Gas Signaling Molecules and Mitochondrial Potassium Channels



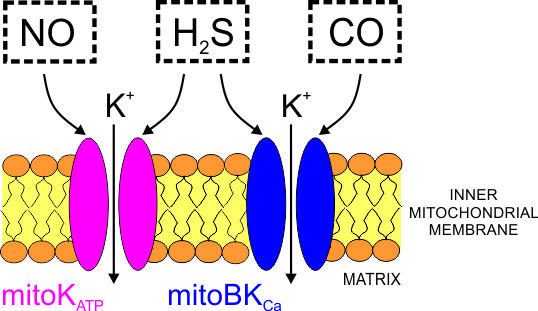
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Carbon Monoxide
2.1. Physiological Sources of Carbon Monoxide-Hemoxygenases
2.2. Signal Transduction via Heme-CO Interaction
2.3. Regulation of BKCa Channels by CO
2.4. KATP Channels
2.5. Pharmacological CO Donors—CORMs
3. Hydrogen Sulfide
3.1. Physiological Sources of Hydrogen Sulfide
3.2. Pharmacological Donors of H2S
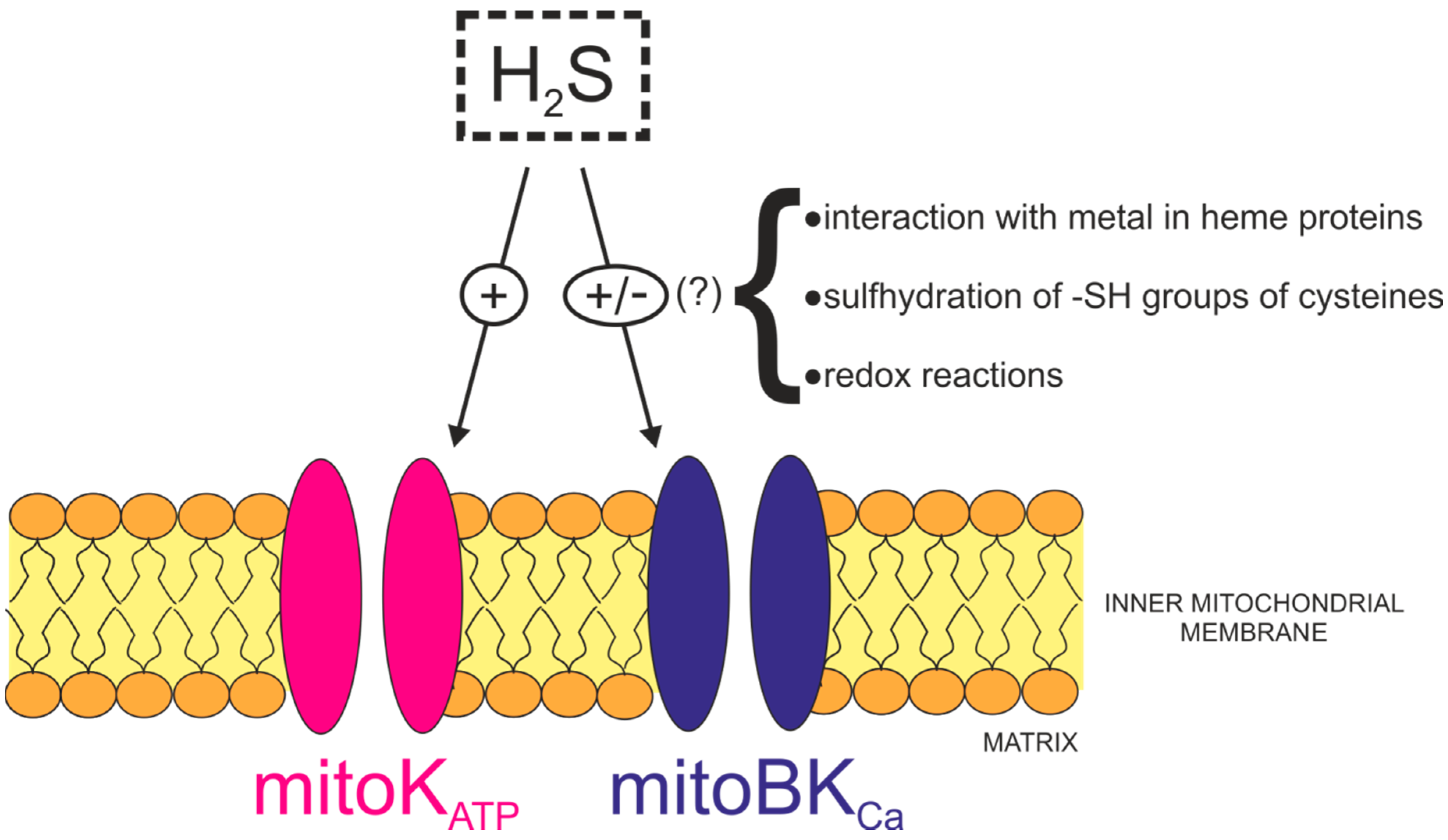
3.3. H2S Regulation of Heme Proteins
3.4. H2S Regulation via Protein S-Sulfhydration
3.5. Potassium Channels as Targets for H2S
4. Nitric Oxide
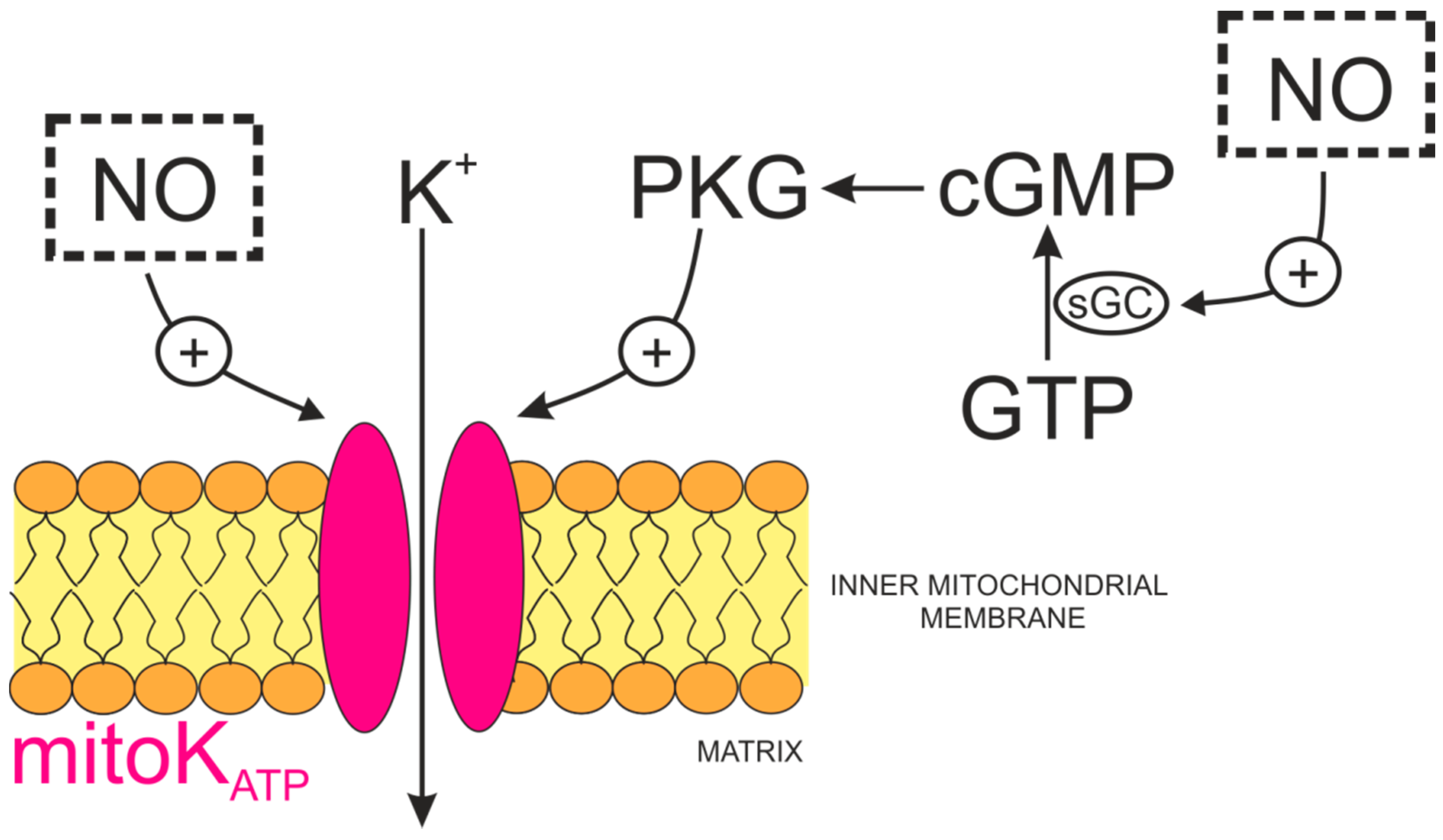
4.1. The Role and Physiological Sources of Nitric Oxide
4.2. Regulation of Mitochondrial KATP Channels by Nitric Oxide
5. Final Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Smith, C.O.; Nehrke, K.; Brookes, P.S. The Slo(w) path to identifying the mitochondrial channels responsible for ischemic protection. Biochem. J. 2017, 474, 2067–2094. [Google Scholar] [CrossRef] [PubMed]
- Krabbendam, I.E.; Honrath, B.; Culmsee, C.; Dolga, A.M. Mitochondrial Ca(2+)-activated K(+) channels and their role in cell life and death pathways. Cell Calcium 2018, 69, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, Q.L.; Wang, G.Z.; Deng, T.Z.; Chen, R.; Liu, M.H.; Wang, S.W. The protective roles of mitochondrial ATP-sensitive potassium channels during hypoxia-ischemia-reperfusion in brain. Neurosci. Lett. 2011, 491, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, Y.; Wang, S.; McDonald, T.; Van Eyk, J.E.; Sidor, A.; O’Rourke, B. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science 2002, 298, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Akao, M.; O’Rourke, B.; Marbán, E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca(2+) overload during simulated ischemia and reperfusion: Possible mechanism of cardioprotection. Circ. Res. 2001, 89, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabò, I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leanza, L.; Trentin, L.; Becker, K.A.; Frezzato, F.; Zoratti, M.; Semenzato, G.; Szabò, I. Clofazimine, Psora-4 and PAP-1, inhibitors of the potassium channel Kv1.3, as a new and selective therapeutic strategy in chronic lymphocytic leukemia. Leukemia 2013, 27, 1782–1785. [Google Scholar] [CrossRef] [PubMed]
- Quast, S.A.; Berger, A.; Buttstadt, N.; Friebel, K.; Schonherr, R.; Eberle, J. General sensitization of melanoma cells for TRAIL-induced apoptosis by the potassium channel inhibitor TRAM-34 depends on release of SMAC. PLoS ONE 2013, 7, e39290. [Google Scholar] [CrossRef] [PubMed]
- Matkovic, K.; Koszela-Piotrowska, I.; Jarmuszkiewicz, W.; Szewczyk, A. Ion conductance pathways in potato tuber (Solanum tuberosum) inner mitochondrial membrane. Biochim. Biophys. Acta 2011, 1807, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Kicinska, A.; Swida, A.; Bednarczyk, P.; Koszela-Piotrowska, I.; Choma, K.; Dolowy, K.; Szewczyk, A.; Jarmuszkiewicz, W. ATP-sensitive potassium channel in mitochondria of the eukaryotic microorganism Acanthamoeba castellanii. J. Biol. Chem. 2007, 282, 17433–17441. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, M.; Kicinska, A.; Szewczyk, A.; Jarmuszkiewicz, W. Mitochondrial large-conductance potassium channel from Dictyostelium discoideum. Int. J. Biochem. Cell Biol. 2015, 60, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Paucek, P.; Yarov-Yarovoy, V.; Murray, H.N.; Darbenzio, R.B.; D’Alonzo, A.J.; Lodge, N.J.; Smith, M.A.; Grover, G.J. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels: Possible mechanism of cardioprotection. Circ. Res. 1997, 81, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Siemen, D.; Loupatatzis, C.; Borecky, J.; Gulbins, E.; Lang, F. Ca2+-activated K channel of the BK-type in the inner mitochondrial membrane of a human glioma cell line. Biochem. Biophys. Res. Commun. 1999, 257, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Skalska, J.; Piwońska, M.; Wyroba, E.; Surmacz, L.; Wieczorek, R.; Koszela-Piotrowska, I.; Zielińska, J.; Bednarczyk, P.; Dołowy, K.; Wilczynski, G.M.; et al. A novel potassium channel in skeletal muscle mitochondria. Biochim. Biophys. Acta 2008, 1777, 651–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaggar, J.H.; Li, A.L.; Parfenova, H.; Liu, J.X.; Umstot, E.S.; Dopico, A.M.; Leffler, C.W. