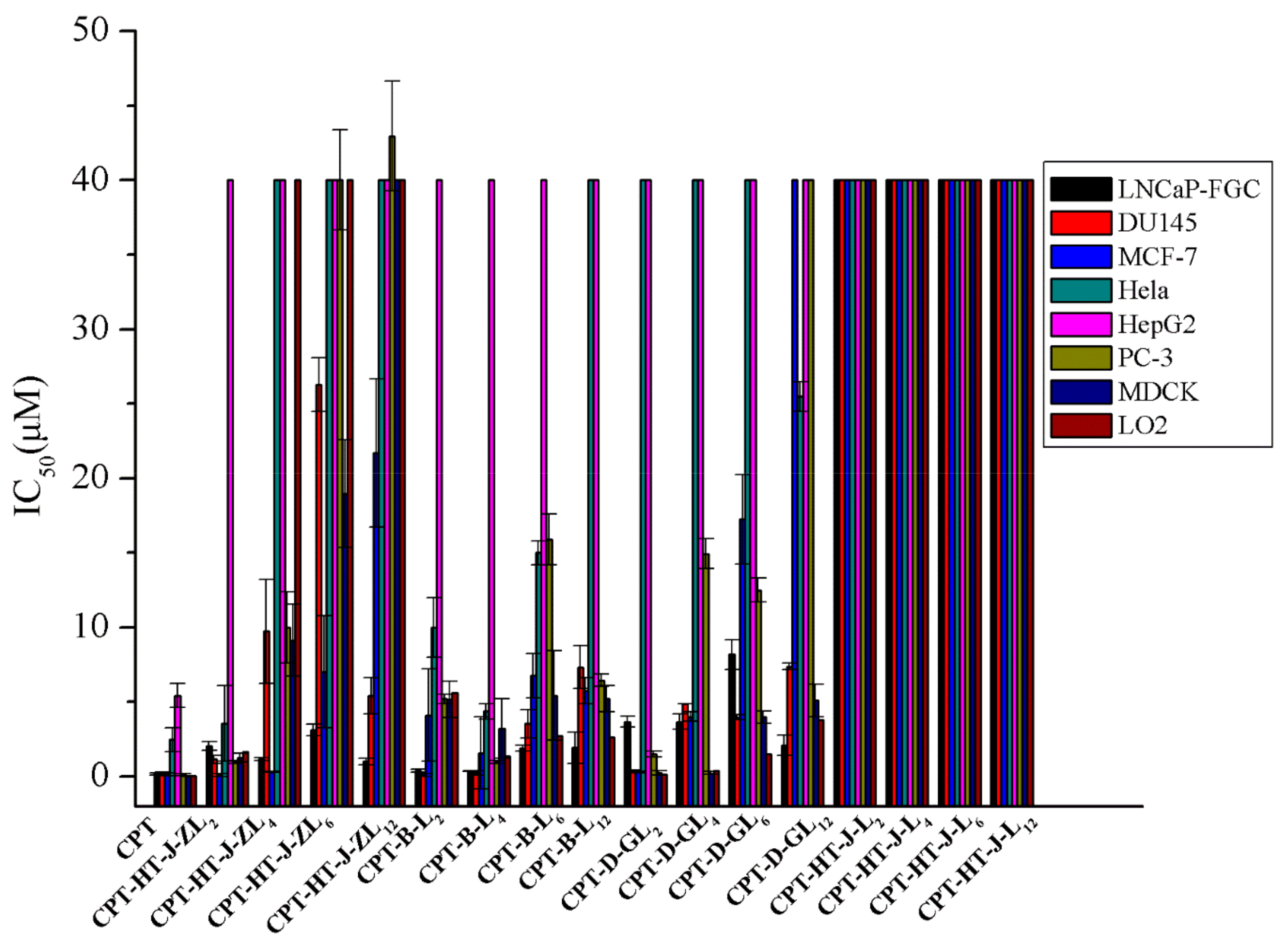
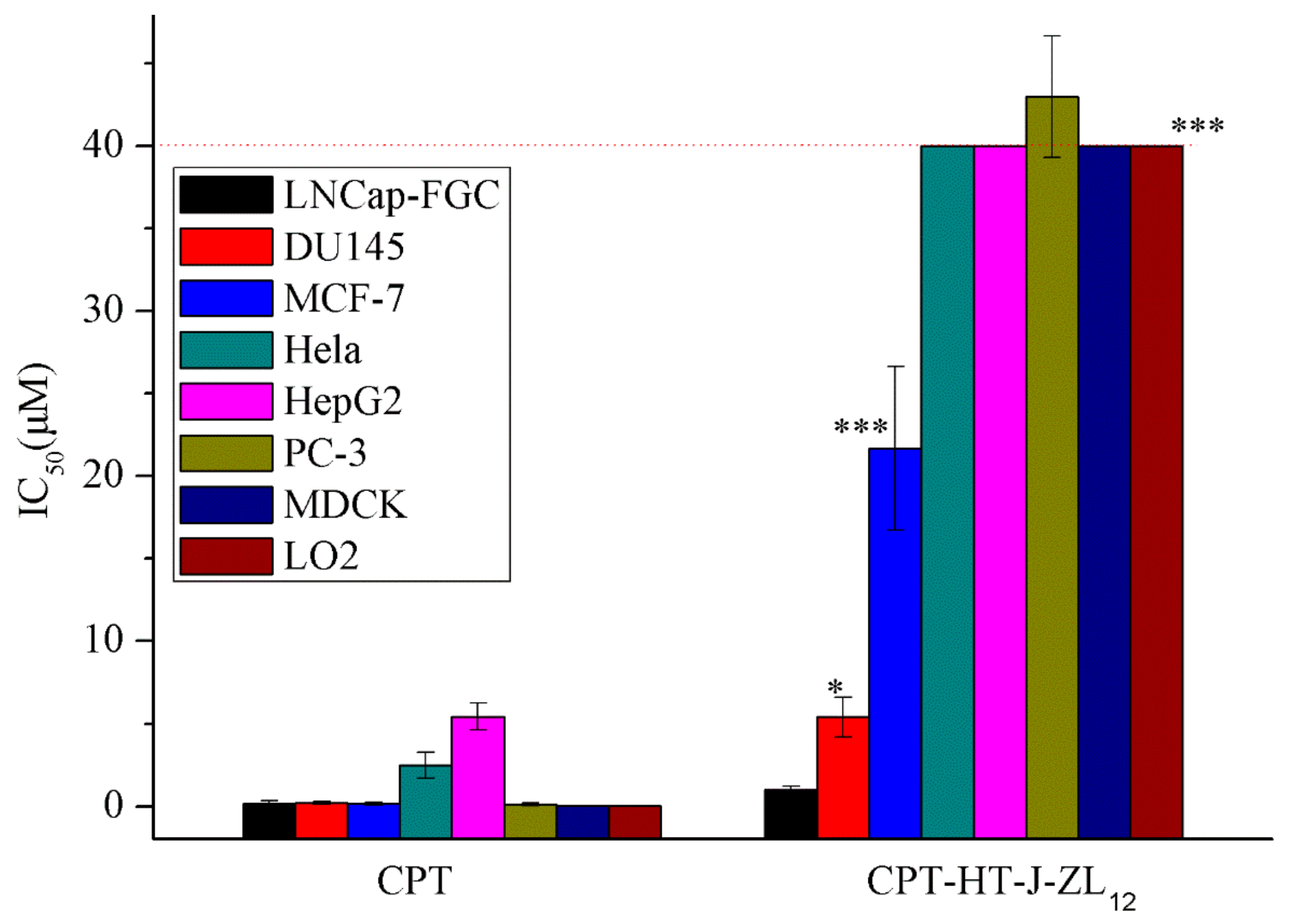
2.2. Cytotoxicity
2.3. Computer Simulation of ClogP and the Aqueous Solubility Study
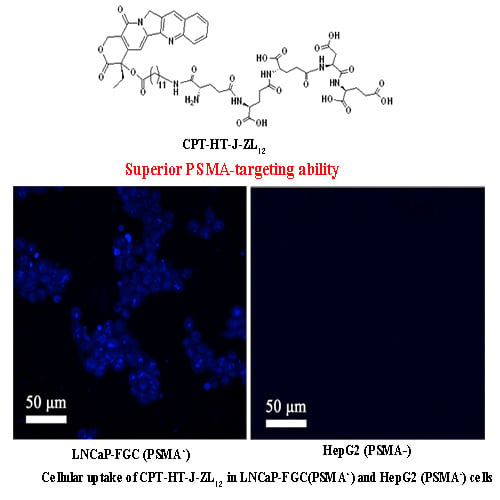
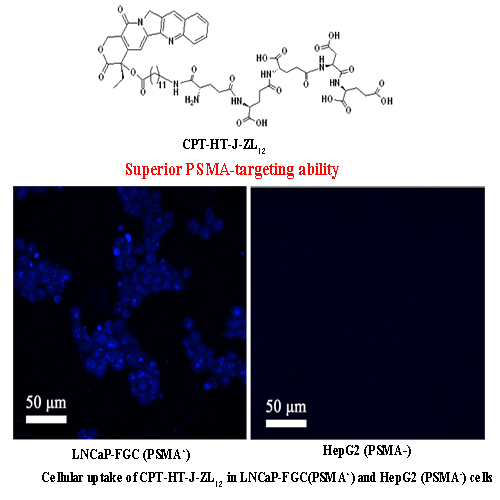
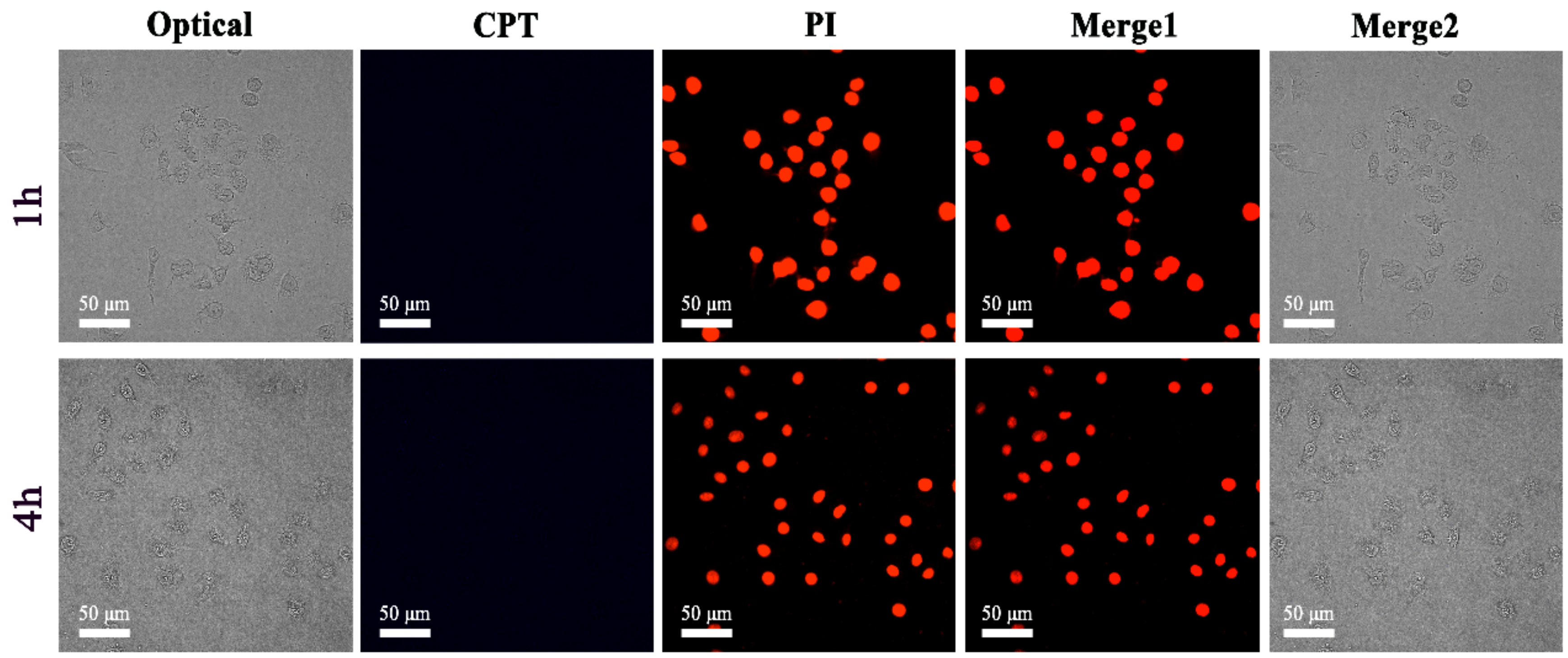
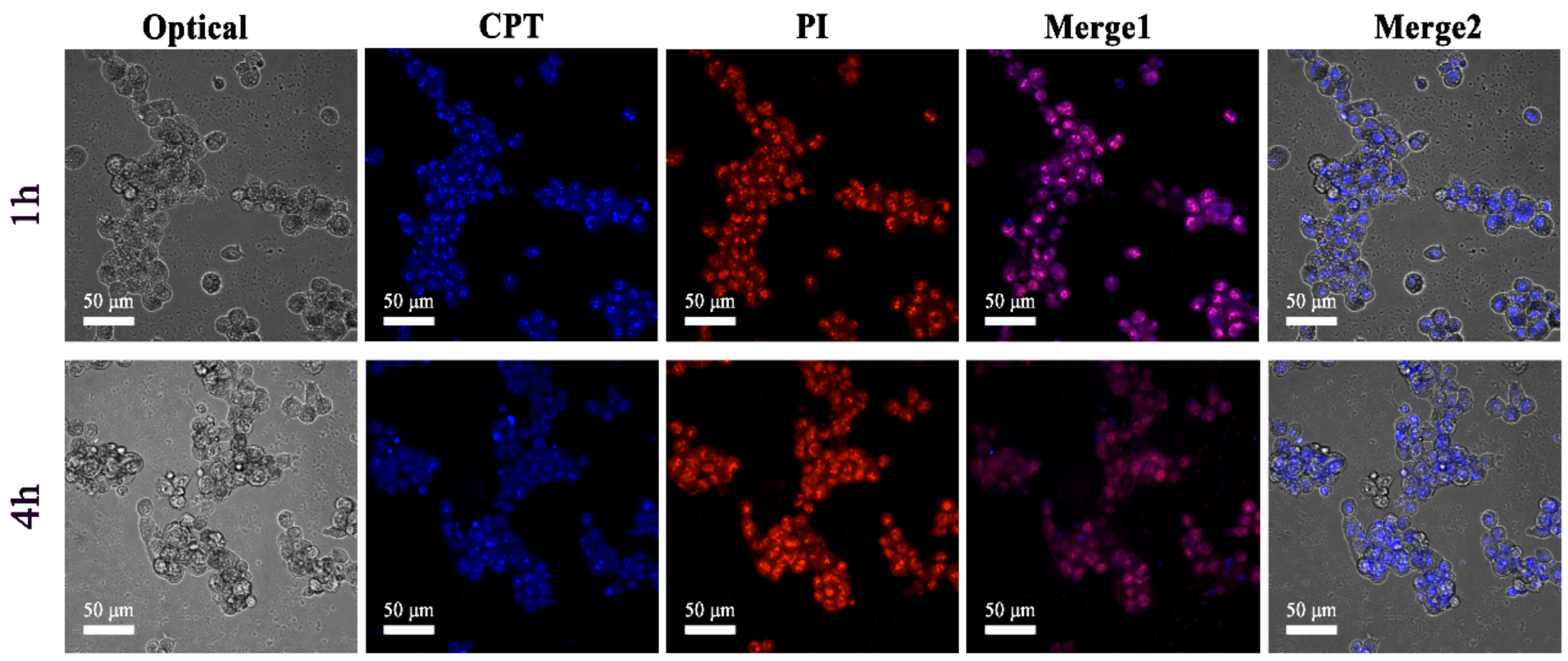
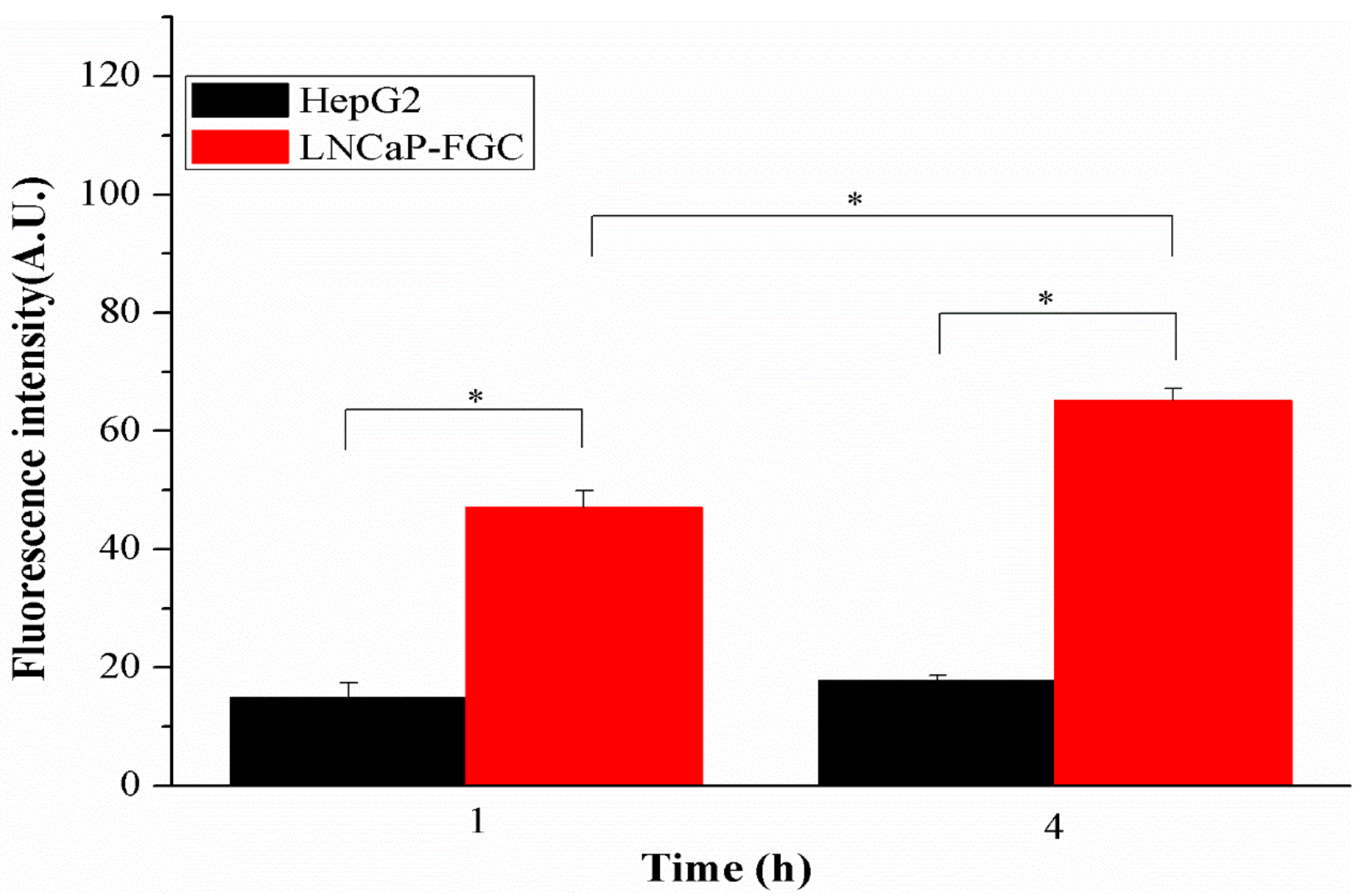
2.4. In Vitro Cellular Uptake
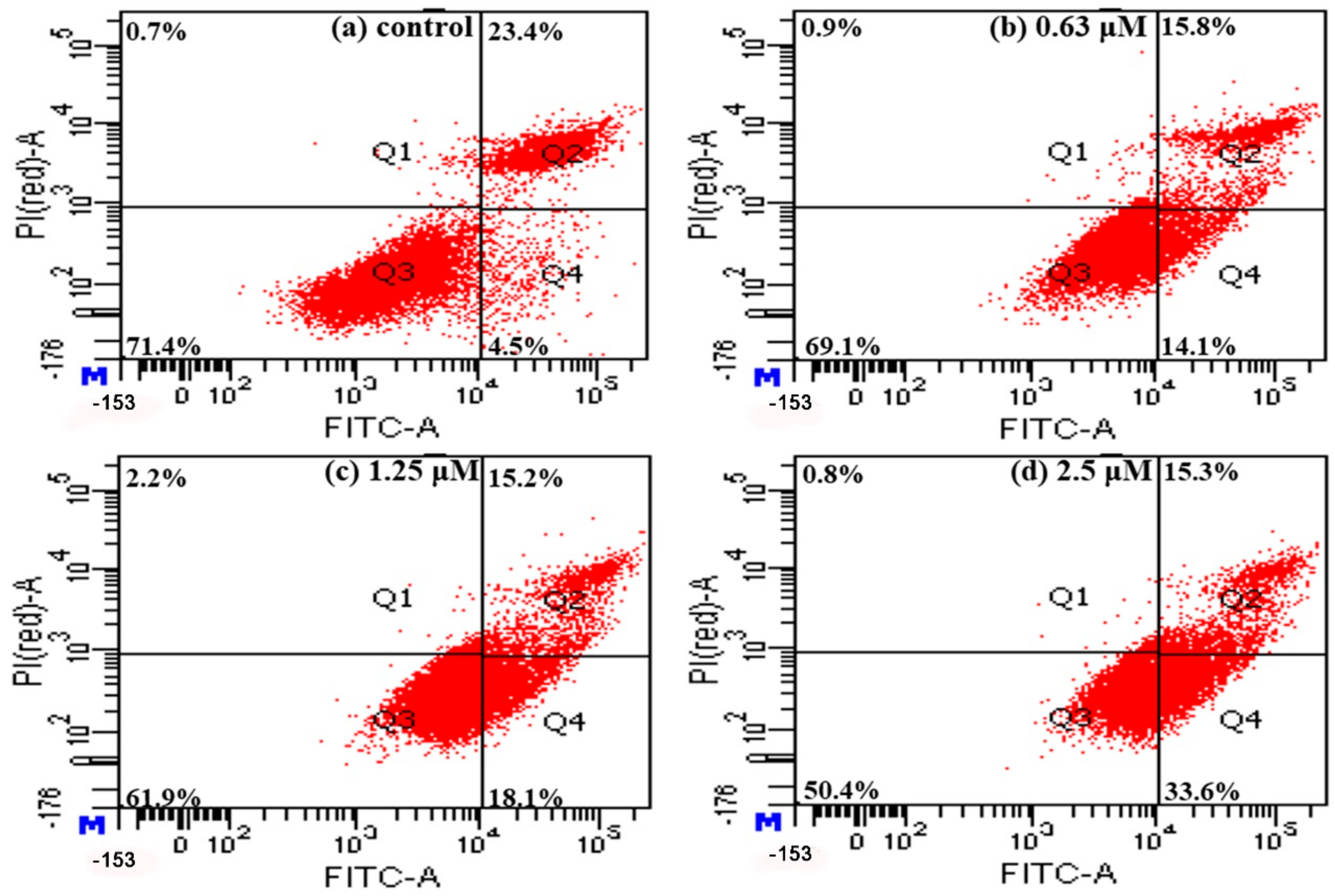
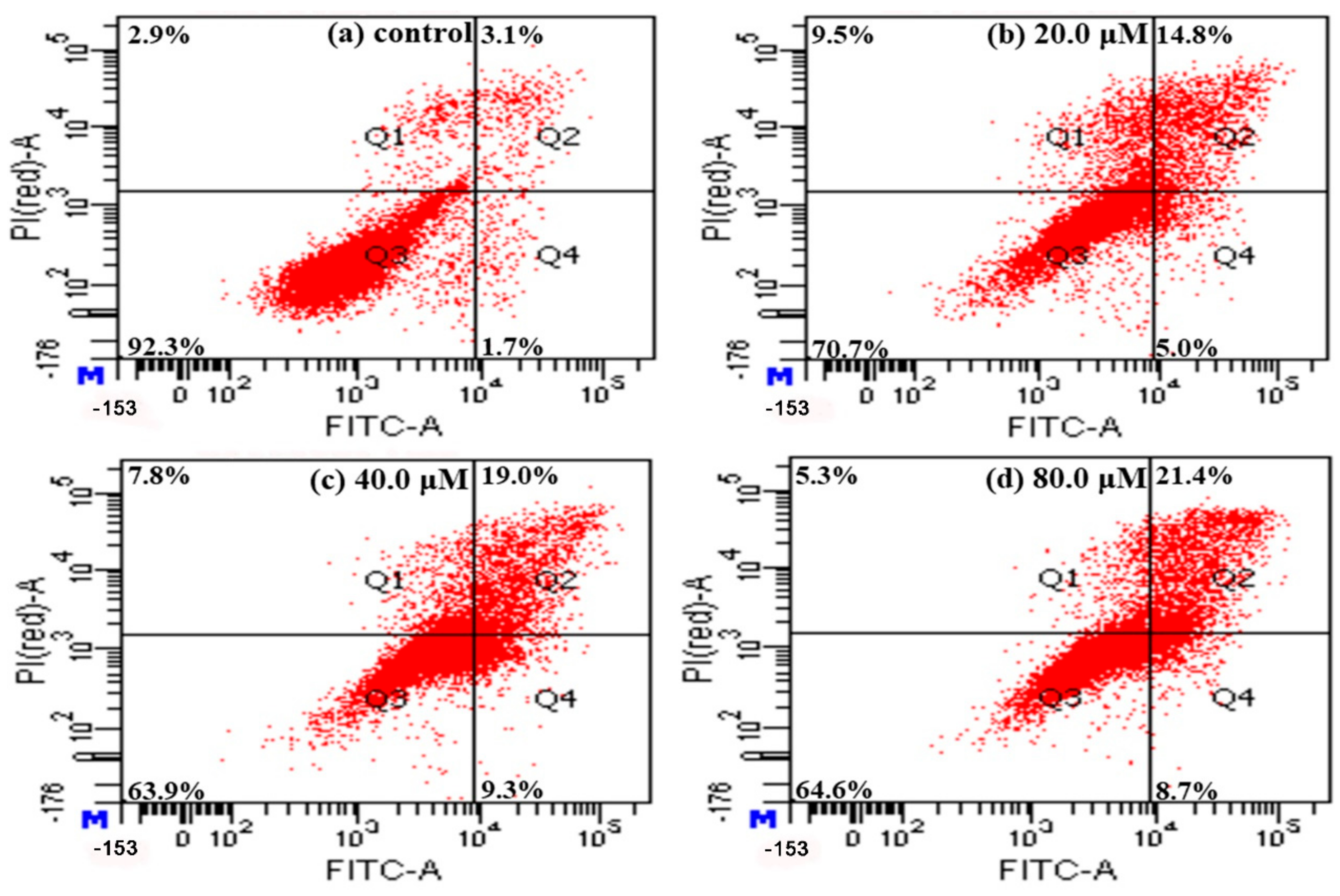
2.5. Detection of Apoptosis Using Annexin V-FITC/PI Staining
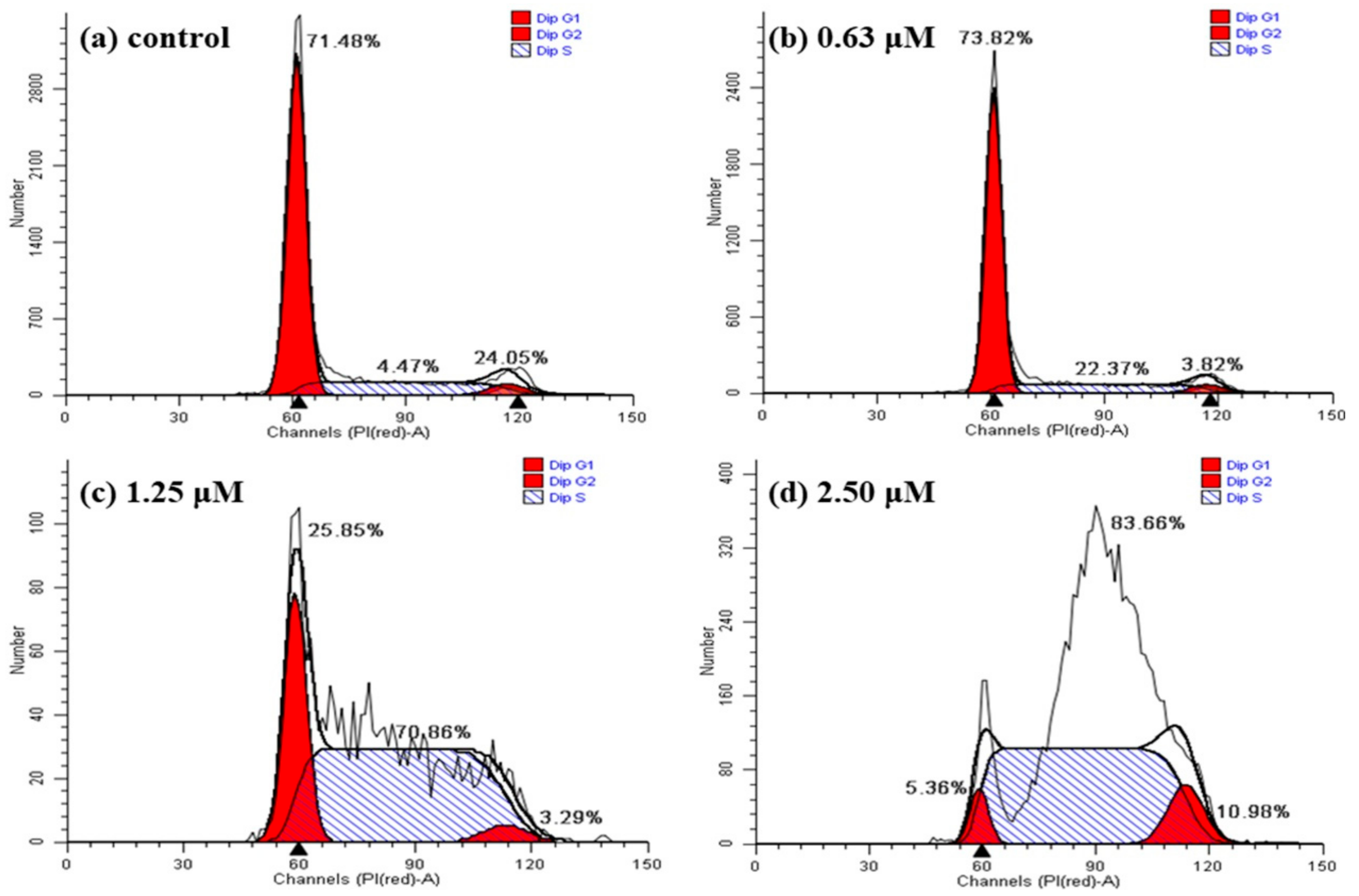
2.6. Detection of Cell Cycle Using PI Staining
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Synthetic Procedure of Condensation Reaction (Method A)
4.1.2. General Synthetic Procedure of Deprotection (Method B)
4.1.3. General Synthetic Procedure of Deprotection (Method C)
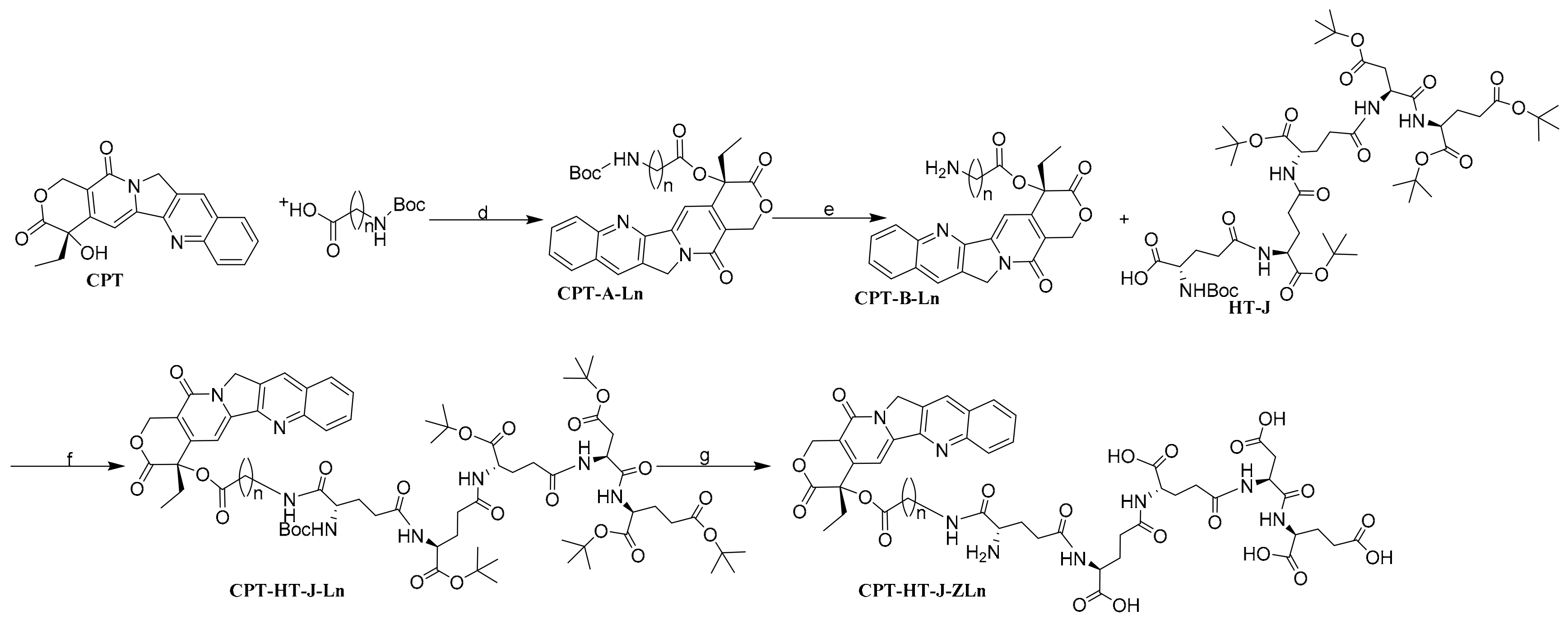
4.1.4. General Synthetic Procedure of the Preparation of PSMA-Activated Prodrug CPT-HT-J-ZLn
General Synthetic Procedure of the Intermediate CPT-A-Ln (Method D)
General Synthetic Procedure of the Intermediate CPT-B-Ln (Method E)
General Synthetic Procedure of the Intermediate CPT-HT-J-Ln (Method F)
General Synthetic Procedure of the Prodrug CPT-HT-J-ZLn (Method G)
4.1.5. General Synthetic Procedure of the Preparation of PSMA Hydrolysate CPT-D-GLn
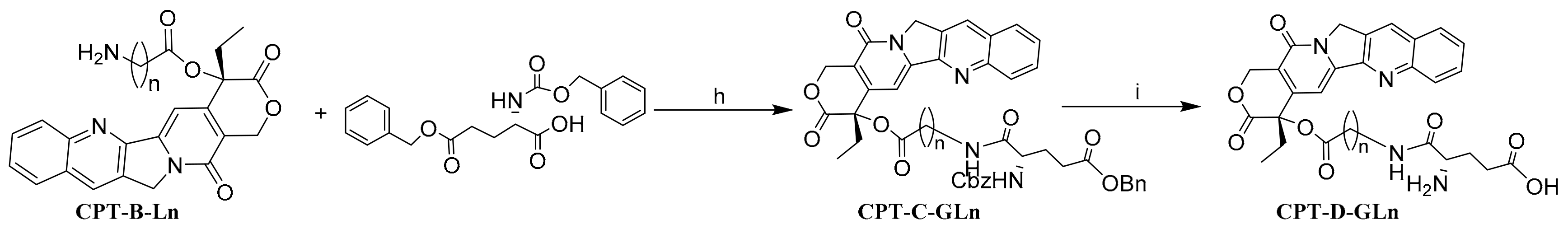
General Synthetic Procedure of the Intermediate CPT-C-GLn (Method H)
General Synthetic Procedure of PSMA Hydrolysate CPT-D-GLn (Method I)
