Histone Deacetylases Enhance Ca2+-Activated K+ Channel KCa3.1 Expression in Murine Inflammatory CD4+ T Cells

 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
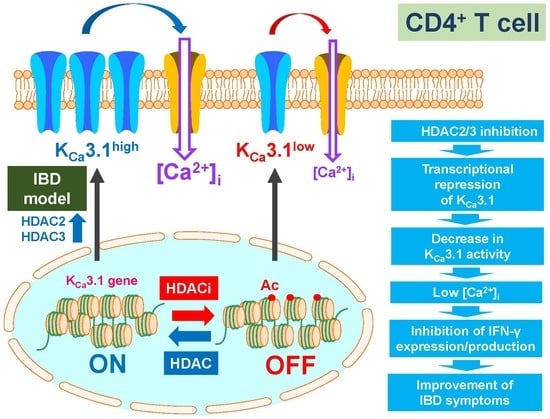
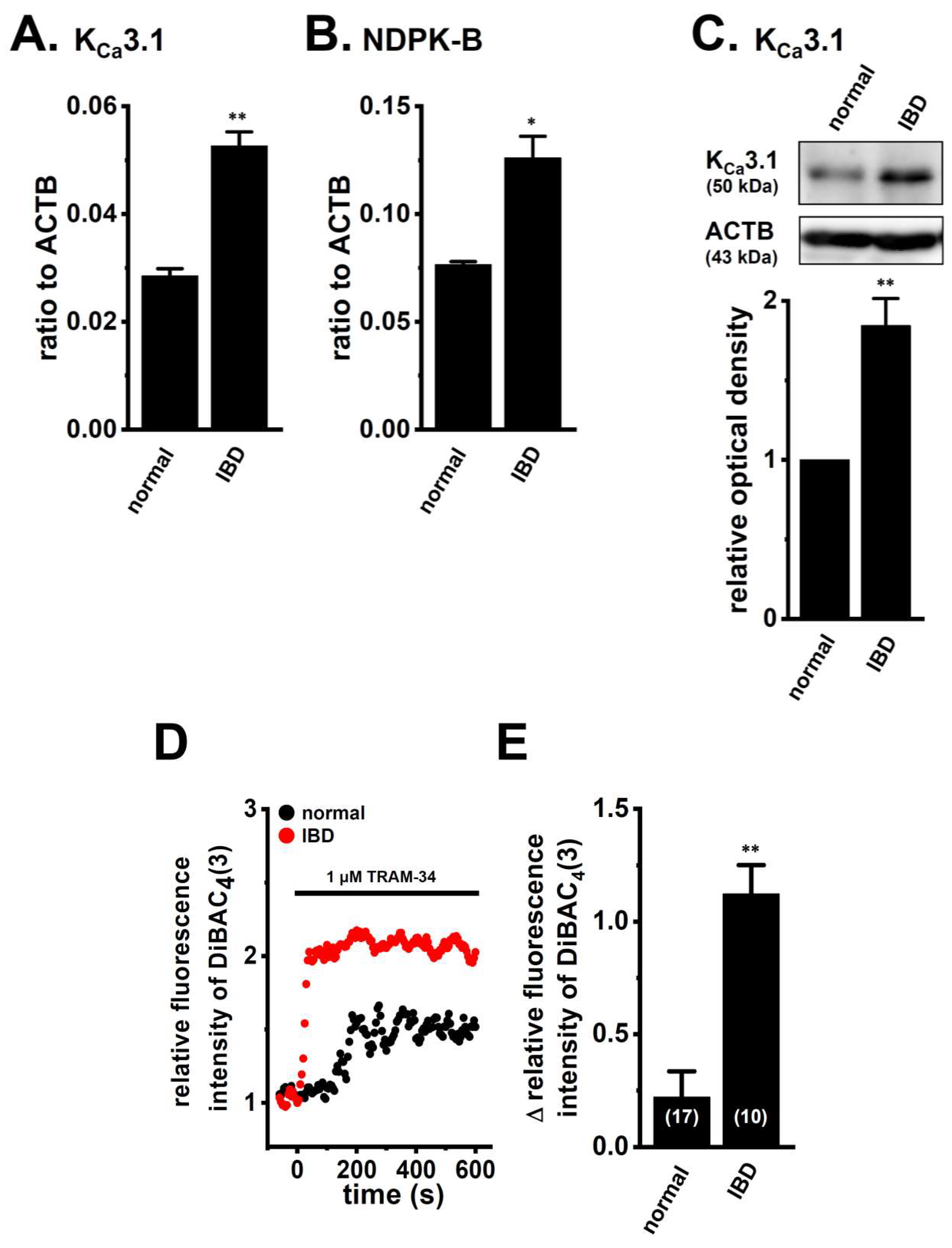
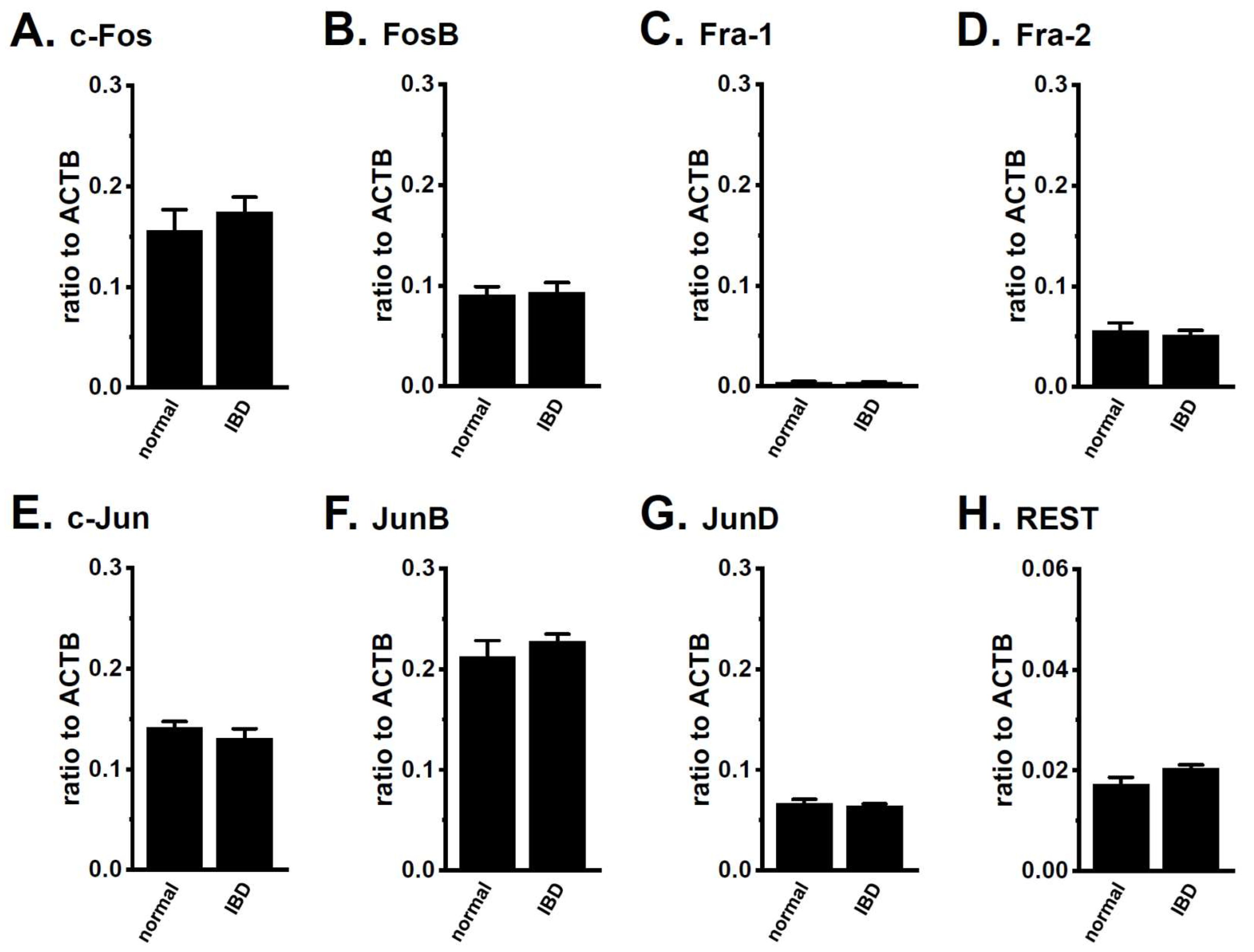
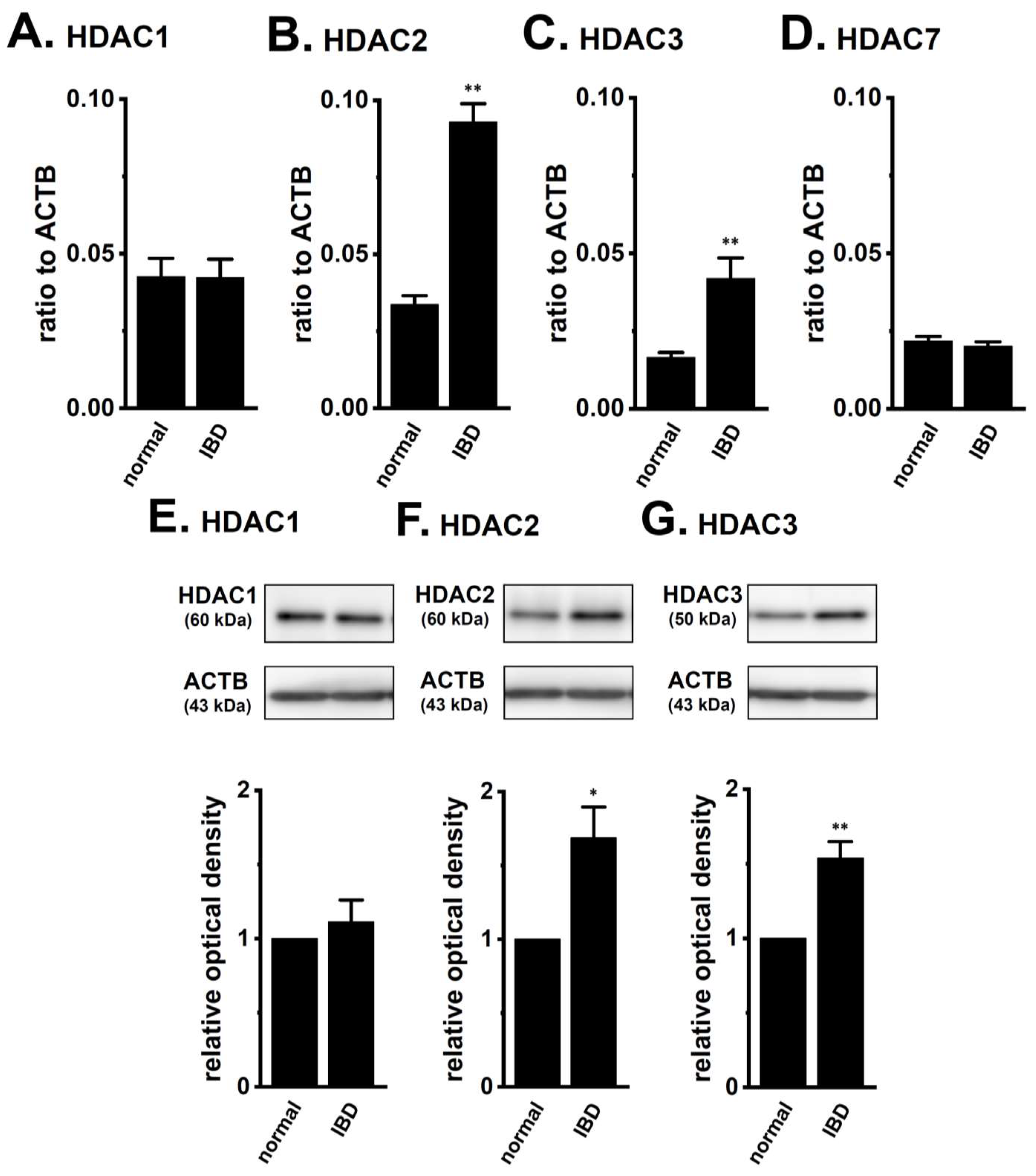
2.1. Up-Regulation of HDAC2 and 3 in CD4+ T Cells of Dextran Sulfate Sodium (DSS)-Induced Inflammatory Bowel Disease (IBD) Model Mice
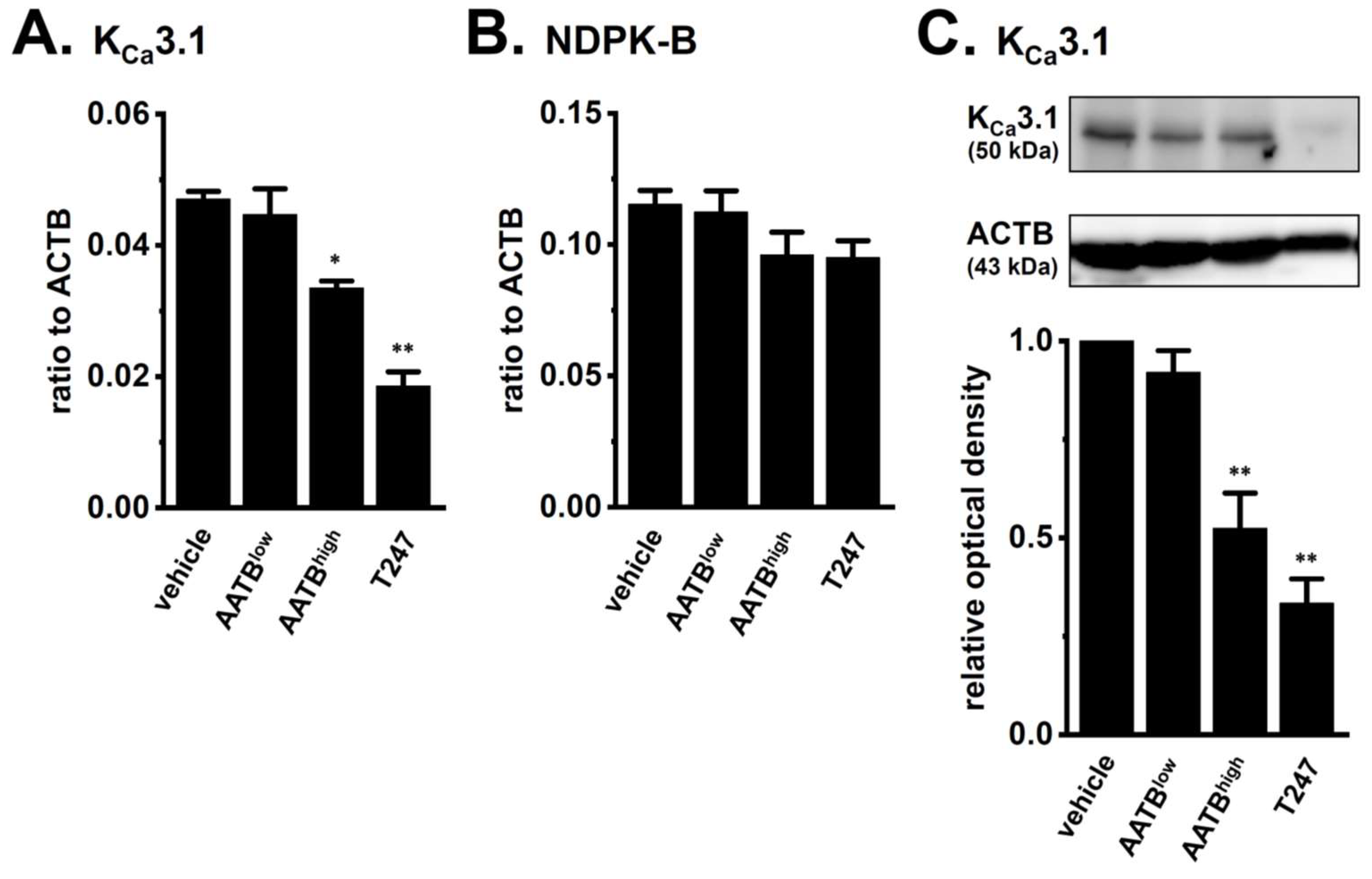
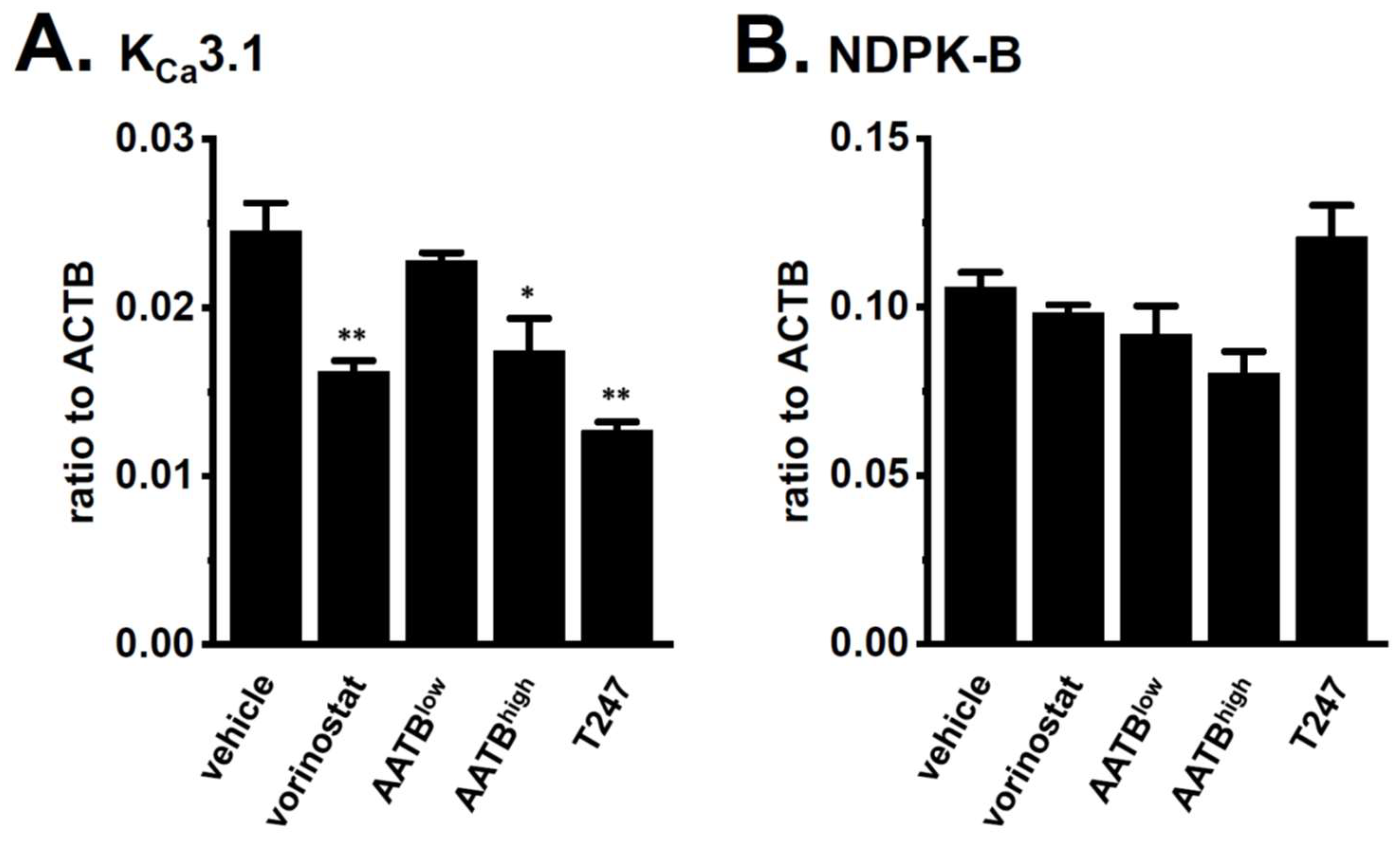
2.2. Decreased Expression and Activity of KCa3.1 by Treatment with the Pan-HDACi, Vorinostat, and HDAC2- and HDAC3-Selective HDACis in CD4+ T Cells of IBD Model Mice
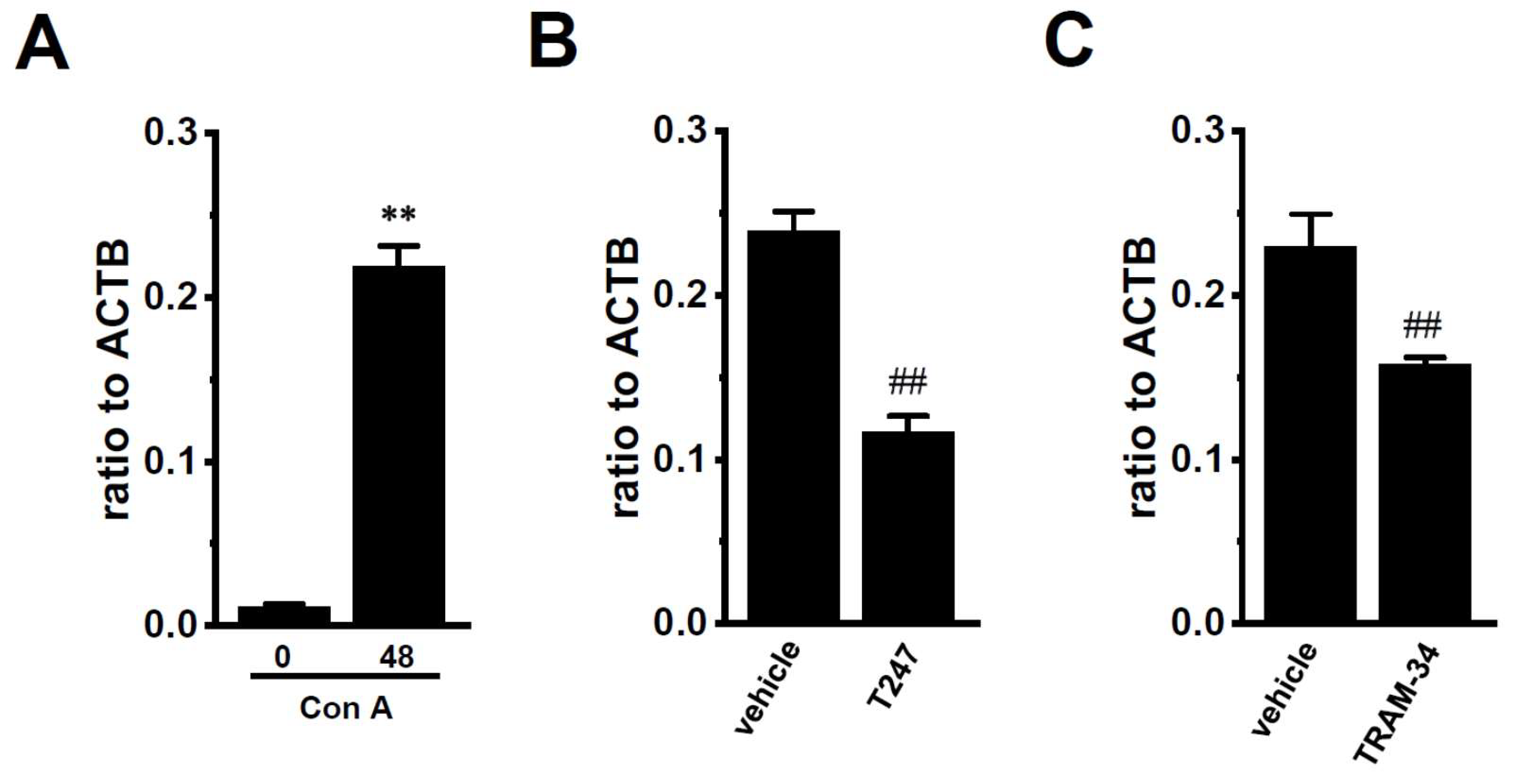
2.3. Decreased Expression Levels of KCa3.1 by the Inhibition of HDAC2 and HDAC3 in Con A-Stimulated Mouse Thymocytes
3. Discussion
4. Materials and Methods
4.1. Preparation of a DSS-Induced Mouse IBD Model and Isolation of CD4+ T Cells Using Dynabeads
4.2. RNA Extraction, Reverse Transcription (RT)-PCR, and Real-Time PCR
4.3. Western Blotting
4.4. Measurement of Membrane Potentials Using Fluorescent Voltage-Sensitive Dyes
4.5. Chemicals
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Cahalan, M.D.; Chandy, K.G. The functional network of ion channels in T lymphocytes. Immunol. Rev. 2009, 231, 59–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Wulff, H. The lymphocyte potassium channels KV1.3 and KCa3.1 as targets for immunosuppression. Drug Dev. Res. 2011, 72, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Kito, H. Ca2+-activated K+ channel KCa3.1 as a therapeutic target for immune disorders. Biol. Pharm. Bull. 2018, 41, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Srivastava, S.; Zhdanova, O.; Ding, Y.; Li, Z.; Wulff, H.; Lafaille, M.; Skolnik, E.Y. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proc. Natl. Acad. Sci. USA 2010, 107, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.K. The role of T cell potassium channels, KV1.3 and KCa3.1, in the inflammatory cascade in ulcerative colitis. Dan. Med. J. 2014, 61, B4946. [Google Scholar] [PubMed]