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ. Res. 2005, 97, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Dallas, M.L.; Boyle, J.P.; Milligan, C.J.; Sayer, R.; Kerrigan, T.L.; McKinstry, C.; Lu, P.; Mankouri, J.; Harris, M.; Scragg, J.L.; et al. Carbon monoxide protects against oxidant-induced apoptosis via inhibition of Kv2.1. FASEB J. 2011, 25, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Duckles, H.; Al-Owais, M.M.; Elies, J.; Johnson, E.; Boycott, H.E.; Dallas, M.L.; Porter, K.E.; Boyle, J.P.; Scragg, J.L.; Peers, C. T-Type Ca2+ channel regulation by CO: A mechanism for control of cell proliferation. Adv. Exp. Med. Biol. 2015, 860, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, W.J.; Gadeberg, H.C.; Harrison, A.W.; Allen, N.D.; Riccardi, D.; Kemp, P.J. Carbon monoxide is a rapid modulator of recombinant and native P2X(2) ligand-gated ion channels. Br. J. Pharmacol. 2009, 158, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, W.J.; Kemp, P.J. Carbon monoxide: An emerging regulator of ion channels. J. Physiol. 2011, 589, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Von Burg, R. Carbon monoxide. J. Appl. Toxicol. 1999, 19, 379–386. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From metabolism to molecular therapy. Am. J. Respir. Cell Mol. Biol. 2009, 41, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharm. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, K.T.; Shang, M.; Wu, R.X.; Chang, H.C.; Khechaduri, A.; Sato, T.; Kamide, C.; Liu, T.; Prasad, S.V.N.; Ardehali, H. Increased heme levels in the heart lead to exacerbated ischemic injury. J. Am. Heart Assoc. 2015, 4, e002272. [Google Scholar] [CrossRef] [PubMed]
- Barañano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Converso, D.P.; Taille, C.; Carreras, M.C.; Jaitovich, A.; Poderoso, J.J.; Boczkowski, J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 2006, 20, 1236–1238. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W. Metals and their scaffolds to promote difficult enzymatic reactions. Chem. Rev. 2006, 106, 3317–3337. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, E.L.; Ray, P.D.; Tsuji, Y. Role and regulation of ferritin H in rotenone-mediated mitochondrial oxidative stress. Free Radic. Biol. Med. 2008, 44, 1762–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity—Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Landar, A.; Zmijewski, J.W.; Dickinson, D.A.; Le Goffe, C.; Johnson, M.S.; Milne, G.L.; Zanoni, G.; Vidari, G.; Morrow, J.D.; Darley-Usmar, V.M. Interaction of electrophilic lipid oxidation products with mitochondria in endothelial cells and formation of reactive oxygen species. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1777–H1787. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Keyse, S.M.; Applegate, L.A.; Tromvoukis, Y.; Tyrrell, R.M. Oxidant stress leads to transcriptional activation of the human hem oxygenase gene in cultured skin fibroblasts. Mol. Cell. Biol. 1990, 10, 4967–4969. [Google Scholar] [CrossRef] [PubMed]
- Paschen, W.; Uto, A.; Djuricic, B.; Schmitt, J. Hemeoxygenase expression after reversible ischemia of rat-brain. Neurosci. Lett. 1994, 180, 5–8. [Google Scholar] [CrossRef]
- Maines, M.D.; Trakshel, G.M.; Kutty, R.K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 1986, 261, 411–419. [Google Scholar] [PubMed]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Higashimoto, Y.; Hara, T.; Sagara, Y.; Noguchi, M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene 2004, 336, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.D.; Chen, C.S.; Nguyen, J.; Thayanithy, V.; Subramanian, S.; Steer, C.J.; Vercellotti, G.M. Regulation of heme oxygenase-1 protein expression by miR-377 in combination with miR-217. J. Biol. Chem. 2011, 286, 3194–3202. [Google Scholar] [CrossRef] [PubMed]
- Applegate, L.A.; Luscher, P.; Tyrrell, R.M. Induction of heme oxygenase—A general response to oxidant stress in cultured-mammalian-cells. Cancer Res. 1991, 51, 974–978. [Google Scholar] [PubMed]