4.2. Bio-Evaluation Methods
4.2.1. Cell Culture
4.2.2. Cytotoxicity Assay
4.2.3. Aqueous Solubility Study
4.2.4. In Vitro Cellular Uptake
4.2.5. Detection of Apoptosis Using Annexin V-FITC/PI Staining
4.2.6. Detection of Cell Cycle Using PI Staining
4.2.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stewart, B.; Wild, C.P. World Cancer Report 2014; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Li, Y.; Lin, J.Y.; Ma, J.Y.; Song, L.; Lin, H.R.; Tang, B.W.; Chen, D.Y.; Su, G.H.; Ye, S.F.; Zhu, X.; et al. Methotrexate–Camptothecin prodrug nanoassemblies as a versatile nanoplatform for biomodal imaging-guided self-active targeted and synergistic chemotherapy. ACS Appl. Mater. Interfaces 2017, 9, 34650–34665. [Google Scholar] [CrossRef] [PubMed]
- Savard, J.; Ivers, H.; Savard, M.H.; Morin, C.M. Cancer treatments and their side effects are associated with aggravation of insomnia: Results of a longitudinal study. Cancer 2015, 121, 1703–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sioka, C.; Kyritsis, A.P. Central and peripheral nervous system toxicity of common chemotherapeutic agents. Cancer Chemother. Pharmacol. 2009, 63, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Cardinale, D.; Suter, T.; Plataniotis, G.; de Azambuja, E.; Sandri, M.T.; Criscitiello, C.; Goldhirsch, A.; Cipolla, C.; Roila, F.; et al. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO clinical practice guidelines. Ann. Oncol. 2012, 23, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Perazella, M.A. Onco-nephrology: Renal toxicities of chemotherapeutic agents. Clin. J. Am. Soc. Nephrol. 