The Proposal of Molecular Mechanisms of Weak Organic Acids Intake-Induced Improvement of Insulin Resistance in Diabetes Mellitus via Elevation of Interstitial Fluid pH


Abstract
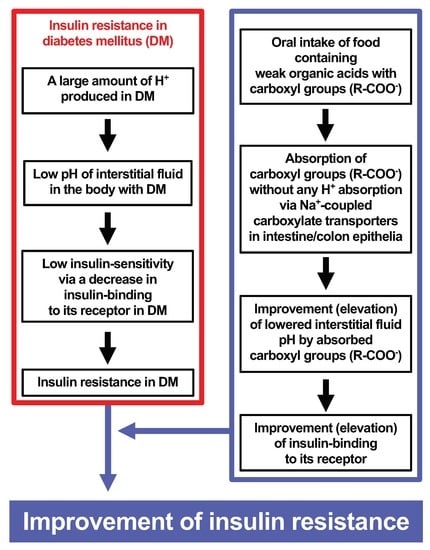
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
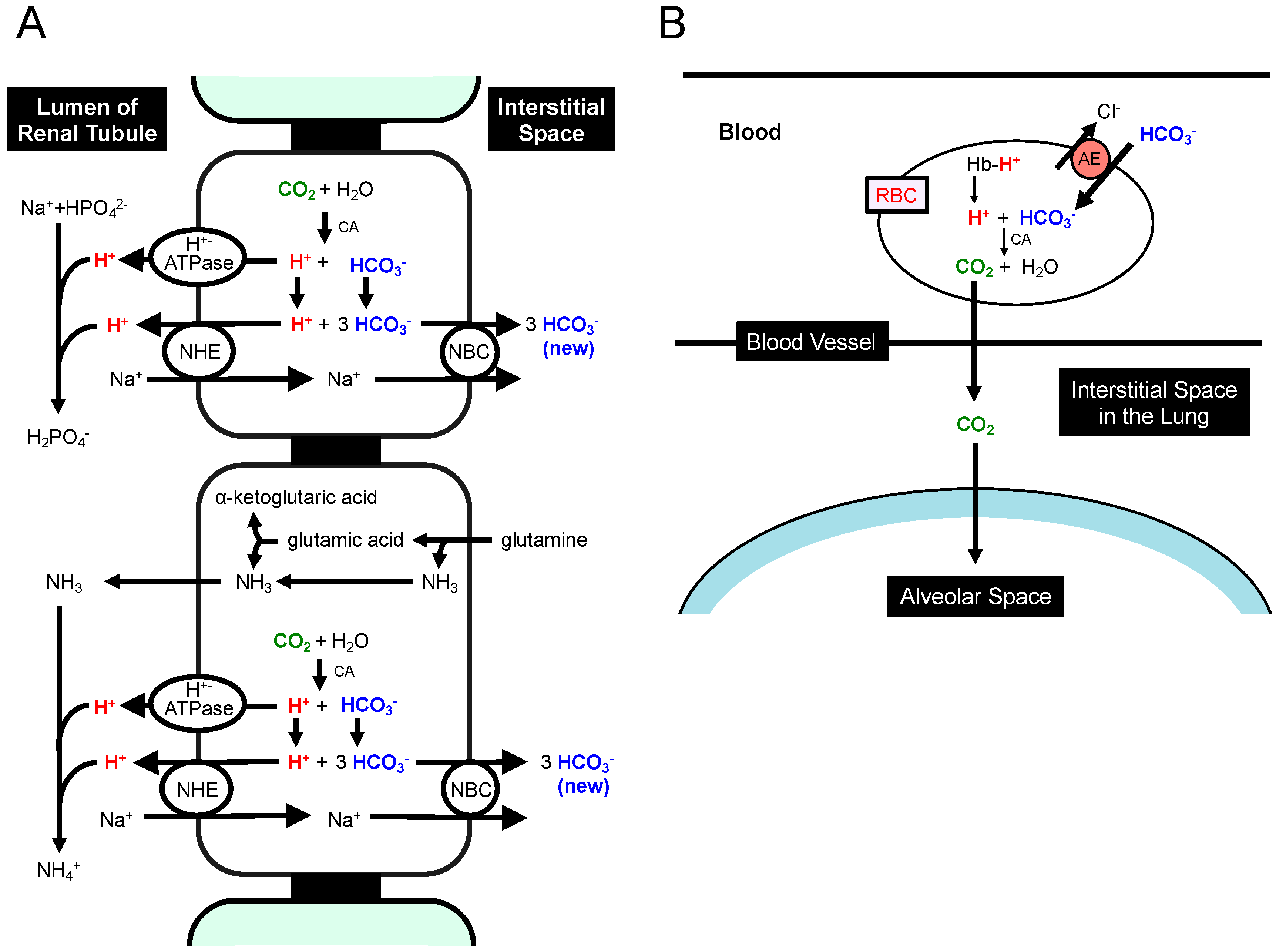{kind=link}
{kind=link}
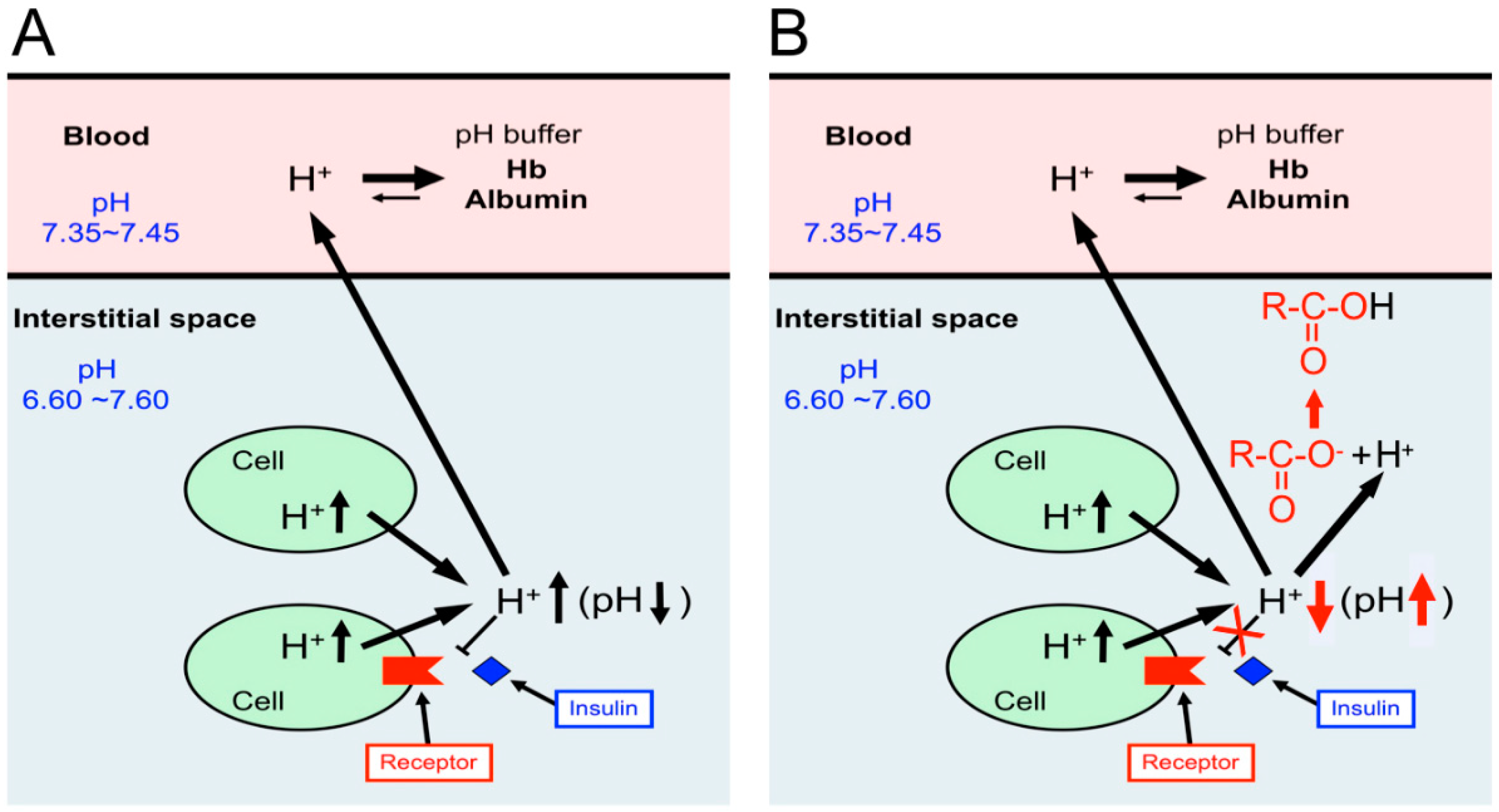{kind=link}
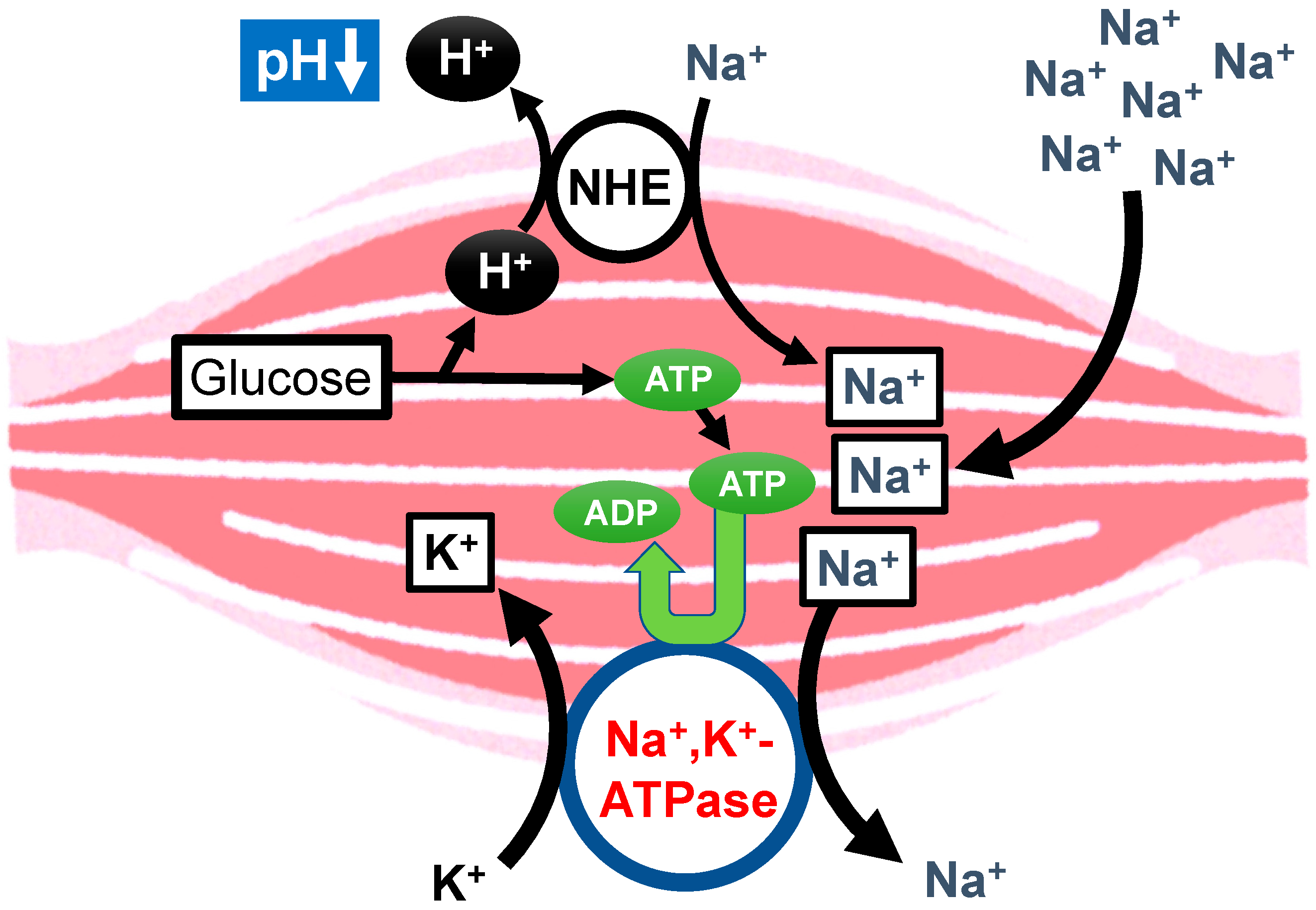{kind=link}
{kind=link}
1. Introduction
2. Variety of the Interstitial Fluid pH
3. Physiological Roles of Little pH Buffering Capacity of Interstitial Fluids in Bodily and Cellular Functions
4. Sources of H+ in Interstitial Fluids
5. Transporting Systems of H+ between the Intracellular and Interstitial Fluid Spaces
6. Roles of Abnormal Interstitial Fluid pH in Diabetes Mellitus
7. Roles of Foods Containing Weak Organic Acids and Carboxylate Transporters in Improvement of Low Interstitial Fluid pH
8. Proposal of Molecular Mechanisms of Diabetes Mellitus Occurrence Caused by High Salt Intake
9. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AE | anion exchanger |
| CA | carbonic anhydrase |
| DM | diabetes mellitus |
| GLP | glucagon-like peptide |
| GLUT4 | glucose transporter 4 |
| [H+] | H+ concentration |
| Hb | hemoglobin |
| HCT | H+-coupled carboxylate transporter |
| IDF | International Diabetes Federation |
| IRS-1 | insulin receptor substrate-1 |
| MC | monocarboxylic acid |
| MCT | monocarboxylate transporter |
| NBC | Na+-HCO3− cotransporter |
| NDCBE | Na+-driven Cl−/HCO3− exchanger |
| NHE | Na+/H+ exchanger |
| OLETF | Otsuka Long-Evans Tokushima Fatty |
| PIP2 | phosphatidylinositol 4,5-diphosphate |
| PPD | dipeptidyl-peptidase |
| PTPR | protein tyrosine phosphatase receptor |
| RBC | red blood cells |
| SCT | sodium-coupled carboxylate transporters |
| SGLT2 | sodium-glucose cotransporter 2 |
References
- Marunaka, Y. Roles of interstitial fluid pH in diabetes mellitus: Glycolysis and mitochondrial function. World J. Diabetes 2015, 6, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y.; Yoshimoto, K.; Aoi, W.; Hosogi, S.; Ikegaya, H. Low pH of interstitial fluid around hippocampus of the brain in diabetic OLETF rats. Mol. Cell. Ther. 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, T. The metabolic syndrome in Japan. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5 (Suppl. 1), S15–S18. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y.; Hagiwara, N.; Tohda, H. Insulin activates single amiloride-blockable Na channels in a distal nephron cell line (A6). Am. J. Physiol. Renal Physiol. 1992, 263, F392–F400. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Kuhn, C.M.; Feinglos, M.N.; Surwit, R. Hypertension in CB57BL/6J mouse model of non-insulin-dependent diabetes mellitus. Am. J. Physiol. 1993, 264, R73–R78. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.G.; Tipton, C.M. Influences of exogenous insulin on arterial blood pressure measurements of the rat. J. Appl. Physiol. 1989, 67, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Meehan, W.P.; Buchanan, T.A.; Hsueh, W. Chronic insulin administration elevates blood pressure in rats. Hypertension 1994, 23, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y.; Niisato, N.; Taruno, A.; Ohta, M.; Miyazaki, H.; Hosogi, S.; Nakajima, K.; Kusuzaki, K.; Ashihara, E.; Nishio, K.; et al. Regulation of epithelial sodium transport via epithelial Na+ channel. J. Biomed. Biotechnol. 2011, 2011, 978196. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y. Characteristics and pharmacological regulation of epithelial Na+ channel (ENaC) and epithelial Na+ transport. J. Pharmacol. Sci. 2014, 126, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Horita, S.; Nakamura, M.; Suzuki, M.; Satoh, N.; Suzuki, A.; Homma, Y.; Nangaku, M. The role of renal proximal tubule transport in the regulation of blood pressure. Kidney Res. Clin. Pract. 