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein-induced in human-skin fibroblasts by UVA radiation, hydrogen-peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Barupala, D.P.; Dzul, S.P.; Riggs-Gelasco, P.J.; Stemmler, T.L. Synthesis, delivery and regulation of eukaryotic heme and Fe-S cluster cofactors. Arch. Biochem. Biophys. 2016, 592, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Pae, H.O.; Chung, H.T. Heme oxygenase-1: Its therapeutic roles in inflammatory diseases. Immune Netw. 2009, 9, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Ng, M.K.C.; Stocker, R. Haem oxygenase-I and cardiovascular disease: mechanisms and therapeutic potential. Clin. Sci. 2011, 120, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Pal, C.; Dey, S.; Goyal, M.; Alam, A.; Iqbal, M.S.; Dutta, S.; Sarkar, S.; Kumar, R.; Maity, P.; et al. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J. Biol. Chem. 2011, 286, 39387–39402. [Google Scholar] [CrossRef] [PubMed]
- Slebos, D.J.; Ryter, S.W.; van der Toorn, M.; Liu, F.; Guo, F.; Baty, C.J.; Karlsson, J.M.; Watkins, S.C.; Kim, H.P.; Wang, X.; et al. Mitochondrial localization and function of heme oxygenase-7 in cigarette smoke-induced cell death. Am. J. Respir. Cell Mol. Biol. 2007, 36, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Bolisetty, S.; Traylor, A.; Zarjou, A.; Johnson, M.S.; Benavides, G.A.; Ricart, K.; Boddu, R.; Moore, R.D.; Landar, A.; Barnes, S.; et al. Mitochondria-targeted heme oxygenase-1 decreases oxidative stress in renal epithelial cells. Am. J. Physiol. Ren. Physiol. 2013, 305, F255–F264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, S.; Biswas, G.; Avadhani, N.G. Mitochondria-targeted heme oxygenase-1 induces oxidative stress and mitochondrial dysfunction in macrophages, kidney fibroblasts and in chronic alcohol hepatotoxicity. Redox Biol. 2014, 2, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Regan, R.F.; Guo, Y.P.; Kumar, N. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci. Lett. 2000, 282, 1–4. [Google Scholar] [CrossRef]
- Dore, S.; Goto, S.; Sampei, K.; Blackshaw, S.; Hester, L.D.; Ingi, T.; Sawa, A.; Traystman, R.J.; Koehler, R.C.; Snyder, S.H. Heme oxygenase-2 acts to prevent neuronal death in brain cultures and following transient cerebral ischemia. Neuroscience 2000, 99, 587–592. [Google Scholar] [CrossRef]
- Yi, L.; Jenkins, P.M.; Leichert, L.I.; Jakob, U.; Martens, J.R.; Ragsdale, S.W. Heme regulatory motifs in heme oxygenase-2 form a thiol/disulfide redox switch that responds to the cellular redox state. J. Biol. Chem. 2009, 284, 20556–20561. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Ragsdale, S.W. Evidence that the heme regulatory motifs in heme oxygenase-2 serve as a thiol/isulfide redox switch regulating heme binding. J. Biol. Chem. 2007, 282, 21056–21067. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Rupp, M.; Hofmann, S.; Mittal, M.; Fuchs, B.; Sommer, N.; Parajuli, N.; Quanz, K.; Schubert, D.; Dony, E.; et al. Heme oxygenase-2 and large-conductance Ca(2+)-activated K(+) channels lung vascular effects of hypoxia. Am. J. Respir. Crit. Care Med. 2009, 180, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 544, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruginsk, S.G.; Mecawi, A.D.; da Silva, M.P.; Reis, W.L.; Coletti, R.; de Lima, J.B.M.; Elias, L.L.K.; Antunes-Rodrigues, J. Gaseous modulators in the control of the hypothalamic neurohypophyseal system. Physiology 2015, 30, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Schafer, A.I. Carbon monoxide and vascular cell function. Int. J. Mol. Med. 1998, 2, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M.K. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Perrella, M.A.; Lee, M.E.; Kourembanas, S. Smooth-muscle cell-derived carbon-monoxide is a regulator of vascular cGMP. Proc. Natl. Acad. Sci. USA 1995, 92, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Brune, B.; Schmidt, K.U.; Ullrich, V. Activation of soluble guanylate-cyclase by carbon-monoxide and inhibition by superoxide anion. Eur. J. Biochem. 1990, 192, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Seya, K.; Motomura, S.; Furukawa, K. Cardiac mitochondrial cGMP stimulates cytochrome c release. Clin. Sci. 2007, 112, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Seya, K.; Ono, K.; Fujisawa, S.; Okumura, K.; Motomura, S.; Furukawa, K. Cytosolic Ca2+-induced apoptosis in rat cardiomyocytes via mitochondrial NO-cGMP-protein kinase G pathway. J. Pharmacol. Exp. Ther. 2013, 344, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Behmenburg, F.; Dorsch, M.; Huhn, R.; Mally, D.; Heinen, A.; Hollmann, M.W.; Berger, M.M. Impact of mitochondrial Ca2+-sensitive potassium (mBK(Ca)) channels in sildenafil-induced cardioprotection in rats. PLoS ONE 2015, 10, e0144737. [Google Scholar] [CrossRef] [PubMed]