- Strøbæk, D.; Brown, D.T.; Jenkins, D.P.; Chen, Y.J.; Coleman, N.; Ando, Y.; Chiu, P.; Jørgensen, S.; Demnitz, J.; Wulff, H.; et al. NS6180, a new KCa3.1 channel inhibitor prevents T-cell activation and inflammation in a rat model of inflammatory bowel disease. Br. J. Pharmacol. 2013, 168, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Fukuyo, Y.; Kito, H.; Shibaoka, R.; Matsui, M.; Niguma, H.; Maeda, Y.; Yamamura, H.; Fujii, M.; Kimura, K.; et al. Up-regulation of KCa3.1 K+ channel in mesenteric lymph node CD4+ T-lymphocytes from a mouse model of dextran sodium sulfate-induced inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G873–G885. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Caioni, M.; Müller, M.; Strauch, U.; Kunst, C.; Woelfel, G. K+ channel inhibition differentially regulates migration of intestinal epithelial cells in inflamed vs. non-inflamed conditions in a PI3K/Akt-mediated manner. PLoS ONE 2016, 11, e0147736. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Srivastava, S.; Zhdanowa, O.; Sun, Y.; Li, Z.; Skolnik, E.Y. Nucleoside diphosphate kinase B knock-out mice have impaired activation of the K+ channel KCa3.1, resulting in defective T cell activation. J. Biol. Chem. 2010, 285, 38765–38771. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Li, Z.; Lin, L.; Liu, G.; Ko, K.; Coetzee, W.A.; Skolnik, E.Y. The phosphatidylinositol 3-phosphate phosphatase myotubularin-related protein 6 (MTMR6) is a negative regulator of the Ca2+-activated K+ channel KCa3.1. Mol. Cell. Biol. 2005, 25, 3630–3638. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Li, Z.; Ko, K.; Choudhury, P.; Albaqumi, M.; Johnson, A.K.; Yan, Y.; Backer, J.M.; Unutmaz, D.; Coetzee, W.A.; et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol. Cell 2006, 24, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Zhdanova, O.; Di, L.; Li, Z.; Albaqumi, M.; Wulff, H.; Skolnik, E.Y. Protein histidine phosphatase 1 negatively regulates CD4 T cells by inhibiting the K+ channel KCa3.1. Proc. Natl. Acad. Sci. USA 2008, 105, 14442–14446. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Srivastava, S.; Li, Z.; Vaeth, M.; Fuhs, S.R.; Hunter, T.; Skolnik, E.Y. Identification of PGAM5 as a mammalian protein histidine phosphatase that plays a central role to negatively regulate CD4+ T cells. Mol. Cell 2016, 63, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Ghanshani, S.; Wulff, H.; Miller, M.J.; Rohm, H.; Neben, A.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J. Biol. Chem. 2000, 275, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.P.; Wang, Y.; Zhao, L.M.; Li, G.R.; Deng, X.L. Angiotensin II upregulates KCa3.1 channels and stimulates cell proliferation in rat cardiac fibroblasts. Biochem. Pharmacol. 2013, 85, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Gole, H.K.; Tharp, D.L.; Bowles, D.K. Upregulation of intermediate-conductance Ca2+-activated K+ channels (KCNN4) in porcine coronary smooth muscle requires NADPH oxidase 5 (NOX5). PLoS ONE 2014, 9, e105337. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Lively, S.; Schlichter, L.C. IL-4 type 1 receptor signaling up-regulates KCNN4 expression, and increases the KCa3.1 current and its contribution to migration of alternative-activated microglia. Front. Cell Neurosci. 2014, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Cheong, A.; Bingham, A.J.; Li, J.; Kumar, B.; Sukumar, P.; Muncsch, C.; Buckley, J.; Neylon, C.B.; Porter, K.E.; Beech, D.J.; et al. Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol. Cell 2005, 20, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Niwa, S.; Kojima, Y.; Sasaki, S.; Sakuragi, M.; Kohri, K.; Imaizumi, Y. Intermediate-conductance Ca2+-activated K+ channel, KCa3.1, as a novel therapeutic target for benign prostatic hyperplasia. J. Pharmacol. Exp. Ther. 2011, 338, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Kanatsuka, S.; Hatano, N.; Kito, H.; Matsui, A.; Fujimoto, M.; Matsuba, S.; Niwa, S.; Zhan, P.; Suzuki, T.; et al. Downregulation of the Ca2+-activated K+ channel KCa3.1 by histone deacetylase inhibition in human breast cancer cells. Pharmacol. Res. Perspect. 2016, 4, e00228. [Google Scholar] [CrossRef] [PubMed]