2017, 36, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimihodimos, V.; Gonzalez-Villalpando, C.; Meigs, J.B.; Ferrannini, E. Hypertension and Diabetes Mellitus: Coprediction and Time Trajectories. Hypertension 2018, 71, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Touati, H.; Ouali-Hassenaoui, S.; Dekar-Madoui, A.; Challet, E.; Pevet, P.; Vuillez, P. Diet-induced insulin resistance state disturbs brain clock processes and alters tuning of clock outputs in the Sand rat, Psammomys obesus. Brain Res. 2018, 1679, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Cederholm, J.; Wibell, L. Glucose intolerance in middle-aged subjects--a cause of hypertension? Acta Med. Scand. 1985, 217, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, K.F.; Lindgarde, F. Contribution of estimated insulin resistance and glucose intolerance to essential hypertension. J. Intern. Med. 1991, 735, 75–83. [Google Scholar]
- Bao, W.; Srinivasan, S.R.; Berenson, G.S. Persistent elevation of plasma insulin levels is associated with increased cardiovascular risk in children and young adults. The Bogalusa Heart Study. Circulation 1996, 93, 54–59. [Google Scholar] [CrossRef]
- Haffner, S.M.; Ferrannini, E.; Hazuda, H.P.; Stern, M.P. Clustering of cardiovascular risk factors in confirmed prehypertensive individuals. Hypertension 1992, 20, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Lahera, V.; de Las Heras, N.; Lopez-Farre, A.; Manucha, W.; Ferder, L. Role of mitochondrial dysfunction in hypertension and obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hamid, A.A.M.; Firgany, A.E.L. Favorable outcomes of hydroxychloroquine in insulin resistance may be accomplished by adjustment of the endothelial dysfunction as well as the skewed balance of adipokines. Acta Histochem. 2016, 118, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Almind, K.; Manieri, M.; Sivitz, W.I.; Cinti, S.; Kahn, C.R. Ectopic brown adipose tissue in muscle provides a mechanism for differences in risk of metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2007, 104, 2366–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagdade, J.D. Disorders of glucose metabolism in uremia. Adv. Nephrol. Necker Hosp. 1979, 8, 87–100. [Google Scholar] [PubMed]
- Benard, O.; Lim, J.; Apontes, P.; Jing, X.; Angeletti, R.H.; Chi, Y. Impact of high-fat diet on the proteome of mouse liver. J. Nutr. Biochem. 2016, 31, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Bitsi, S.; Ali, H.; Maskell, L.; Ounzain, S.; Mohamed-Ali, V.; Budhram-Mahadeo, V.S. Profound hyperglycemia in knockout mutant mice identifies novel function for POU4F2/Brn-3b in regulating metabolic processes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E303–E312. [Google Scholar] [CrossRef] [PubMed]
- Bruce, K.D.; Cagampang, F.R. Epigenetic priming of the metabolic syndrome. Toxicol. Mech. Methods 2011, 21, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin resistance in nonalcoholic fatty liver disease. Curr. Pharm. Des. 2010, 16, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Dhurandhar, E.J.; Krishnapuram, R.; Hegde, V.; Dubuisson, O.; Tao, R.; Dong, X.C.; Ye, J.; Dhurandhar, N.V. E4orf1 improves lipid and glucose metabolism in hepatocytes: A template to improve steatosis & hyperglycemia. PLoS ONE 2012, 7, e47813. [Google Scholar]