2012, 7, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. DNA topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.L.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.L.; Gao, C.; Cui, L.G.; Wang, S.M.; Wang, J.R.; Dai, Z.F. Self-Assembly of an amphiphilic janus camptothecin–floxuridine conjugate into liposome-like nanocapsules for more efficacious combination chemotherapy in cancer. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Chen, X.; Ding, J.M.; Yu, L.; Ma, D.; Ding, J.D. Improved solubility and bioactivity of camptothecin family antitumor drugs with supramolecular encapsulation by water-soluble pillar[6]arene. ACS Omega 2017, 2, 5283–5288. [Google Scholar] [CrossRef] [PubMed]
- Sawant, R.R.; Sawant, R.M.; Torchilin, V.P. Mixed PEG–PE/vitamin E tumor-targeted immunomicelles as carriers for poorly soluble anti-cancer drugs: Improved drug solubilization and enhanced in vitro cytotoxicity. Eur. J. Pharm. Biopharm. 2008, 70, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Lee, C.; Sai, P.; Choe, Y.H.; Boro, M.; Pendri, A.; Guan, S.Y.; Greenwald, R.B. 20-O-acylcamptothecin derivatives: Evidence for lactone stabilization. J. Org. Chem. 2000, 65, 4601–4606. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Y.; Zu, Y.G.; Shi, R.Z.; Yao, L.P. Review camptothecin: Current perspectives. Curr. Med. Chem. 2006, 13, 2021–2039. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Xue, P.; Gao, Y.E.; Ma, X.; Bai, S.; Kang, Y.; Xu, Z. Gemcitabine–camptothecin conjugates: A hybrid prodrug for controlled drug release and synergistic therapeutics. Biomater. Sci. 2017, 5, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.A.; Doorneweerd, D.D.; Hilgenbrink, A.R.; Kularatne, S.A.; Low, P.S. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg. Med. Chem. Lett. 2006, 16, 5350–5355. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Heston, W.D. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J. Cell. Biochem. 2004, 91, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Bennett, M.J.; Thomas, L.M.; Bjorkman, P.J. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc. Natl. Acad. Sci. USA 2005, 102, 5981–5986. [Google Scholar] [CrossRef] [PubMed]
- Mhaka, A.; Gady, A.M.; Rosen, D.M.; Lo, K.M.; Gillies, S.D.; Denmeade, S.R. Use of methotrexate-based peptide substrates to characterize the substrate specificity of prostate-specific membrane antigen (PSMA). Cancer Biol. Ther. 2004, 3, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen–activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.T. Prodrug Targeting Based upon the NAALADase Activity to Prostate Specific Membrane Antigen by Prostate Cancer Cells; Johns Hopkins Univ Baltimore Md School of Medicine: Baltimore, MD, USA, 2001. [Google Scholar]
- Kularatne, S.A.; Venkatesh, C.; Santhapuram, H.K.; Wang, K.; Vaitilingam, B.; Henne, W.A.; Low, P.S. Synthesis and biological analysis of prostate-specific membrane antigen-targeted anticancer prodrugs. J. Med. Chem. 2010, 53, 7767–7777. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.Z.; Yang, J.C.; Zhang, R.S.; Yang, Z.M.; Yang, Z.T.; Wang, Y.J.; Xu, Y.J.; He, Z.G. Prostate-specific membrane antigen targeted therapy of prostate cancer using a DUPA–paclitaxel conjugate. Mol. Pharm. 2018, 15, 1842–1852. [Google Scholar] [CrossRef] [PubMed]
- Wullner, U.; Neef, I.; Eller, A.; Kleines, M.; Tur, M.K.; Barth, S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr. Cancer Drug Targets 2008, 8, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, H.S.; Yu, R.; Lee, K.; Gardner, T.A.; Jung, C.; Jeng, M.H.; Yeung, F.; Cheng, L.; Kao, C. Novel prostate-specific promoter derived from PSA and PSMA enhancers. Mol. Ther. 2002, 6, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Uchida, A.; O’Keefe, D.S.; Bacich, D.J.; Molloy, P.L.; Heston, W.D.W. In vivo suicide gene therapy model using a newly discovered prostate-specific membrane antigen promoter/enhancer: A potential alternative approach to androgen deprivation therapy. Urology 2001, 58, 132–139. [Google Scholar] [CrossRef]
- Wolf, P.; Gierschner, D.; Bühler, P.; Wetterauer, U.; Elsässer-Beile, U. A recombinant PSMA-specific single-chain immunotoxin has potent and selective toxicity against prostate cancer cells. Cancer Immunol. Immunother. 2006, 55, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.X.; Liu, G.H.; Li, Y.; Ma, J.Y.; Lin, J.Y.; Guo, F.Q.; Hou, Z.Q.; Xie, L.Y. Self-assembly of the active lactone form of a camptothecin–phospholipid complex for sustained nuclear drug delivery. RSC Adv. 2016, 6, 82949–82960. [Google