- Glauben, R.; Siegmund, B. Inhibition of histone deacetylases in inflammatory bowel diseases. Mol. Med. 2011, 17, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.N.; Choijookhuu, N.; Takagi, H.; Srisowanna, N.; Nguyen Nhat Huynh, M.; Yamaguchi, Y.; Synn Oo, P.; Tin Htwe Kyaw, M.; Sato, K.; Yamaguchi, R.; et al. The HDAC inhibitor, SAHA, prevents colonic inflammation by suppressing pro-inflammatory cytokines and chemokines in DSS-induced colitis. Acta Histochem. Cytochem. 2018, 51, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Tsaprouni, L.G.; Ito, K.; Powell, J.J.; Adcock, I.M.; Punchard, N. Differential patterns of histone acetylation in inflammatory bowel diseases. J. Inflamm. (Lond.) 2011, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zoeten, E.F.; Wang, L.; Sai, H.; Dillmann, W.H.; Hancock, W.W. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 2010, 138, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Akimova, T.; Xiao, H.; Liu, Y.; Bhatti, T.R.; Jiao, J.; Eruslanov, E.; Singhal, S.; Wang, L.; Han, R.; Zacharia, K.; et al. Targeting surtuin-1 alleviates experimental autoimmune colitis by induction of Foxp3+ T-regulatory cells. Mucosal Immunol. 2014, 7, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Han, R.; Beier, U.H.; Bhatti, T.R.; Akimova, T.; Greene, M.I.; Hiebert, S.W.; Hancock, W.W. FOXP3+ regulatory T cell development and function require histone/protein deacetylase 3. J. Clin. Investig. 