- Di Marzio, D.; Mohn, A.; Mokini, Z.H.; Giannini, C.; Chiarelli, F. Macroangiopathy in adults and children with diabetes: From molecular mechanisms to vascular damage (part 1). Horm. Metab. Res. 2006, 38, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Fougerat, A.; Pan, X.; Smutova, V.; Heveker, N.; Cairo, C.W.; Issad, T.; Larrivee, B.; Medin, J.A.; Pshezhetsky, A.V. Neuraminidase 1 activates insulin receptor and reverses insulin resistance in obese mice. Mol. Metab. 2018, 12, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Hamann, C.; Goettsch, C.; Mettelsiefen, J.; Henkenjohann, V.; Rauner, M.; Hempel, U.; Bernhardt, R.; Fratzl-Zelman, N.; Roschger, P.; Rammelt, S.; et al. Delayed bone regeneration and low bone mass in a rat model of insulin-resistant type 2 diabetes mellitus is due to impaired osteoblast function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1220–E1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, G.; Ghadieh, H.E.; Ghanem, S.S.; Muturi, H.T.; Rezaei, K.; Al-Share, Q.Y.; Bowman, T.A.; Zhang, D.; Garofalo, R.S.; Yin, L.; et al. Loss of Hepatic CEACAM1: A Unifying Mechanism Linking Insulin Resistance to Obesity and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. (Lausanne) 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Houdali, B.; Nguyen, V.; Ammon, H.P.; Haap, M.; Schechinger, W.; Machicao, F.; Rett, K.; Haring, H.U.; Schleicher, E.D. Prolonged glucose infusion into conscious rats inhibits early steps in insulin signalling and induces translocation of GLUT4 and protein kinase C in skeletal muscle. Diabetologia 2002, 45, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Jesmin, J.; Rashid, M.S.; Jamil, H.; Hontecillas, R.; Bassaganya-Riera, J. Gene regulatory network reveals oxidative stress as the underlying molecular mechanism of type 2 diabetes and hypertension. BMC Med. Genomics 2010, 3, 45. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Palomares, M.; Ramos-Rodriguez, J.J.; Lopez-Acosta, J.F.; Pacheco-Herrero, M.; Lechuga-Sancho, A.M.; Perdomo, G.; Garcia-Alloza, M.; Cozar-Castellano, I. Increased Abeta production prompts the onset of glucose intolerance and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1373–E1380. [Google Scholar] [CrossRef] [PubMed]
- Julve, J.; Escola-Gil, J.C.; Marzal-Casacuberta, A.; Ordonez-Llanos, J.; Gonzalez-Sastre, F.; Blanco-Vaca, F. Increased production of very-low-density lipoproteins in transgenic mice overexpressing human apolipoprotein A-II and fed with a high-fat diet. Biochim. Biophys. Acta 2000, 1488, 233–244. [Google Scholar] [CrossRef]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.H. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 2008, 7, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, A.; Samal, K.C.; Sanjeevi, C.B. Molecular mechanisms involved in the etiopathogenesis of malnutrition-modulated diabetes mellitus. Ann. N. Y. Acad. Sci. 2002, 958, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Katsanos, C.S. Lipid-induced insulin resistance in the liver: Role of exercise. Sports Med. 2004, 34, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Kellerer, M.; Rett, K.; Renn, W.; Groop, L.; Haring, H.U. Circulating TNF-alpha and leptin levels in offspring of NIDDM patients do not correlate to individual insulin sensitivity. Horm. Metab. Res. 1996, 28, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.L.; Tang, Y.; Li, H.; Guertin, D.A. Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol. Metab. 2016, 5, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, P.; Mahesh Kumar, M.J.; Venkatesan, R.; Majundar, S.S.; Juyal, R.C. Genetically modified mouse models for the study of nonalcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Tanaka, T. Insulin resistance in obesity and its molecular control. Obes. Res. 1995, 3 (Suppl. 2), 195s–198s. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Khil, L.Y.; Cho, K.A.; Ryu, S.J.; Ha, M.K.; Cheon, G.J.; Lee, T.S.; Yoon, J.W.; Jun, H.S.; Park, S.C. A potential role for skeletal muscle caveolin-1 as an insulin sensitivity modulator in ageing-dependent non-obese type 2 diabetes: Studies in a new mouse model. Diabetologia 2008, 51, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Perie, L.; Parente, A.; Baraige, F.; Magnol, L.; Blanquet, V. Alterations in Adiposity and Glucose Homeostasis in Adult Gasp-1 Overexpressing Mice. Cell. Physiol. Biochem. 2017, 44, 1896–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, D.J.; Turban, S.; Gray, A.; Hajduch, E.; Hundal, H.S. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem. J. 2004, 382 Pt 2, 619–629. [Google Scholar] [CrossRef]