- Nara, M.; Dhulipala, P.D.K.; Ji, G.J.; Kamasani, U.R.; Wang, Y.X.; Matalon, S.; Kotlikoff, M.I. Guanylyl cyclase stimulatory coupling to K-Ca channels. Am. J. Physiol. Cell Physiol. 2000, 279, C1938–C1945. [Google Scholar] [CrossRef] [PubMed]
- Nara, M.; Dhulipala, P.D.K.; Kotlikoff, M.I. cGMP-dependent protein kinase (PKG) modulation of maxi-K channels expressed in Xenopus oocytes. Am. J. Respir. Crit. Care Med. 1999, 159, A723. [Google Scholar]
- Fukao, M.; Mason, H.S.; Britton, F.C.; Kenyon, J.L.; Horowitz, B.; Keef, K.D. Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J. Biol. Chem. 1999, 274, 10927–10935. [Google Scholar] [CrossRef] [PubMed]
- Frankenreiter, S.; Bednarczyk, P.; Kniess, A.; Bork, N.I.; Straubinger, J.; Koprowski, P.; Wrzosek, A.; Mohr, E.; Logan, A.; Murphy, M.P.; et al. cGMP-elevating compounds and ischemic conditioning provide cardioprotection against ischemia and reperfusion injury via cardiomyocyte-specific BKCa channels. Circulation 2017, 136, 2337–2355. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.D.; Xu, R.; Reynolds, M.F.; Garcia, M.L.; Heinemann, S.H.; Hoshi, T. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature 2003, 425, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Bowman, S.E.J.; Bren, K.L. The chemistry and biochemistry of heme c: Functional bases for covalent attachment. Nat. Prod. Rep. 2008, 25, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.E.J.; Wootton, P.; Mason, H.S.; Bould, J.; Iles, D.E.; Riccardi, D.; Peers, C.; Kemp, P.J. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science 2004, 306, 2093–2097. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Morgan, J.T.; Ragsdale, S.W. Identification of a thiol/disulfide redox switch in the human BK channel that controls its affinity for heme and CO. J. Biol. Chem. 2010, 285, 20117–20127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhong, H.; Vollmer, C.; Nurse, C.A. Co-release of ATP and ACh mediates hypoxic signalling at rat carotid body chemoreceptors. J. Physiol. 2000, 525, 143–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczara, P.; Motterlini, R.; Rosen, G.M.; Augustynek, B.; Bednarczyk, P.; Szewczyk, A.; Foresti, R.; Chlopicki, S. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: A role for mitoBKCa channels. Biochim. Biophys. Acta 2015, 1847, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.E.; Naughton, P.; Shurey, S.; Green, C.J.; Johnson, T.R.; Mann, B.E.; Foresti, R.; Motterlini, R. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ. Res. 2003, 93, e2–e8. [Google Scholar] [CrossRef] [PubMed]
- Soni, H.; Patel, P.; Rath, A.C.; Jain, M.; Mehta, A.A. Cardioprotective effect with carbon monoxide releasing molecule-2 (CORM-2) in isolated perfused rat heart: Role of coronary endothelium and underlying mechanism. Vasc. Pharmacol. 2010, 53, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Burton, M.J.; Kapetanaki, S.M.; Chernova, T.; Jamieson, A.G.; Dorlet, P.; Santolini, J.; Moody, P.C.E.; Mitcheson, J.S.; Davies, N.W.; Schmid, R.; et al. A heme-binding domain controls regulation of ATP-dependent potassium channels. Proc. Natl. Acad. Sci. USA 2016, 113, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Noma, A. ATP-regulated K+ channels in cardiac muscle. Nature 1983, 305, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Zingman, L.V.; Hodgson, D.M.; Bast, P.H.; Kane, G.C.; Perez-Terzic, C.; Gumina, R.J.; Pucar, D.; Bienengraeber, M.; Dzeja, P.P.; Miki, T.; et al. Kir6.2 is required for adaptation to stress. Proc. Natl. Acad. Sci. USA 2002, 99, 13278–13283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapetanaki, S.M.; Burton, M.J.; Basran, J.; Uragami, C.; Moody, P.C.E.; Mitcheson, J.S.; Schmid, R.; Davies, N.W.; Dorlet, P.; Vos, M.H.; et al. A mechanism for CO regulation of ion channels. Nat. Commun. 2018, 9, 907. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Kroboth, S.L.; Pu, J.L.; Sims, J.J.; Aggarwal, N.T.; McNally, E.M.; Makielski, J.C.; Shi, N.Q. Molecular identification and functional characterization of a mitochondrial sulfonylurea receptor 2 splice variant generated by intraexonic splicing. Circ. Res. 2009, 105, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Tanaka, O.; Sekiguchi, M.; He, H.J.; Yasuoka, Y.; Itoh, H.; Kawahara, K.; Abe, H. ATP-sensitive K+-channel subunits on the mitochondria and endoplasmic reticulum of rat cardiomyocytes. J. Histochem. Cytochem. 