BGJ398, A Pan-FGFR Inhibitor, Overcomes Paclitaxel Resistance in Urothelial Carcinoma with FGFR1 Overexpression

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
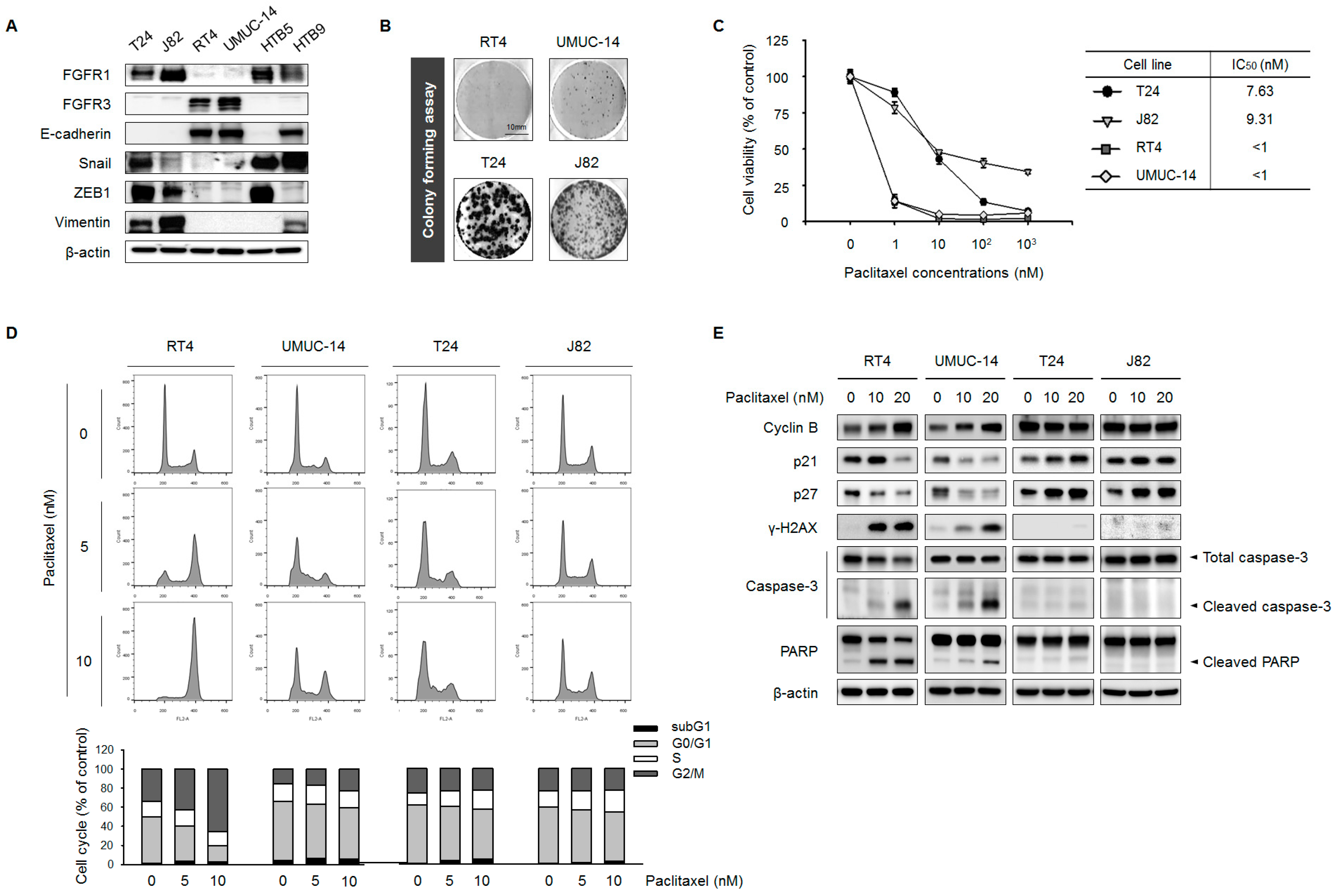
2.1. FGFR1 Overexpression Is Correlated with EMT and PTX Resistance in UC Cell Lines
2.2. Expression Levels of FGFR1 and EMT Markers Are Correlated
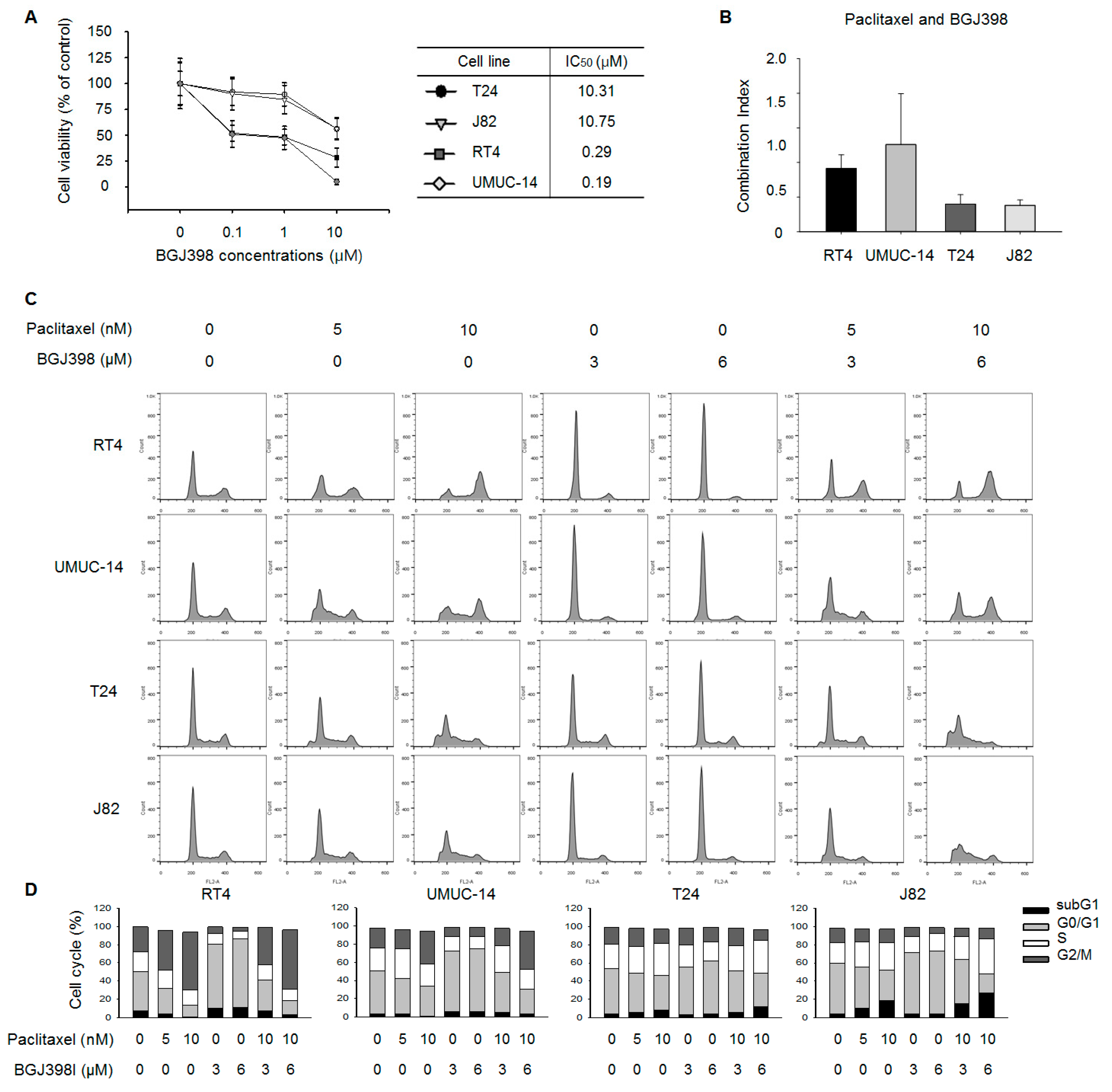
2.3. FGFR Inhibition Overcomes PTX Resistance in UC Cell Lines
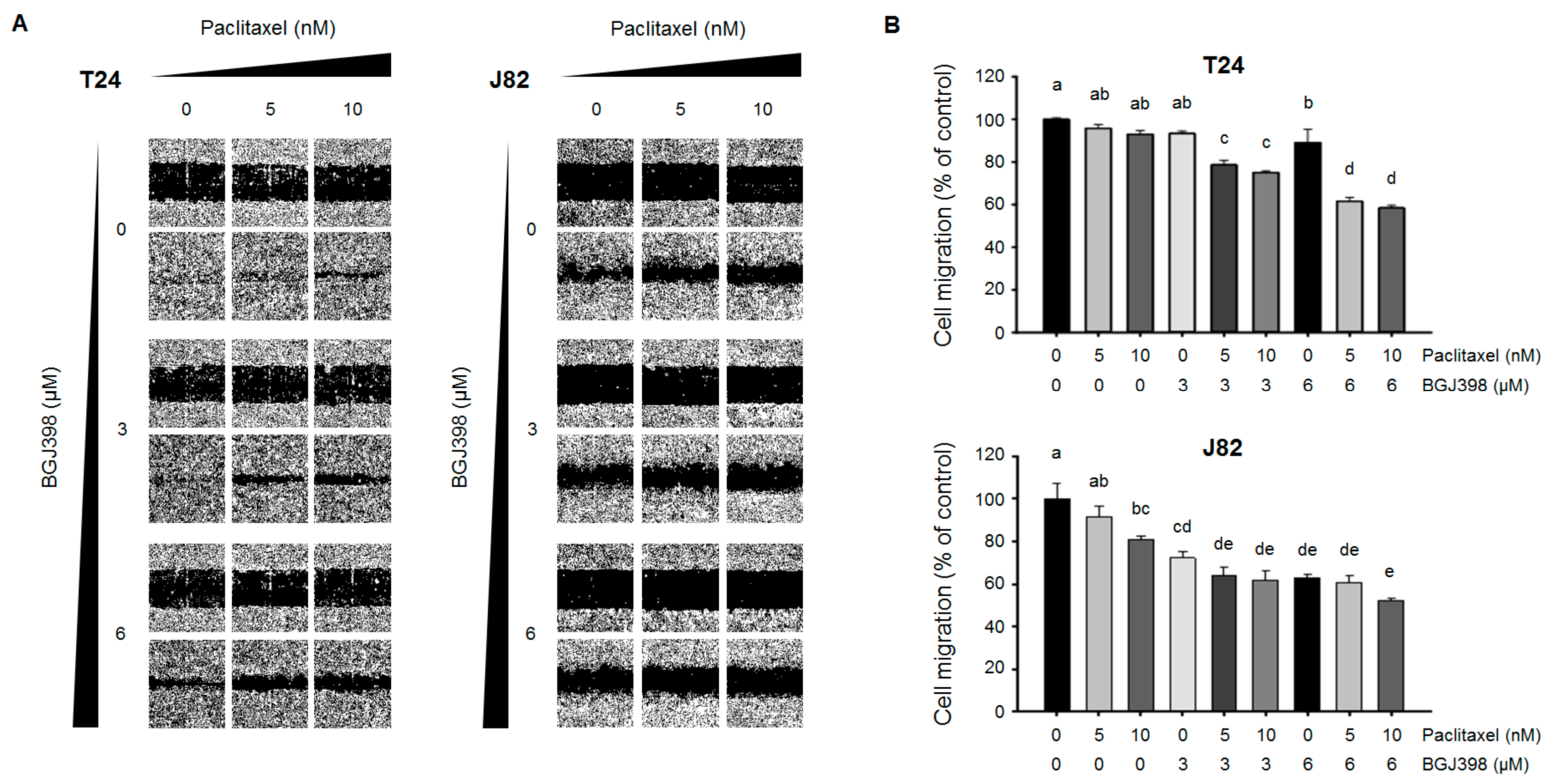