- Pruznak, A.M.; Hong-Brown, L.; Lantry, R.; She, P.; Frost, R.A.; Vary, T.C.; Lang, C.H. Skeletal and cardiac myopathy in HIV-1 transgenic rats. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E964–E973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, H.; John, A.; Howarth, F.C. Increased oxidative stress and mitochondrial dysfunction in zucker diabetic rat liver and brain. Cell. Physiol. Biochem. 2015, 35, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Printz, R.L. Diabetes in Zucker diabetic fatty rat. Methods Mol. Biol. 2012, 933, 103–123. [Google Scholar] [PubMed]
- Staudinger, J.L.; Lichti, K. Cell signaling and nuclear receptors: New opportunities for molecular pharmaceuticals in liver disease. Mol. Pharm. 2008, 5, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Suk, H.Y.; Zhou, C.; Yang, T.T.; Zhu, H.; Yu, R.Y.; Olabisi, O.; Yang, X.; Brancho, D.; Kim, J.Y.; Scherer, P.E.; et al. Ablation of calcineurin Abeta reveals hyperlipidemia and signaling cross-talks with phosphodiesterases. J. Biol. Chem. 2013, 288, 3477–3488. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Xie, M.H.; Lei, Y.; Zhou, L.; Xu, Y.P.; Cai, J.G. The roles of aerobic exercise training and suppression IL-6 gene expression by RNA interference in the development of insulin resistance. Cytokine 2013, 61, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Thomson, M.J.; Williams, M.G.; Frost, S.C. Development of insulin resistance in 3T3-L1 adipocytes. J. Biol. Chem. 1997, 272, 7759–7764. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xiong, X.; Li, Y.; Liu, X.; Wang, T.; Zhang, H.; Jiao, Y.; Jiang, J.; Zhang, H.; Tang, Q.; et al. Hepatic F-Box Protein FBXW7 Maintains Glucose Homeostasis Through Degradation of Fetuin-A. Diabetes 2018, 67, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.G.; Zhou, Y.P.; Zhang, X.; Thu, P.M.; Xie, Z.S.; Lu, C.; Pang, T.; Xue, B.; Xu, D.Q.; Chen, Y.; et al. Anhydroicaritin improves diet-induced obesity and hyperlipidemia and alleviates insulin resistance by suppressing SREBPs activation. Biochem. Pharmacol. 2016, 122, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Zochodne, D.W. Sensory Neurodegeneration in Diabetes: Beyond Glucotoxicity. Int. Rev. Neurobiol. 2016, 127, 151–180. [Google Scholar] [PubMed]
- Utriainen, T.; Takala, T.; Luotolahti, M.; Ronnemaa, T.; Laine, H.; Ruotsalainen, U.; Haaparanta, M.; Nuutila, P.; Yki-Jarvinen, H. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia 1998, 41, 555–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, L.M.; Traxinger, R.; Kraus, A.P. Uremia and insulin resistance: N-carbamoyl-asparagine decreases insulin-sensitive glucose uptake in rat adipocytes. Kidney Int. 2004, 65, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, M.; Chuprun, J.K.; Rengo, G.; Gao, E.; Wei, Z.; Peroutka, R.J.; Gold, J.I.; Gumpert, A.; Chen, M.; Otis, N.J.; et al. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation 2011, 123, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, M.J.; Wierts, R.; Hoeks, J.; Gemmink, A.; Brans, B.; Mottaghy, F.M.; Schrauwen, P.; van Marken Lichtenbelt, W.D. Glucose uptake in human brown adipose tissue is impaired upon fasting-induced insulin resistance. Diabetologia 2015, 58, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Malkki, H. Alzheimer disease: Insulin resistance could be linked to risk of AD via reduced glucose uptake. Nat. Rev. Neurol. 2015, 11, 485. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of Insulin Resistance with Cerebral Glucose Uptake in Late Middle-Aged Adults at Risk for Alzheimer Disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- IDF Diabetes Atlas. Available online: http://www.diabetesatlas.org/key-messages.html (accessed on 18 October 2018).