Scholar] [CrossRef]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of molecular lipophilicity: State-of-the-art and comparison of log P methods on more than 96,000 compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Dohta, Y.; Nakamura, T.; Fukami, T. High-speed solubility screening assay using ultra-performance liquid chromatography/mass spectrometry in drug discovery. J. Chromatogr. A 2008, 1182, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Dohta, Y.; Yamashita, T.; Horiike, S.; Nakamura, T.; Fukami, T. A system for LogD screening of 96-well plates using a water-plug aspiration/injection method combined with high-performance liquid chromatography-mass spectrometry. Anal. Chem. 2007, 79, 8312–8315. [Google Scholar] [CrossRef] [PubMed]
- Alelyunas, Y.W.; Pelosi-Kilby, L.; Turcotte, P.; Kary, M.B.; Spreena, R.C. A high throughput dried DMSO LogD lipophilicity measurement based on 96-well shake-flask and atmospheric pressure photoionization mass spectrometry detection. J. Chromatogr. A 2010, 1217, 1950–1955. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | CLogP | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MCF-7 | PC-3 | DU145 | LNCaP-FGC | HepG2 | HeLa | LO2 | MDCK | ||
| CPT | 0.16 ± 0.10 | 0.13 ± 0.09 | 0.21 ± 0.09 | 0.18 ± 0.17 | 5.43 ± 0.81 | 2.48 ± 0.80 | 0.04 ± 0.01 | 0.02 ± 0.01 | 0.9 |
| CPT-HT-J-ZL2 | 0.11 ± 0.08 | 1.00 ± 0.10 | 1.16 ± 0.28 | 2.04 ± 0.30 | >40.00 | 3.54 ± 2.54 | 1.68 ± 0.45 | 1.27 ± 0.30 | −7.45 |
| CPT-HT-J-ZL4 | 0.32 ± 0.01 | 9.98 ± 2.38 | 9.73 ± 3.49 | 1.18 ± 0.10 | >40.00 | >40.00 | >40.00 | 9.13 ± 2.40 | −6.86 |