2015, 125, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Felice, C.; Lewis, A.; Armuzzi, A.; Lindsay, J.O.; Silver, A. Review article: Selective histone deacetylase isoforms as potential therapeutic targets in inflammatory bowel diseases. Aliment Pharmacol. Ther. 2015, 41, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Takaku, F.; Hayashi, M.; Tanaka, I.; Abe, Y.; Nagai, Y.; Ishii, S. A role of valency of concanavalin A and its chemically modified derivatives in lymphocyte activation. Monovalent monomeric concanavalin A derivative can stimulate lymphocyte blastoid transformation. J. Biol. Chem. 1983, 258, 7499–7505. [Google Scholar] [PubMed]
- Tagishi, K.; Shimizu, A.; Endo, K.; Kito, H.; Niwa, S.; Fujii, M.; Ohya, S. Defective splicing of the background K+ channel K2P5.1 by the pre-mRNA splicing inhibitor, pladienolide B in lectin-activated mouse splenic CD4+ T cells. J. Pharmacol. Sci. 2016, 132, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Methot, J.L.; Chakravarty, P.K.; Chenard, M.; Close, J.; Cruz, J.C.; Dahlberg, W.K.; Fleming, J.; Hamblett, C.L.; Hamill, J.E.; Harrington, P.; et al. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kasuya, Y.; Itoh, Y.; Ota, Y.; Zhan, P.; Asamitsu, K.; Nakagawa, H.; Okamoto, T.; Miyata, N. Identification of highly selective and potent histone deacetylase 3 inhibitors using click chemistry-based combinatorial fragment assembly. PLoS ONE 2013, 8, e68669. [Google Scholar] [CrossRef] [PubMed]
- Matsuba, S.; Niwa, S.; Muraki, K.; Kanatsuka, S.; Nakazono, Y.; Hatano, N.; Fujii, M.; Suzuki, T.; Ohya, S. Downregulation of Ca2+-activated Cl− channel TMEM16A by the inhibition of histone deacetylase in TMEM16A-expressing cancer cells. J. Pharmacol. Exp. Ther. 2014, 351, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.J.; Pender, S.L. Histone deacetylase inhibitors and their potential role in inflammatory bowel diseases. Biochem. Soc. Trans. 2011, 39, 1092–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Wang, R.; Xu, H.; Song, Y.; Qi, Y. A high potent and selective histone deacetylase 6 inhibitor prevents DSS-induced colitis in mice. Biol. Pharm. Bull. 2017, 40, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, I.; Ishihara, S.; Rumi, M.A.; Aziz, M.D.; Mishima, Y.; Oshima, N.; Kadota, C.; Kadowaki, Y.; Amano, Y.; Kinoshita, Y. Decoy oligodeoxynucleotide targeting activator protein-1 (AP-1) attenuates intestinal inflammation in murine experimental colitis. Lab. Investig. 