- Chen, G.; Yang, X.; Yang, X.; Li, L.; Luo, J.; Dong, H.; Xu, L.; Yi, P.; Wang, K.; Zou, X.; et al. Jia-Wei-Jiao-Tai-Wan ameliorates type 2 diabetes by improving beta cell function and reducing insulin resistance in diabetic rats. BMC Complement. Altern. Med. 2017, 17, 507. [Google Scholar] [CrossRef] [PubMed]
- Olivera Santa-Catalina, M.; Redondo, P.C.; Cantonero, C.; Granados, M.P.; Sanchez-Collado, J.; Albarran, L.; Lopez, J.J. New insights into adipokines as potential biomarkers for type-2 diabetes mellitus. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef]
- Othman, N.A.; Docherty, P.D.; Krebs, J.D.; Bell, D.A.; Chase, J.G. The Need to Calculate Target Glucose Levels When Measuring Changes in Insulin Sensitivity During Interventions for Individuals with Type 2 Diabetes. J. Diabetes Sci. Technol. 2018, 12, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Rehman, K.; Akash, M.S.H.; Liaqat, A.; Kamal, S.; Qadir, M.I.; Rasul, A. Role of Interleukin-6 in Development of Insulin Resistance and Type 2 Diabetes Mellitus. Crit. Rev. Eukaryot. Gene Expr. 2017, 27, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Labra, R.; Silva, L.; Subiabre, M.; Araos, J.; Salsoso, R.; Fuenzalida, B.; Saez, T.; Toledo, F.; Gonzalez, M.; Quezada, C.; et al. Akt/mTOR Role in Human Foetoplacental Vascular Insulin Resistance in Diseases of Pregnancy. J. Diabetes Res. 2017, 2017, 5947859. [Google Scholar] [CrossRef] [PubMed]
- Riddy, D.M.; Delerive, P.; Summers, R.J.; Sexton, P.M.; Langmead, C.J. G Protein-Coupled Receptors Targeting Insulin Resistance, Obesity, and Type 2 Diabetes Mellitus. Pharmacol. Rev. 2018, 70, 39–67. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.P.; Duan, L.; Li, S. Effect of vitamin K2 on type 2 diabetes mellitus: A review. Diabetes Res. Clin. Pract. 2018, 136, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.N.; Hussein, U.K.; Yassa, H.D.; Hassan, S.S.; Rashed, L.A. Synergistic actions of Vitamin D and metformin on skeletal muscles and insulin resistance of Type 2 Diabetic Rats. J. Cell. Physiol. 2018, 233, 5768–5779. [Google Scholar] [CrossRef] [PubMed]
- Awazawa, M.; Gabel, P.; Tsaousidou, E.; Nolte, H.; Kruger, M.; Schmitz, J.; Ackermann, P.J.; Brandt, C.; Altmuller, J.; Motameny, S.; et al. A microRNA screen reveals that elevated hepatic ectodysplasin A expression contributes to obesity-induced insulin resistance in skeletal muscle. Nat. Med. 2017, 23, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Puddu, A.; Sanguineti, R.; Montecucco, F.; Viviani, G.L. Evidence for the gut microbiota short-chain fatty acids as key pathophysiological molecules improving diabetes. Mediat. Inflamm. 2014, 2014, 162021. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, Y.; Kogiso, H.; Hosogi, S.; Tanaka, S.; Shimamoto, C.; Inui, T.; Nakahari, T.; Marunaka, Y. Measurement of [Cl−]i unaffected by the cell volume change using MQAE-based two-photon microscopy in airway ciliary cells of mice. J. Physiol. Sci. 2018, 68, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Shiozaki, A.; Nako, Y.; Ichikawa, D.; Kosuga, T.; Shoda, K.; Arita, T.; Konishi, H.; Komatsu, S.; Kubota, T.; et al. Chloride intracellular channel 1 as a switch among tumor behaviors in human esophageal squamous cell carcinoma. Oncotarget 2018, 9, 23237–23252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogiso, H.; Hosogi, S.; Ikeuchi, Y.; Tanaka, S.; Inui, T.; Marunaka, Y.; Nakahari, T. [Ca2+]i modulation of cAMP-stimulated ciliary beat frequency via PDE1 in airway ciliary cells of mice. Exp. Physiol. 2018, 103, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Kogiso, H.; Hosogi, S.; Ikeuchi, Y.; Tanaka, S.; Shimamoto, C.; Matsumura, H.; Nakano, T.; Sano, K.I.; Inui, T.; Marunaka, Y.; et al. A low [Ca2+]i-induced enhancement of cAMP-activated ciliary beating by PDE1A inhibition in mouse airway cilia. Pflugers Arch. 2017, 469, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Kogiso, H.; Ikeuchi, Y.; Sumiya, M.; Hosogi, S.; Tanaka, S.; Shimamoto, C.; Inui, T.; Marunaka, Y.; Nakahari, T. Seihai-to (TJ-90)-Induced Activation of Airway Ciliary Beatings of Mice: Ca2+ Modulation of cAMP-Stimulated Ciliary Beatings via PDE1. Int. J. Mol. Sci. 2018, 19, 658. [Google Scholar] [CrossRef] [PubMed]
- Kosuga, T.; Shiozaki, A.; Kudou, M.; Yamazato, Y.; Ichikawa, D.; Komatsu, S.; Konishi, H.; Okamoto, K.; Shoda, K.; Arita, T.; et al. Blockade of potassium ion transports enhances hypotonicity-induced cytocidal effects in gastric cancer. Oncotarget 2017, 8, 101394–101405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudou, M.; Shiozaki, A.; Kosuga, T.; Shimizu, H.; Ichikawa, D.; Konishi, H.; Morimura, R.; Komatsu, S.; Ikoma, H.; Fujiwara, H.; et al. Heat shock exerts anticancer effects on liver cancer via autophagic degradation of aquaporin 5. Int. J. Oncol. 2017, 50, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Taruno, A.; Ohmoto, M.; Jyotaki, M.; Lim, J.C.; Miyazaki, H.; Niisato, N.; Marunaka, Y.; Lee, R.J.; Hoff, H.; et al. CALHM3 Is Essential for Rapid Ion Channel-Mediated Purinergic Neurotransmission of GPCR-Mediated Tastes. Neuron 2018, 98, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y. The mechanistic links between insulin and cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. Int. J. Mol. Sci. 2017, 18, 1767. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y. Actions of quercetin, a flavonoid, on ion transporters: Its physiological roles. Ann. N. Y. Acad. Sci. 2017, 1398, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Zhang, J.; Zhang, X.; Liu, J.; Jiang, J.X.; Yamaguchi, K.; Taruno, A.; Katsuyama, M.; Iwata, K.; Ibi, M.; et al. The NOX1 isoform of NADPH oxidase is involved in dysfunction of liver sinusoids in nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2018, 115, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Taruno, A.; Shiraishi, M.; Nakahari, T.; Inui, T.; Sokabe, M.; Eaton, D.C.; Marunaka, Y. Current-direction/amplitude-dependent single channel gating kinetics of mouse pannexin 1 channel: A new concept for gating kinetics. Sci. Rep. 2017, 7, 10512. [Google Scholar] [CrossRef] [PubMed]