| CPT-HT-J-ZL6 | 7.03 ± 3.76 | 40.00 ± 3.37 | 26.28 ± 1.81 | 3.13 ± 0.40 | >40.00 | >40.00 | >40.00 | 18.96 ± 3.60 | −6.60 |
| CPT-HT-J-ZL12 | 21.68 ± 4.96 | 42.96 ± 3.69 | 5.40 ± 1.22 | 1.00 ± 0.20 | >40.00 | >40.00 | >40.00 | >40.00 | −3.42 |
| CPT-B-L2 | 4.11 ± 3.09 | 5.19 ± 0.30 | 0.12 ± 0.09 | 0.38 ± 0.10 | >40.00 | 10.00 ± 2.00 | 5.61 ± 1.40 | 5.16 ± 1.20 | 0.54 |
| CPT-B-L4 | 1.58 ± 2.42 | 1.00 ± 0.10 | 0.25 ± 0.13 | 0.37 ± 0.02 | >40.00 | 4.37 ± 0.50 | 1.36 ± 0.30 | 3.20 ± 2.00 | 1.26 |
| CPT-B-L6 | 6.76 ± 1.47 | 15.89 ± 1.73 | 3.55 ± 0.95 | 1.89 ± 0.20 | >40.00 | 15.0 ± 0.80 | 2.71 ± 0.38 | 5.44 ± 3.00 | 1.76 |
| CPT-B-L12 | 5.76 ± 0.86 | 6.45 ± 0.42 | 7.32 ± 1.43 | 1.93 ± 1.03 | >40.00 | >40.00 | 2.63 ± 0.84 | 5.2 ± 0.88 | 4.94 |
| CPT-D-GL2 | 0.30 ± 0.05 | 1.5 ± 0.20 | 0.40 ± 0.05 | 3.68 ± 0.36 | >40.00 | >40.00 | 0.16 ± 0.07 | 0.26 ± 0.14 | −2.88 |
| CPT-D-GL4 | 4.03 ± 0.30 | 14.93 ± 1.00 | 4.89 ± 0.01 | 3.68 ± 0.50 | >40.00 | >40.00 | 0.40 ± 0.15 | 0.23 ± 0.10 | −2.29 |
| CPT-D-GL6 | 17.25 ± 3.00 | 12.50 ± 0.80 | 3.97 ± 0.16 | 8.17 ± 1.00 | >40.00 | >40.00 | 1.5 ± 0.60 | 4.0 ± 0.42 | −2.03 |
| CPT-D-GL12 | >40.00 | >40.00 | 7.38 ± 0.20 | 2.10 ± 0.68 | >40.00 | 25.47 ± 1.0 | 3.8 ± 0.80 | 5.1 ± 1.10 | 1.15 |
| CPT-HT-J-L2 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | 4.04 |
| CPT-HT-J-L4 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | 4.62 |
| CPT-HT-J-L6 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | 4.89 |