2008, 88, 652–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, C.M.; Mah, N.; Häsler, R.; Rosenstiel, P.; Waetzig, G.H.; Hahn, A.; Lu, T.; Gurbuz, Y.; Nikolaus, S.; Albrecht, M.; et al. Dissection of the inflammatory bowel disease transcriptome using genome-wide cDNA microarrays. PLoS Med. 2005, 2, e199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Chen, Z.; Xu, Z.; Lv, F.; Zhang, K.; Yang, Y. The effects of dihydroartemisinin on inflammatory bowel disease-related bone loss in a rat model. Exp. Biol. Med. (Maywood) 2018, 243, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Alexander, R.J.; Panja, A.; Kaplan-Liss, E.; Mayer, L.; Raicht, R.F. Expression of protooncogene-encoded mRNA by colonic epithelial cells in inflammatory bowel disease. Dig. Dis. Sci. 1996, 41, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Do, A.; Reid, R.C.; Lohman, R.J.; Sweet, M.J.; Fairlie, D.P.; Iyer, A. An HDAC6 inhibitor confers protection and selectivity inhibits B-cell infiltration in DSS-induced colitis in mice. J. Pharmacol. Exp. Ther. 2017, 360, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, S.; Lv, C.; Sun, X.; Wang, J.; Zheng, W.; Wang, D. Analysis of murine and human Treg subsets in inflammatory bowel disease. Mol. Med. Rep. 2017, 16, 2893–2898. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, J.; Savic, S.; Wong, C.; Hempshall, A.; Inman, M.; Emery, P.; Grigg, R.; McDermott, M.F. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum. 2012, 64, 418–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J.; Adcock, I.M.; Ito, K. Histone acetylation and deacetylation: Important in inflammatory lung disease. Eur. Respir. J. 2005, 25, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Leus, N.G.; van den Bosch, T.; van der Wouden, P.E.; Krist, K.; Ourailidou, M.E.; Eleftheriadis, N.; Kistemaker, L.E.; Bos, S.; Gjaltema, R.A.; Mekonnen, S.A.; et