- Puppulin, L.; Pezzotti, G.; Sun, H.; Hosogi, S.; Nakahari, T.; Inui, T.; Kumamoto, Y.; Tanaka, H.; Marunaka, Y. Raman micro-spectroscopy as a viable tool to monitor and estimate the ionic transport in epithelial cells. Sci. Rep. 2017, 7, 3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiozaki, A.; Hikami, S.; Ichikawa, D.; Kosuga, T.; Shimizu, H.; Kudou, M.; Yamazato, Y.; Kobayashi, T.; Shoda, K.; Arita, T.; et al. Anion exchanger 2 suppresses cellular movement and has prognostic significance in esophageal squamous cell carcinoma. Oncotarget 2018, 9, 25993–26006. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, A.; Ichikawa, D.; Kosuga, T.; Marunaka, Y.; Otsuji, E. Regulation of osmolality for cancer treatment. J. Physiol. Sci. 2017, 67, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, A.; Kudou, M.; Ichikawa, D.; Fujiwara, H.; Shimizu, H.; Ishimoto, T.; Arita, T.; Kosuga, T.; Konishi, H.; Komatsu, S.; et al. Esophageal cancer stem cells are suppressed by tranilast, a TRPV2 channel inhibitor. J. Gastroenterol. 2018, 53, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, A.; Kudou, M.; Ichikawa, D.; Shimizu, H.; Arita, T.; Kosuga, T.; Konishi, H.; Komatsu, S.; Fujiwara, H.; Okamoto, K.; et al. Expression and role of anion exchanger 1 in esophageal squamous cell carcinoma. Oncotarget 2017, 8, 17921–17935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taruno, A.; Marunaka, Y. Hypotonicity activates a voltage-dependent membrane conductance in N2a neuroblastoma cells. Biochem. Biophys. Res. Commun. 2017, 484, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Taruno, A.; Sun, H.; Nakajo, K.; Murakami, T.; Ohsaki, Y.; Kido, M.A.; Ono, F.; Marunaka, Y. Post-translational palmitoylation controls the voltage gating and lipid raft association of the CALHM1 channel. J. Physiol. 2017, 595, 6121–6145. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hashimoto, Y.; Tanaka, M.; Asano, M.; Yamazaki, M.; Oda, Y.; Toda, H.; Marunaka, Y.; Nakamura, N.; Fukui, M. Sodium-chloride Difference and Metabolic Syndrome: A Population-based Large-scale Cohort Study. Intern. Med. 2016, 55, 3085–3090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudou, M.; Shiozaki, A.; Kosuga, T.; Ichikawa, D.; Konishi, H.; Morimura, R.; Komatsu, S.; Ikoma, H.; Fujiwara, H.; Okamoto, K.; et al. Inhibition of regulatory volume decrease enhances the cytocidal effect of hypotonic shock in hepatocellular carcinoma. J. Cancer 2016, 7, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Ariyoshi, Y.; Shiozaki, A.; Ichikawa, D.; Shimizu, H.; Kosuga, T.; Konishi, H.; Komatsu, S.; Fujiwara, H.; Okamoto, K.; Kishimoto, M.; et al. Na+/H+ exchanger 1 has tumor suppressive activity and prognostic value in esophageal squamous cell carcinoma. Oncotarget 2017, 8, 2209–2223. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y.; Marunaka, R.; Sun, H.; Yamamoto, T.; Kanamura, N.; Taruno, A. Na+ homeostasis by epithelial Na+ channel (ENaC) and Nax channel (Nax): Cooperation of ENaC and Nax. Ann. Transl. Med. 2016, 4 (Suppl. 1), S11. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y.; Marunaka, R.; Sun, H.; Yamamoto, T.; Kanamura, N.; Inui, T.; Taruno, A. Actions of quercetin, a polyphenol, on blood pressure. Molecules 2017, 22, 209. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y.; Niisato, N.; Miyazaki, H.; Nakajima, K.I.; Taruno, A.; Sun, H.; Marunaka, R.; Okui, M.; Yamamoto, T.; Kanamura, N.; et al. Quercetin is a useful medicinal compound showing various actions including control of blood pressure, neurite elongation and epithelial ion transport. Curr. Med. Chem. 2016, 23, 1–12. [Google Scholar] [CrossRef]
- Nakajima, K.I.; Marunaka, Y. Intracellular chloride ion concentration in differentiating neuronal cell and its role in growing neurite. Biochem. Biophys. Res. Commun. 2016, 479, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Sasamoto, K.; Marunaka, R.; Niisato, N.; Sun, H.; Taruno, A.; Pezzotti, G.; Yamamoto, T.; Kanamura, N.; Zhu, W.; Nishio, K.; et al. Analysis of aprotinin, a protease inhibitor, action on the trafficking of epithelial Na+ Channels (ENaC) in renal epithelial cells using a mathematical model. Cell. Physiol. Biochem. 2017, 41, 1865–1880. [Google Scholar] [CrossRef] [PubMed]
- Sasamoto, K.; Niisato, N.; Taruno, A.; Marunaka, Y. Simulation of Cl− secretion in epithelial tissues: New methodology estimating activity of electro-neutral Cl− transporter. Front. Physiol. 2015, 6, 370. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Hosogi, S.; Sawabe, Y.; Shimamoto, C.; Matsumura, H.; Inui, T.; Marunaka, Y.; Nakahari, T. PPARalpha induced NOS1 phosphorylation via PI3K/Akt in guinea pig antral mucous cells: NO-enhancement in Ca2+-regulated exocytosis. Biomed. Res. 2016, 37, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Miyazaki, H.; Shiozaki, A.; Ichikawa, D.; Otsuji, E.; Marunaka, Y. Cytosolic Cl− affects the anticancer activity of paclitaxel in the gastric cancer cell line, MKN28 cell. Cell. Physiol. Biochem. 2017, 42, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Taruno, A.; Marunaka, Y. Neurotransmission of taste mediated by calcium homeostasis modulator ion channels. J. Physiol. Sci. 2017, 67 (Suppl. 1), S15. [Google Scholar]
- Miyazaki, H.; Marunaka, Y. The molecular mechanism of Cl− regulating S-phase cell cycle transition via regulating DNA polymerase activity. J. Physiol. Sci. 2017, 67 (Suppl. 1), S179. [Google Scholar]
- Nakajima, K.; Marunaka, Y. Intracellular chloride ion concentration in growing neurite and its role in neurite outgrowth. J. Physiol. Sci. 2017, 67 (Suppl. 1), S142. [Google Scholar]
- Kuwahara, A.; Kuwahara, Y.; Inui, T.; Marunaka, Y. Regulation of Ion Transport in the Intestine by Free Fatty Acid Receptor 2 and 3: Possible Involvement of the Diffuse Chemosensory System. Int. J. Mol. Sci. 2018, 19, 735. [Google Scholar] [CrossRef] [PubMed]
- Alwood, J.S.; Tran, L.H.; Schreurs, A.S.; Shirazi-Fard, Y.; Kumar, A.; Hilton, D.; Tahimic, C.G.T.; Globus, R.K. Dose- and ion-dependent effects in the oxidative stress response to space-like radiation exposure in the skeletal system. Int. J. Mol. Sci. 2017, 18, 2117. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, T.; Yoshikawa, Y.; Yasui, H. Anti-diabetic effect of organo-chalcogen (sulfur and selenium) Zinc complexes with hydroxy-pyrone derivatives on leptin-deficient Type 2 diabetes model ob/ob mice. Int. J. Mol. Sci. 2017, 18, 2647. [Google Scholar] [CrossRef] [PubMed]
- Aoi, W.; Marunaka, Y. The importance of regulation of body fluid pH in the development and progression of metabolic diseases. In Advances in Medicine and Biology; Berhardt, L.V., Ed.; Nova Publishers: Hauppauge, NY, USA, 2014; Volume 77, pp. 177–189. [Google Scholar]
- Aoi, W.; Marunaka, Y. Importance of pH homeostasis in metabolic health and diseases: Crucial role of membrane proton transport. BioMed Res. Int. 2014, 2014, 598986. [Google Scholar] [CrossRef] [PubMed]
- Hayata, H.; Miyazaki, H.; Niisato, N.; Yokoyama, N.; Marunaka, Y. Lowered extracellular pH is involved in the pathogenesis of skeletal muscle insulin resistance. Biochem. Biophys. Res. Commun. 2014, 445, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.H. Calculating pH and salt dependence of protein-protein binding. Curr. Pharm. Biotechnol. 2008, 9, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; De Franceschi, G.; Frare, E.; Spolaore, B.; Zambonin, M.; Chiti, F.; de Laureto, P.P.; Fontana, A. Amyloid fibril formation and disaggregation of fragment 1–29 of apomyoglobin: Insights into the effect of pH on protein fibrillogenesis. J. Mol. Biol. 2007, 367, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Aoi, W.; Hosogi, S.; Niisato, N.; Yokoyama, N.; Hayata, H.; Miyazaki, H.; Kusuzaki, K.; Fukuda, T.; Fukui, M.; Nakamura, N.; et al. Improvement of insulin resistance, blood pressure and interstitial pH in early developmental stage of insulin resistance in OLETF rats by intake of propolis extracts. Biochem. Biophys. Res. Commun. 2013, 432, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Marunaka, Y.; Aoi, W.; Hosogi, S.; Niisato, N.; Yokoyama, N.; Hayata, H.; Miyazaki, H.; Kusuzaki, K.; Taruno, A.; Nomura, T. What is the role of interstitial pH in diabetes mellitus? Improving action of propolis on type II diabetes mellitus via pH regulation. Int. J. Mol. Med. 2013, 32 (Suppl. 1), S50. [Google Scholar]