2.4. PTX Combined with BGJ398 Reduces the Migratory Capacity of Mesenchymal-Type UC Cells
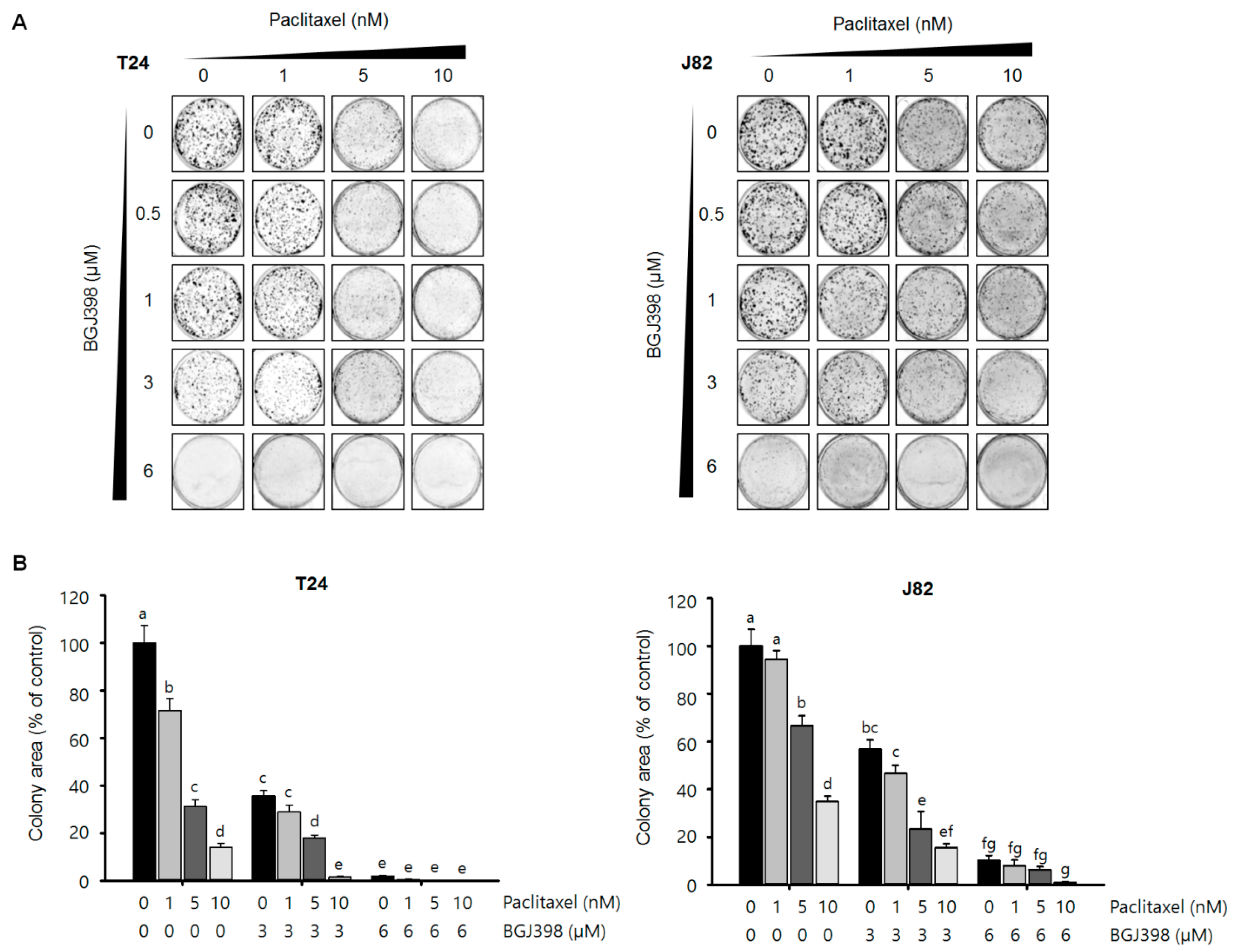
2.5. BGJ398 Enhances the Effect of PTX on Colony Formation by Mesenchymal-Type UC Cells
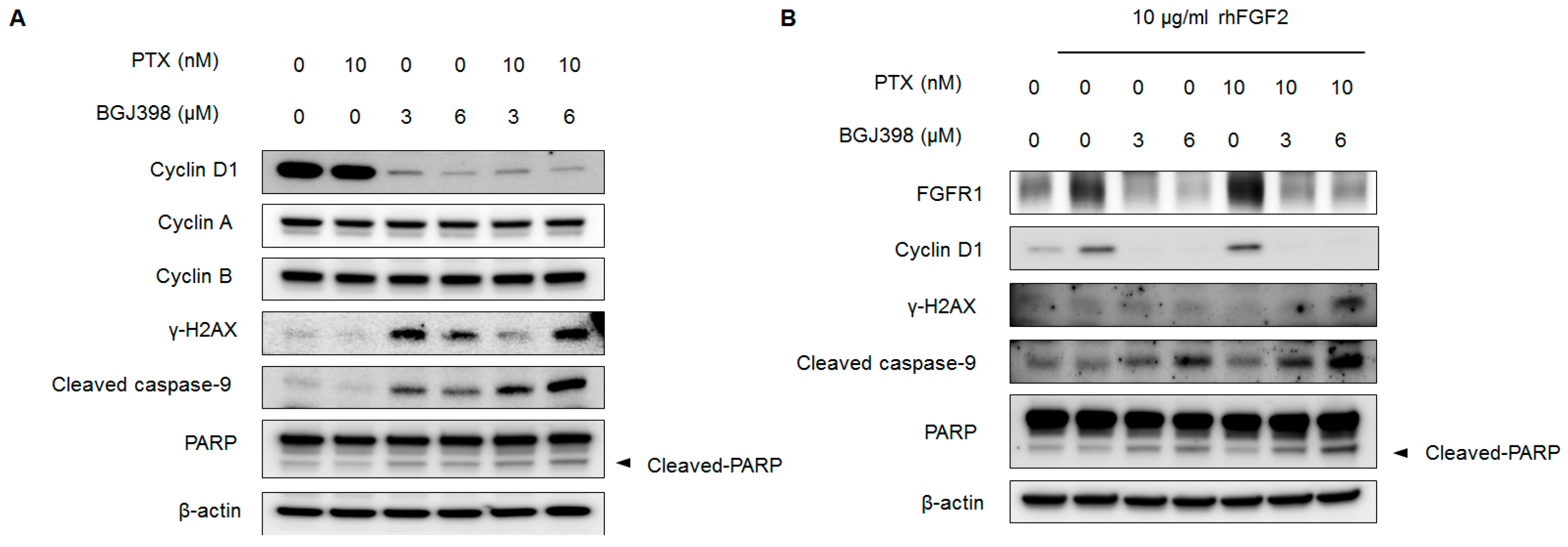
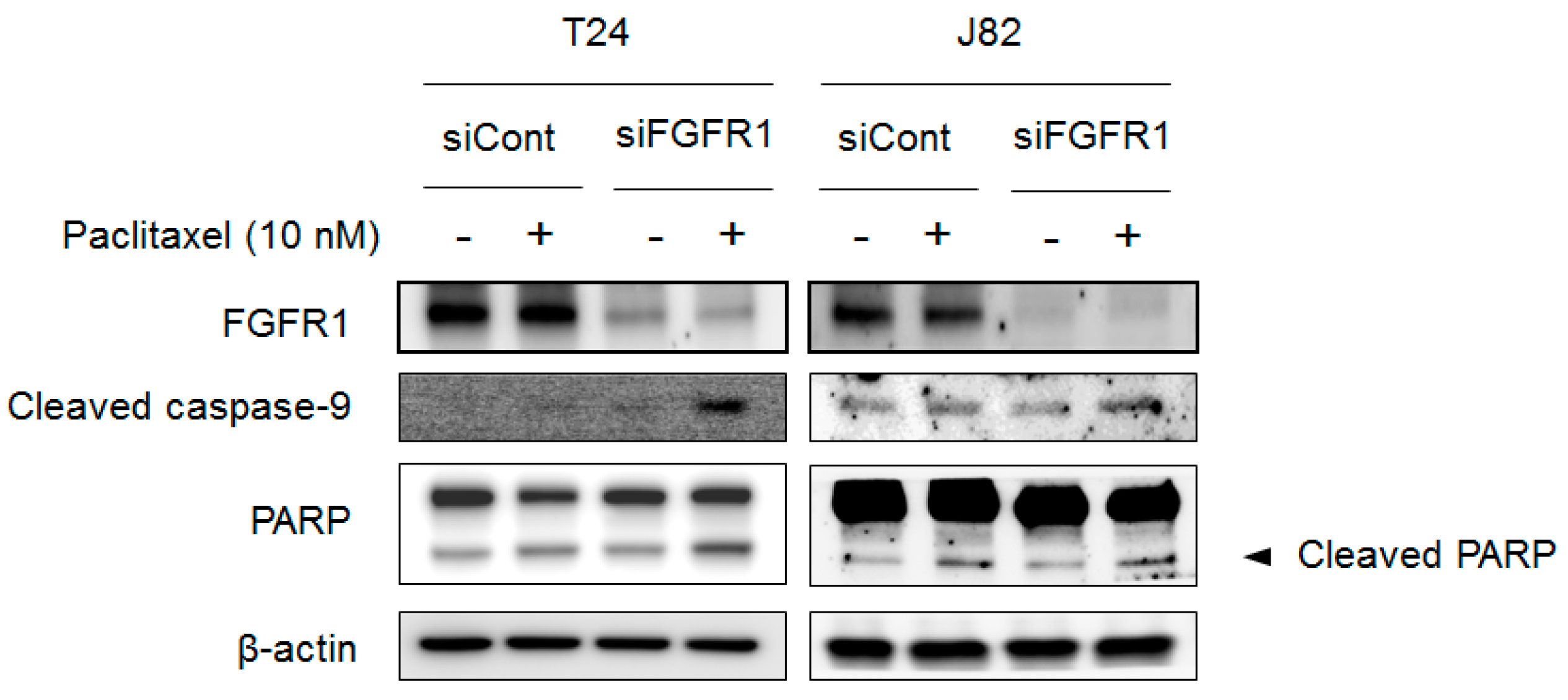
2.6. Blocking FGFR1 Signaling Stimulates Apoptosis in PTX-Resistant UC Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Analysis of Combination Index
4.5. Western Blot Analysis