2005, 53, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.E.; Schwarzer, S.; Duchen, M.R.; Tinker, A. The intracellular localization and function of the ATP-sensitive K+ channel subunit Kir6.1. J. Membr. Biol. 2010, 234, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Kuniyasu, A.; Kaneko, K.; Kawahara, K.; Nakayama, H. Molecular assembly and subcellular distribution of ATP-sensitive potassium channel proteins in rat hearts. FEBS Lett. 2003, 552, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Foster, D.B.; Rucker, J.J.; Marban, E. Is Kir6.1 a subunit of mitoK(ATP)? Biochem. Biophys. Res. Commun. 2008, 366, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.B.; Ho, A.S.; Rucker, J.; Garlid, A.O.; Chen, L.; Sidor, A.; Garlid, K.D.; O’Rourke, B. Mitochondrial ROMK channel is a molecular component of mitoK(ATP). Circ. Res. 2012, 111, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Kottelat, E.; Zobi, F. Visible light-activated photoCORMs. Inorganics 2017, 5, 24. [Google Scholar] [CrossRef]
- Kottelat, E.; Ruggi, A.; Zobi, F. Red-light activated photoCORMs of Mn(I) species bearing electron deficient 2,2′-azopyridines. Dalton Trans. 2016, 45, 6920–6927. [Google Scholar] [CrossRef] [PubMed]
- Ruggi, A.; Zobi, F. Quantum-CORMs: Quantum dot sensitized CO releasing molecules. Dalton Trans. 2015, 44, 10928–10931. [Google Scholar] [CrossRef] [PubMed]
- Zobi, F. CO and CO-releasing molecules in medicinal chemistry. Future Med. Chem. 2013, 5, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Zobi, F.; Quaroni, L.; Santoro, G.; Zlateva, T.; Blacque, O.; Sarafimov, B.; Schaub, M.C.; Bogdanova, A.Y. Live-fibroblast IR imaging of a cytoprotective photoCORM activated with visible light. J. Med. Chem. 2013, 56, 6719–6731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, K.; Men, F.; Wang, W.C.; Zhou, Y.Q.; Zhang, H.W.; Ye, D.W. Carbon monoxide and its controlled release: Therapeutic application, detection, and development of carbon monoxide releasing molecules (CORMs). J. Med. Chem. 2018, 61, 2611–2635. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Wareham, L.K.; McLean, S.; Begg, R.; Greaves, S.; Mann, B.E.; Sanguinetti, G.; Poole, R.K. CO-releasing molecules have nonheme targets in bacteria: Transcriptomic, mathematical modeling and biochemical analyses of CORM-3 Ru(CO)(3)Cl(glycinate) actions on a heme-deficient mutant of Escherichia coli. Antioxid. Redox Signal. 2015, 23, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.F.A.; Seixas, J.D.; Coelho, A.C.; Mukhopadhyay, A.; Reis, P.M.; Romao, M.J.; Romao, C.C.; Santos-Silva, T. New insights into the chemistry of fac- Ru(CO)(3) (2+) fragments in biologically relevant conditions: The CO releasing activity of Ru(CO)(3)Cl-2(1,3-thiazole), and the X-ray crystal structure of its adduct with lysozyme. J. Inorg. Biochem. 2012, 117, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Riddle, M.A.; Walker, B.R. Regulation of endothelial BK channels by heme oxygenase-derived carbon monoxide and caveolin-1. Am. J. Physiol. Cell Physiol. 2012, 303, C92–C101. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.L.; Zhang, Y.; Lin, D.H.; Chen, J.; Patschan, S.; Goligorsky, M.S.; Nasjletti, A.; Yang, B.F.; Wang, W.H. Carbon monoxide stimulates the Ca2+-activated big conductance K channels in cultured human endothelial cells. Hypertension 2007, 50, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Yue, P.; Lin, D.H.; Wang, W.H. Carbon monoxide stimulates Ca2+-dependent big-conductance K channels in the cortical collecting duct. Am. J. Physiol. Ren. Physiol. 2013, 304, F543–F552. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.C.; Liu, B.L.; Zhang, Q.Y.; Wu, X.A.; Yu, Y.; Jesse, F.F.; Li, L.M. Carbon monoxide augments electrical signaling in cultured neural networks of hippocampal neurons partly through activation of BKCa channels. Acta Biochim. Biophys. Sin. 2015, 47, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.E.; Brazier, S.P.; Baban, N.; Telezhkin, V.; Muller, C.T.; Riccardi, D.; Kemp, P.J. A structural motif in the C-terminal tail of slo1 confers carbon monoxide sensitivity to human BK(Ca) channels. Pflug. Arch. Eur. J. Physiol. 2008, 456, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.W.; Xu, R.; Heinemann, S.H.; Hoshi, T. The RCK1 high-affinity Ca2+ sensor confers carbon monoxide sensitivity to Slo1 BK channels. Proc. Natl. Acad. Sci. USA 2008, 105, 4039–4043. [Google Scholar] [CrossRef] [PubMed]