- Marunaka, Y. Cl− and H+ as mediators of biofunction and biodysfunction in health and disease. J. Physiol. Sci. 2016, 66, S4. [Google Scholar]
- Marunaka, Y. Regulation of body environments by epithelial ion transport. J. Physiol. Sci. 2017, 67, S6. [Google Scholar]
- Curry, F.R. Microvascular solute and water transport. Microcirculation 2005, 12, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Levick, J.R.; Michel, C.C. Microvascular fluid exchange and the revised Starling principle. Cardiovasc. Res. 2010, 87, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, R.K.; Rubin, K. Transcapillary exchange: Role and importance of the interstitial fluid pressure and the extracellular matrix. Cardiovasc. Res. 2010, 87, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, C.H. Volume-targeted therapy of increased intracranial pressure. Acta Neurochir. Suppl. 2003, 86, 355–360. [Google Scholar] [PubMed]
- Li, R.; Guan, M.X. Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol. Cell. Biol. 2010, 30, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, S.; Yang, F. Mitochondria in the pathogenesis of diabetes: A proteomic view. Protein Cell. 2012, 3, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Candeias, E.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Insulin signaling, glucose metabolism and mitochondria: Major players in Alzheimer’s disease and diabetes interrelation. Brain Res. 2012, 1441, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P.; Gaudel, C.; Krause, M. Mitochondria and diabetes. An intriguing pathogenetic role. Adv. Exp. Med. Biol. 2012, 942, 235–247. [Google Scholar] [PubMed]
- Martin, S.D.; McGee, S.L. The role of mitochondria in the aetiology of insulin resistance and type 2 diabetes. Biochim. Biophys. Acta 2014, 1840, 1303–1312. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Emrick, L.T.; Hsu, J.W.; Chanprasert, S.; Jahoor, F.; Scaglia, F.; Craigen, W.J. Glucose metabolism derangements in adults with the MELAS m.3243A>G mutation. Mitochondrion 2014, 18, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeva-Andany, M.; Lopez-Ojen, M.; Funcasta-Calderon, R.; Ameneiros-Rodriguez, E.; Donapetry-Garcia, C.; Vila-Altesor, M.; Rodriguez-Seijas, J. Comprehensive review on lactate metabolism in human health. Mitochondrion 2014, 17c, 76–100. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Tocchetti, C.G.; Bhatt, N.; Paolocci, N.; Cortassa, S. Protective mechanisms of mitochondria and heart function in diabetes. Antioxid. Redox Signal. 2015, 22, 1563–1586. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Dolinsky, V.W.; Mughal, W.; Gordon, G.R.; McGavock, J. Targeting skeletal muscle mitochondria to prevent type 2 diabetes in youth. Biochem. Cell Biol. 2015, 93, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Flemming, N.B.; Gallo, L.A.; Ward, M.S.; Forbes, J.M. Tapping into mitochondria to find novel targets for diabetes complications. Curr. Drug Targets 2016, 17, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Hesselink, M.K.; Schrauwen-Hinderling, V.; Schrauwen, P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Persson, B. Determination of plasma acetoacetate and D-beta-hydroxybutyrate in new-born infants by an enzymatic fluorometric micro-method. Scand. J. Clin. Lab. Investig. 1970, 25, 9–18. [Google Scholar] [CrossRef]
- Gosmanov, A.R.; Gosmanova, E.O.; Dillard-Cannon, E. Management of adult diabetic ketoacidosis. Diabetes Metab. Syndr. Obes. 2014, 7, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Role of beta-hydroxybutyrate, its polymer poly-beta-hydroxybutyrate and inorganic polyphosphate in mammalian health and disease. Front. Physiol. 2014, 5, 260. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.A.; Moffat, M.P. Modulation of sodium-hydrogen exchange activity in cardiac myocytes during acidosis and realkalinisation: Effects on calcium, pHi, and cell shortening. Cardiovasc. Res. 1995, 29, 247–253. [Google Scholar] [CrossRef]
- Park, C.O.; Xiao, X.H.; Allen, D.G. Changes in intracellular Na+ and pH in rat heart during ischemia: Role of Na+/H+ exchanger. Am. J. Physiol. 1999, 276, H1581–H1590. [Google Scholar] [CrossRef] [PubMed]
- Loh, S.H.; Chen, W.H.; Chiang, C.H.; Tsai, C.S.; Lee, G.C.; Jin, J.S.; Cheng, T.H.; Chen, J.J. Intracellular pH regulatory mechanism in human atrial myocardium: Functional evidence for Na+/H+ exchanger and Na+/HCO3− symporter. J. Biomed. Sci. 2002, 9, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Hosogi, S.; Miyazaki, H.; Nakajima, K.; Ashihara, E.; Niisato, N.; Kusuzaki, K.; Marunaka, Y. An inhibitor of Na+/H+ exchanger (NHE), ethyl-isopropyl amiloride (EIPA), diminishes proliferation of MKN28 human gastric cancer cells by decreasing the cytosolic Cl− concentration via DIDS-sensitive pathways. Cell. Physiol. Biochem. 2012, 30, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Hosogi, S.; Kusuzaki, K.; Inui, T.; Wang, X.; Marunaka, Y. Cytosolic chloride ion is a key factor in lysosomal acidification and function of autophagy in human gastric cancer cell. J. Cell. Mol. Med. 2014, 18, 1124–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marunaka, Y. Roles of ionic environments in growth of human cancer cell and potentials of ion transporter blockers in cancer therapies. J. Gastrointest. Digest. Syst. 2014, 3, 163. [Google Scholar] [CrossRef]
- Bonen, A. Lactate transporters (MCT proteins) in heart and skeletal muscles. Med. Sci. Sports Exerc. 2000, 32, 778–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juel, C. Lactate-proton cotransport in skeletal muscle. Physiol. Rev. 1997, 77, 321–358. [Google Scholar] [CrossRef] [PubMed]
- Kourtoglou, G.I. Insulin therapy and exercise. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. 1), S73–S77. [Google Scholar] [CrossRef]
- Garcia, C.K.; Goldstein, J.L.; Pathak, R.K.; Anderson, R.G.; Brown, M.S. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: Implications for the Cori cycle. Cell 1994, 76, 865–873. [Google Scholar] [CrossRef]
- Xu, A.S.; Kuchel, P.W. Characterisation of erythrocyte transmembrane exchange of trifluoroacetate using 19F-NMR: Evidence for transport via the monocarboxylate transporter. Biochim. Biophys. Acta 1993, 1150, 35–44. [Google Scholar] [CrossRef]
- Poole, R.C.; Halestrap, A.P. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am. J. Physiol. 1993, 264, C761–C782. [Google Scholar] [CrossRef] [PubMed]