al. HDAC1-3 inhibitor MS-275 enhances IL10 expression in RAW264.7 macrophages and reduces cigarette smoke-induced airway inflammation in mice. Sci. Rep. 2017, 7, 45047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leus, N.G.; van der Wouden, P.E.; van den Bosch, T.; Hooghiemstra, W.T.; Ourailidou, M.E.; Kistemaker, L.E.; Bischoff, R.; Gosense, R.; Haisma, H.J.; Dekker, F.J. HDAC3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slides by attenuating NF-κB p65 transcriptional activity. Biochem. Pharmacol. 2016, 106, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.X.; Zhou, T.; Wang, X.A.; Tong, X.H.; Ding, J.W. Histone deacetylases and atherosclerosis. Atherosclerosis 2015, 240, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Hoeksema, M.A.; Gijbels, M.J.; van den Bossche, J.; van der Velden, S.; Sijm, A.; Neele, A.E.; Seijkens, T.; Stöger, J.L.; Meiler, S.; Boschuizen, M.C.; et al. Targeting macrophage histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol. Med. 2014, 6, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Oh, E.Y.; Seo, J.Y.; Yu, M.R.; Kim, Y.O.; Ha, H.; Lee, H.B. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am. J. Physiol. Ren. Physiol. 2009, 297, F729–F739. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, X.Q.; Xu, T.; Li, X.F.; Yang, Y.; Li, W.X.; Huang, C.; Meng, X.M.; Li, J. Role of histone deacetylases (HDACs) in progression and reversal of liver fibrosis. Toxicol. Appl. Pharmacol. 2016, 306, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Shen, S.; Ma, Q.; Chen, J.; Gill, A.; Pollock, C.A.; Chen, X.M. Blockade of KCa3.1 ameliorates renal fibrosis through the TGF-β1/Smad pathway in diabetic mice. Diabetes 2013, 62, 2923–2934. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Lin, M.Z.; Cheng, D.; Braet, F.; Pollock, C.A.; Chen, X.M. KCa3.1 mediates dysfunction of tubular autophagy in diabetic kidneys via PI3k/Akt/mTOR signaling pathways. Sci. Rep. 2016, 6, 23884. [Google Scholar] [CrossRef] [PubMed]