- Gessner, G.; Sahoo, N.; Swain, S.M.; Hirth, G.; Schonherr, R.; Mede, R.; Westerhausen, M.; Brewitz, H.H.; Heimer, P.; Imhof, D.; et al. CO-independent modification of K+ channels by tricarbonyldichlororuthenium(II) dimer (CORM-2). Eur. J. Pharmacol. 2017, 815, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Jara-Oseguera, A.; Ishida, I.G.; Rangel-Yescas, G.E.; Espinosa-Jalapa, N.; Perez-Guzman, J.A.; Elias-Vinas, D.; Le Lagadec, R.; Rosenbaum, T.; Islas, L.D. Uncoupling charge movement from channel opening in voltage-gated potassium channels by ruthenium complexes. J. Biol. Chem. 2011, 286, 16414–16425. [Google Scholar] [CrossRef] [PubMed]
- Horrigan, F.T.; Heinemann, S.H.; Hoshi, T. Heme regulates allosteric activation of the Slo1 BK channel. J. Gen. Physiol. 2005, 126, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; McLean, S.; Begg, R.; Rana, N.; Ali, S.; Kendall, J.J.; Sanguinetti, G.; Mann, B.E.; Poole, R.K. The broad-spectrum antimicrobial potential of [Mn(CO)4(S2CNMe(CH2CO2H))], a water-soluble CO-Releasing Molecule (CORM-401): Intracellular accumulation, transcriptomic and statistical analyses, and membrane polarization. Antioxid. Redox Signal. 2018, 28, 1286–1308. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathna, N.; Washington, K.; Bashur, C.; Liao, Y. Nonmetallic carbon monoxide releasing molecules (CORMs). Org. Biomol. Chem. 2017, 15, 8692–8699. [Google Scholar] [CrossRef] [PubMed]
- Winyard, M.W.; Paul, G. Hydrogen sulfide and inflammation: The good, the bad, the ugly and the promising. Expert Rev. Clin. Pharmacol. 2011, 4, 13–32. [Google Scholar] [CrossRef]
- Dunn, W.R.; Alexander, S.P.; Ralevic, V.; Roberts, R.E. Effects of hydrogen sulphide in smooth muscle. Pharmacol. Ther. 2016, 158, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, Q.; Liu, X.; Pan, L.; Xiong, Q.; Zhu, Y.Z. Hydrogen sulfide protects against apoptosis under oxidative stress through SIRT1 pathway in H9c2 cardiomyocytes. Nitric Oxide 2015, 46, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, Q.; Guo, W.; Zhu, Y.Z. Hydrogen sulfide attenuates cardiac dysfunction in a rat model of heart failure: A mechanism through cardiac mitochondrial protection. Biosci. Rep. 2011, 31, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Maclean, K.; Kraus, J. Hydrogen sulfide production and metabolism in mammalian tissues. Signal Transduct. Gasotransmitters 2004, 275–292. [Google Scholar] [CrossRef]
- Shibuya, N.; Koike, S.; Tanaka, M.; Ishigami-Yuasa, M.; Kimura, Y.; Ogasawara, Y.; Fukui, K.; Nagahara, N.; Kimura, H. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 2013, 4, 1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Zhang, W.; Wu, L.; Yang, G.; Li, H.; Wang, R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc. Natl. Acad. Sci. USA 2012, 109, 2943–2948. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Keller, G.A.; Subramani, S. Identification of peroxisomal targeting signals located at the carboxy terminus of four peroxisomal proteins. J. Cell Biol. 1988, 107, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H(2)S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2017, 149, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Sodha, N.R.; Clements, R.T.; Feng, J.; Liu, Y.; Bianchi, C.; Horvath, E.M.; Szabo, C.; Sellke, F.W. The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia-reperfusion injury. Eur. J. Cardio Thorac. Surg. 2008, 33, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Kloesch, B.; Liszt, M.; Steiner, G.; Broll, J. Inhibitors of p38 and ERK1/2 MAPkinase and hydrogen sulphide block constitutive and IL-1beta-induced IL-6 and IL-8 expression in the human chondrocyte cell line C-28/I2. Rheumatol. Int. 2012, 32, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Esechie, A.; Kiss, L.; Olah, G.; Horvath, E.M.; Hawkins, H.; Szabo, C.; Traber, D.L. Protective effect of hydrogen sulfide in a murine model of acute lung injury induced by combined burn and smoke inhalation. Clin. Sci. 2008, 115, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Biggs, T.D.; Xian, M. Hydrogen sulfide (H2S) releasing agents: Chemistry and biological applications. Chem. Commun. 2014, 50, 11788–11805. [Google Scholar] [CrossRef] [PubMed]