- Juel, C.; Holten, M.K.; Dela, F. Effects of strength training on muscle lactate release and MCT1 and MCT4 content in healthy and type 2 diabetic humans. J. Physiol. 2004, 556, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opitz, D.; Lenzen, E.; Schiffer, T.; Hermann, R.; Hellmich, M.; Bloch, W.; Brixius, K.; Brinkmann, C. Endurance training alters skeletal muscle MCT Contents in T2DM men. Int. J. Sports Med. 2014, 35, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Brenachot, X.; Ramadori, G.; Ioris, R.M.; Veyrat-Durebex, C.; Altirriba, J.; Aras, E.; Ljubicic, S.; Kohno, D.; Fabbiano, S.; Clement, S.; et al. Hepatic protein tyrosine phosphatase receptor gamma links obesity-induced inflammation to insulin resistance. Nat. Commun. 2017, 8, 1820. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Stockli, J.; Fazakerley, D.J.; James, D.E. GLUT4 exocytosis. J. Cell Sci. 2011, 124, 4147–4159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, B.J.; Chai, S.Y.; Albiston, A.L. GLUT4 associated proteins as therapeutic targets for diabetes. Recent Pat. Endocr. Metab. Immune Drug Discov. 2011, 5, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Souto, G.; Donapetry, C.; Calvino, J.; Adeva, M.M. Metabolic acidosis-induced insulin resistance and cardiovascular risk. Metab. Syndr. Relat. Disord. 2011, 9, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Mandel, E.I.; Curhan, G.C.; Hu, F.B.; Taylor, E.N. Plasma bicarbonate and risk of type 2 diabetes mellitus. Cmaj 2012, 184, E719–E725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuki, M.; Kitamura, T.; Goya, K.; Saito, H.; Mukai, M.; Kasayama, S.; Shimomura, I.; Koga, M. Association of urine acidification with visceral obesity and the metabolic syndrome. Endocr. J. 2011, 58, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maalouf, N.M.; Cameron, M.A.; Moe, O.W.; Adams-Huet, B.; Sakhaee, K. Low urine pH: A novel feature of the metabolic syndrome. Clin. J. Am. Soc. Nephrol. 2007, 2, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, N.M.; Cameron, M.A.; Moe, O.W.; Sakhaee, K. Metabolic basis for low urine pH in type 2 diabetes. Clin. J. Am. Soc. Nephrol. 2010, 5, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.A.; Maalouf, N.M.; Adams-Huet, B.; Moe, O.W.; Sakhaee, K. Urine composition in type 2 diabetes: Predisposition to uric acid nephrolithiasis. J. Am. Soc. Nephrol. 2006, 17, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Farwell, W.R.; Taylor, E.N. Serum bicarbonate, anion gap and insulin resistance in the National Health and Nutrition Examination Survey. Diabet. Med. 2008, 25, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.S. Beware the low urine pH―The major cause of the increased prevalence of nephrolithiasis in the patient with type 2 diabetes. Diabetes Obes. Metab. 2012, 14, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Dwyer, B.J.; Forbes, S.; Thiel, D.H.; Lewis, P.J.; Ramadori, G. Insulin production and resistance in different models of diet-Induced obesity and metabolic syndrome. Int. J. Mol. Sci. 2017, 18, 285. [Google Scholar] [CrossRef] [PubMed]
- Bellini, S.; Barutta, F.; Mastrocola, R.; Imperatore, L.; Bruno, G.; Gruden, G. Heat shock proteins in vascular diabetic complications: Review and future perspective. Int. J. Mol. Sci. 2017, 18, 2709. [Google Scholar] [CrossRef] [PubMed]
- Devarshi, P.P.; McNabney, S.M.; Henagan, T.M. Skeletal muscle nucleo-mitochondrial crosstalk in obesity and type 2 diabetes. Int. J. Mol. Sci. 2017, 18, 831. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, S.; Kuwabara, T. Diabetes-induced dysfunction of mitochondria and stem cells in skeletal muscle and the nervous system. Int. J. Mol. Sci. 2017, 18, 2147. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Cao, H.; Li, Z.; Fang, J.; Wei, X.; Cheng, P.; Jiao, R.; Liu, X.; Li, Y.; Xing, Y.; et al. A novel multi-epitope vaccine based on urate transporter 1 alleviates streptozotocin-induced diabetes by producing anti-URAT1 antibody and an immunomodulatory effect in C57BL/6J mice. Int. J. Mol. Sci. 2017, 18, 2137. [Google Scholar] [CrossRef] [PubMed]
- Buoite Stella, A.; Gortan Cappellari, G.; Barazzoni, R.; Zanetti, M. Update on the impact of omega 3 fatty acids on inflammation, insulin resistance and sarcopenia: A review. Int. J. Mol. Sci. 2018, 19, 218. [Google Scholar] [CrossRef] [PubMed]
- Hayata, H.; Miyazaki, H.; Niisato, N.; Yokoyama, N.; Marunaka, Y. Involvement of the extracellular pH in skeletal muscle insulin resistance. J. Physiol. Sci. 2013, 63 (Suppl. 1), S199. [Google Scholar]
- Gancheva, S.; Jelenik, T.; Alvarez-Hernandez, E.; Roden, M. Interorgan Metabolic Crosstalk in Human Insulin Resistance. Physiol. Rev. 2018, 98, 1371–1415. [Google Scholar] [CrossRef] [PubMed]
- Pajor, A.M. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflugers Arch. 2014, 466, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Irving, B.A.; Wood, G.C.; Bennotti, P.N.; Babu, E.; Deshpande, A.; Lent, M.R.; Petrick, A.; Gabrielsen, J.; Strodel, W.; Gerhard, G.S.; et al. Nutrient transporter expression in the jejunum in relation to body mass index in patients undergoing bariatric surgery. Nutrients 2016, 8, 683. [Google Scholar] [CrossRef] [PubMed]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-Chain Fatty Acid Transporters: Role in Colonic Homeostasis. Compr. Physiol. 2017, 8, 299–314. [Google Scholar] [PubMed]
- Wang, J.; Qin, C.; He, T.; Qiu, K.; Sun, W.; Zhang, X.; Jiao, N.; Zhu, W.; Yin, J. Alfalfa-containing diets alter luminal microbiota structure and short chain fatty acid sensing in the caecal mucosa of pigs. J. Anim. Sci. Biotechnol. 2018, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, A.R.; Crisma, A.R.; Masi, L.N.; Amaral, C.L.; Marzuca-Nassr, G.N.; Bomfim, L.H.M.; Teodoro, B.G.; Queiroz, A.L.; Serdan, T.D.A.; Torres, R.P.; et al. Attenuation of obesity and insulin resistance by fish oil supplementation is associated with improved skeletal muscle mitochondrial function in mice fed a high-fat diet. J. Nutr. Biochem. 2018, 55, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Ebisu, G.; Hosogi, S.; Marunaka, Y. Improvement of blood glucose level in diabetic rats by intake of citrate. In Proceedings of the 7th Asian Congress of Diabetes: The Rise of Nutrition and Diabetes in Asia, Hong Kong, China, 6–8 July 2018; p. 170. [Google Scholar]
- Marunaka, Y. Effects of Ninjin’yoeito on insulin resistance via improvement of the interstitial fluid pH. Jpn. J. Geriatr. 2018, 55 (Suppl. 1), S188. [Google Scholar]








© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marunaka, Y. The Proposal of Molecular Mechanisms of Weak Organic Acids Intake-Induced Improvement of Insulin Resistance in Diabetes Mellitus via Elevation of Interstitial Fluid pH. Int. J. Mol. Sci. 2018, 19, 3244. https://doi.org/10.3390/ijms19103244
Marunaka Y. The Proposal of Molecular Mechanisms of Weak Organic Acids Intake-Induced Improvement of Insulin Resistance in Diabetes Mellitus via Elevation of Interstitial Fluid pH. International Journal of Molecular Sciences. 2018; 19(10):3244. https://doi.org/10.3390/ijms19103244
Chicago/Turabian StyleMarunaka, Yoshinori. 2018. "The Proposal of Molecular Mechanisms of Weak Organic Acids Intake-Induced Improvement of Insulin Resistance in Diabetes Mellitus via Elevation of Interstitial Fluid pH" International Journal of Molecular Sciences 19, no. 10: 3244. https://doi.org/10.3390/ijms19103244