- Anumanthan, G.; Gupta, S.; Fink, M.K.; Hesemann, N.P.; Bowles, D.K.; McDaniel, L.M.; Muhammad, M.; Mohan, R.R. KCa3.1 ion channel: A novel therapeutic target for corneal fibrosis. PLoS ONE 2018, 13, e0192145. [Google Scholar] [CrossRef] [PubMed]
- Organ, L.; Bacci, B.; Koumoundouros, E.; Kimpton, W.G.; Samuel, C.S.; Mowell, C.J.; Gradding, P.; Roach, K.M.; Jaffar, J.; Snibson, K.J. Inhibition of the KCa3.1 channel alleviates established pulmonary fibrosis in a large animal model. Am. J. Respir. Cell Mol. Biol. 2017, 56, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.H.; Xu, J.R.; Wang, Y.X.; Xu, G.N.; Xu, Z.P.; Yang, K.; Wu, D.Z.; Cui, Y.Y.; Chen, H.Z. Targeted inhibition of KCa3.1 channel attenuates airway inflammation and remodeling in allergic asthma. Am. J. Respir. Cell Mol. Biol. 2013, 48, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Kito, H.; Hatano, N.; Muraki, K. Recent advances in therapeutic strategies that focus on the regulation of ion channel expression. Pharmacol. Ther. 2016, 160, 11–43. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsui, M.; Terasawa, K.; Kajikuri, J.; Kito, H.; Endo, K.; Jaikhan, P.; Suzuki, T.; Ohya, S. Histone Deacetylases Enhance Ca2+-Activated K+ Channel KCa3.1 Expression in Murine Inflammatory CD4+ T Cells. Int. J. Mol. Sci. 2018, 19, 2942. https://doi.org/10.3390/ijms19102942
Matsui M, Terasawa K, Kajikuri J, Kito H, Endo K, Jaikhan P, Suzuki T, Ohya S. Histone Deacetylases Enhance Ca2+-Activated K+ Channel KCa3.1 Expression in Murine Inflammatory CD4+ T Cells. International Journal of Molecular Sciences. 2018; 19(10):2942. https://doi.org/10.3390/ijms19102942
Chicago/Turabian StyleMatsui, Miki, Kyoko Terasawa, Junko Kajikuri, Hiroaki Kito, Kyoko Endo, Pattaporn Jaikhan, Takayoshi Suzuki, and Susumu Ohya. 2018. "Histone Deacetylases Enhance Ca2+-Activated K+ Channel KCa3.1 Expression in Murine Inflammatory CD4+ T Cells" International Journal of Molecular Sciences 19, no. 10: 2942. https://doi.org/10.3390/ijms19102942