- Gerő, D.; Torregrossa, R.; Perry, A.; Waters, A.; Le-Trionnaire, S.; Whatmore, J.L.; Wood, M.; Whiteman, M. The novel mitochondria-targeted hydrogen sulfide (H2S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol. Res. 2016, 113, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen sulfide and polysulfides as signaling molecules. Proc. Jpn. Acad. Ser. B 2015, 91, 131–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collman, J.P.; Ghosh, S.; Dey, A.; Decréau, R.A. Using a functional enzyme model to understand the chemistry behind hydrogen sulfide induced hibernation. Proc. Natl. Acad. Sci. USA 2009, 106, 22090–22095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modis, K.; Coletta, C.; Erdelyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013, 27, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. H2S: A novel gasotransmitter that signals by sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [PubMed]
- Módis, K.; Ju, Y.; Ahmad, A.; Untereiner, A.A.; Altaany, Z.; Wu, L.; Szabo, C.; Wang, R. S-sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol. Res. 2016, 113, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Du, J.; Tang, C.; Huang, Y.; Jin, H. H2S-induced sulfhydration: Biological function and detection methodology. Front. Pharmacol. 2017, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Greiner, R.; Pálinkás, Z.; Bäsell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.Z. Hydrogen sulfide—A potent multichannel anti-arrhythmic drug. J. Cardiovasc. Dis. Res. 2010, 1, 37–39. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Xu, Y.; Kawano, T.; Hendon, T.; Baki, L.; Garai, S.; Papapetropoulos, A.; Thakur, G.A.; Plant, L.D.; Logothetis, D.E. Hydrogen sulfide inhibits Kir2 and Kir3 channels by decreasing sensitivity to the phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2). J. Biol. Chem. 2018, 293, 3546–3561. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Wu, L.; Liang, W.; Wang, R. Direct stimulation of K(ATP) channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol. Pharmacol. 2005, 68, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Testai, L.; Marino, A.; Piano, I.; Brancaleone, V.; Tomita, K.; Di Cesare Mannelli, L.; Martelli, A.; Citi, V.; Breschi, M.C.; Levi, R.; et al. The novel H2S-donor 4-carboxyphenyl isothiocyanate promotes cardioprotective effects against ischemia/reperfusion injury through activation of mitoKATP channels and reduction of oxidative stress. Pharmacol. Res. 2016, 113, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Sivarajah, A.; Collino, M.; Yasin, M.; Benetti, E.; Gallicchio, M.; Mazzon, E.; Cuzzocrea, S.; Fantozzi, R.; Thiemermann, C. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 2009, 31, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Sitdikova, G.F.; Weiger, T.M.; Hermann, A. Hydrogen sulfide increases calcium-activated potassium (BK) channel activity of rat pituitary tumor cells. Pflug. Arch. Eur. J. Physiol. 2010, 459, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Sitdikova, G.F.; Fuchs, R.; Kainz, V.; Weiger, T.M.; Hermann, A. Phosphorylation of BK channels modulates the sensitivity to hydrogen sulfide (H2S). Front. Physiol. 2014, 5, 431. [Google Scholar] [CrossRef] [PubMed]
- Telezhkin, V.; Brazier, S.P.; Cayzac, S.; Muller, C.T.; Riccardi, D.; Kemp, P.J. Hydrogen sulfide inhibits human BK(Ca) channels. Adv. Exp. Med. Biol. 2009, 648, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Dempsey, S.K.; Daneva, Z.; Azam, M.; Li, N.; Li, P.L.; Ritter, J.K. Role of nitric oxide in the cardiovascular and renal systems. Int. J. Mol. Sci. 2018, 19, 2605. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Knaub, L.A.; Olivera-Fragoso, L.F.; Keller, A.C.; Balasubramaniam, V.; Watson, P.A.; Reusch, J.E. Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1624–H1633. [Google Scholar] [CrossRef] [PubMed]
- Elfering, S.L.; Sarkela, T.M.; Giulivi, C. Biochemistry of mitochondrial nitric-oxide synthase. J. Biol. Chem. 2002, 277, 38079–38086. [Google Scholar] [CrossRef] [PubMed]
- Haynes, V.; Elfering, S.; Traaseth, N.; Giulivi, C. Mitochondrial nitric-oxide synthase: enzyme expression, characterization, and regulation. J. Bioenerg. Biomembr. 2004, 36, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995, 369, 136–139. [Google Scholar] [CrossRef] [Green Version]