| CPT-HT-J-L12 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | >40.00 | 8.07 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, B.; Zhou, F.; Yan, M.-M.; Cai, D.-S.; Guo, W.-B.; Yang, Y.-Q.; Jia, X.-H.; Zhang, W.-X.; Li, T.; Ma, T.; et al. PSMA-Oriented Target Delivery of Novel Anticancer Prodrugs: Design, Synthesis, and Biological Evaluations of Oligopeptide-Camptothecin Conjugates. Int. J. Mol. Sci. 2018, 19, 3251. https://doi.org/10.3390/ijms19103251
Xu B, Zhou F, Yan M-M, Cai D-S, Guo W-B, Yang Y-Q, Jia X-H, Zhang W-X, Li T, Ma T, et al. PSMA-Oriented Target Delivery of Novel Anticancer Prodrugs: Design, Synthesis, and Biological Evaluations of Oligopeptide-Camptothecin Conjugates. International Journal of Molecular Sciences. 2018; 19(10):3251. https://doi.org/10.3390/ijms19103251
Chicago/Turabian StyleXu, Bing, Fei Zhou, Meng-Meng Yan, De-Sheng Cai, Wen-Bo Guo, Yu-Qin Yang, Xiao-Hui Jia, Wen-Xi Zhang, Tong Li, Tao Ma, and et al. 2018. "PSMA-Oriented Target Delivery of Novel Anticancer Prodrugs: Design, Synthesis, and Biological Evaluations of Oligopeptide-Camptothecin Conjugates" International Journal of Molecular Sciences 19, no. 10: 3251. https://doi.org/10.3390/ijms19103251
APA StyleXu, B., Zhou, F., Yan, M.-M., Cai, D.-S., Guo, W.-B., Yang, Y.-Q., Jia, X.-H., Zhang, W.-X., Li, T., Ma, T., Wang, P.-L., & Lei, H.-M. (2018). PSMA-Oriented Target Delivery of Novel Anticancer Prodrugs: Design, Synthesis, and Biological Evaluations of Oligopeptide-Camptothecin Conjugates. International Journal of Molecular Sciences, 19(10), 3251. https://doi.org/10.3390/ijms19103251