- Packer, M.A.; Porteous, C.M.; Murphy, M.P. Superoxide production by mitochondria in the presence of nitric oxide forms peroxynitrite. Biochem. Mol. Biol. Int. 1996, 40, 527–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radi, R.; Rodriguez, M.; Castro, L.; Telleri, R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch. Biochem. Biophys. 1994, 308, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Holmuhamedov, E.L.; Wang, L.; Terzic, A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J. Physiol. 1999, 519, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Katakam, P.V.; Dutta, S.; Sure, V.N.; Grovenburg, S.M.; Gordon, A.O.; Peterson, N.R.; Rutkai, I.; Busija, D.W. Depolarization of mitochondria in neurons promotes activation of nitric oxide synthase and generation of nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1097–H1106. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Sato, T.; Ohler, A.; O’Rourke, B.; Marban, E. Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation 2000, 101, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Ljubkovic, M.; Shi, Y.; Cheng, Q.; Bosnjak, Z.; Jiang, M.T. Cardiac mitochondrial ATP-sensitive potassium channel is activated by nitric oxide in vitro. FEBS Lett. 2007, 581, 4255–4259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlem, Y.A.; Horn, T.F.; Buntinas, L.; Gonoi, T.; Wolf, G.; Siemen, D. The human mitochondrial KATP channel is modulated by calcium and nitric oxide: A patch-clamp approach. Biochim. Biophys. Acta 2004, 1656, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Zoga, V.; Kimura, M.; Liang, M.Y.; Wu, H.E.; Gemes, G.; McCallum, J.B.; Kwok, W.M.; Hogan, Q.H.; Sarantopoulos, C.D. Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: action by direct S-nitrosylation. Mol. Pain 2009, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Duarte, E.; Trujillo, X.; Huerta, M.; Ortiz-Mesina, M.; Cortés-Rojo, C.; Manzo-Ávalos, S.; Saavedra-Molina, A.; Montoya-Pérez, R. Mitochondrial KATP channels in skeletal muscle are protein kinases C and G and nitric oxide synthase involved in the fatigue process. Open Access Anim. Physiol. 2012, 4, 21–28. [Google Scholar] [CrossRef]
- Zhang, D.M.; Chai, Y.P.; Erickson, J.R.; Brown, J.H.; Bers, D.M.; Lin, Y.F. Intracellular signalling mechanism responsible for modulation of sarcolemmal ATP sensitive potassium channels by nitric oxide in ventricular cardiomyocytes. J. Physiol. 2014, 592, 971–990. [Google Scholar] [CrossRef] [PubMed]
- Mio, Y.; Shim, Y.H.; Richards, E.; Bosnjak, Z.J.; Pagel, P.S.; Bienengraeber, M. Xenon preconditioning: The role of prosurvival signaling, mitochondrial permeability transition and bioenergetics in rats. Anesth. Analg. 2009, 108, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lian, C.; Zhou, R.; Li, T.; Xiang, X.; Liu, B. Pretreatment with xenon protected immature rabbit heart from ischaemia/reperfusion injury by opening of the mitoKATP channel. Heart Lung Circ. 2013, 22, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Cui, H.; Du, J.B. Sulfur dioxide: A physiologic endothelium derived relaxing factor. Histol. Histopathol. 2017, 32, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.B.; Du, J.B.; Cui, H. Signal pathways involved in the biological effects of sulfur dioxide. Eur. J. Pharmacol. 2015, 764, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Gasotransmitters: Growing pains and joys. Trends Biochem. Sci. 2014, 39, 227–232. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walewska, A.; Szewczyk, A.; Koprowski, P. Gas Signaling Molecules and Mitochondrial Potassium Channels. Int. J. Mol. Sci. 2018, 19, 3227. https://doi.org/10.3390/ijms19103227
Walewska A, Szewczyk A, Koprowski P. Gas Signaling Molecules and Mitochondrial Potassium Channels. International Journal of Molecular Sciences. 2018; 19(10):3227. https://doi.org/10.3390/ijms19103227
Chicago/Turabian StyleWalewska, Agnieszka, Adam Szewczyk, and Piotr Koprowski. 2018. "Gas Signaling Molecules and Mitochondrial Potassium Channels" International Journal of Molecular Sciences 19, no. 10: 3227. https://doi.org/10.3390/ijms19103227
APA StyleWalewska, A., Szewczyk, A., & Koprowski, P. (2018). Gas Signaling Molecules and Mitochondrial Potassium Channels. International Journal of Molecular Sciences, 19(10), 3227. https://doi.org/10.3390/ijms19103227