4.6. Cell Migration
4.7. RNAi
4.8. Cell Cycle Analysis
4.9. Colony Formation Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Hahn, N.M.; Rosenberg, J.; Sonpavde, G.; Hutson, T.; Oh, W.K.; Dreicer, R.; Vogelzang, N.; Sternberg, C.N.; Bajorin, D.F. Treatment of patients with metastatic urothelial cancer “unfit” for cisplatin-based chemotherapy. J. Clin. Oncol. 2011, 29, 2432–2438. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; Van Der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Sonpavde, G.; Sternberg, C.N.; Rosenberg, J.E.; Hahn, N.M.; Galsky, M.D.; Vogelzang, N.J. Second-line systemic therapy and emerging drugs for metastatic transitional-cell carcinoma of the urothelium. Lancet Oncol. 2010, 11, 861–870. [Google Scholar] [CrossRef]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Rev. Cancer 2015, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.C.; Lamont, F.R.; Shnyder, S.D.; Knowles, M.A. Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen-activated protein kinase pathway in bladder cancer. Cancer Res. 2009, 69, 4613–4620. [Google Scholar] [CrossRef] [PubMed]
- Lamont, F.; Tomlinson, D.; Cooper, P.A.; Shnyder, S.D.; Chester, J.; Knowles, M. Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. Br. J. Cancer 2011, 104, 75. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Roth, B.; Choi, W.; Black, P.C.; Dinney, C.; McConkey, D.J. Fibroblast growth factor receptors-1 and-3 play distinct roles in the regulation of bladder cancer growth and metastasis: Implications for therapeutic targeting. PLoS ONE 2013, 8, e57284. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.-P. Efficacy of BGJ398, a fibroblast growth factor receptor 1-3 inhibitor, in patients with previously treated advanced urothelial carcinoma with FGFR3 alterations. Cancer Discov. 2018, 8, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.C.; Baxter, E.W.; Loadman, P.M.; Hull, M.A.; Knowles, M.A. FGFR1-induced epithelial to mesenchymal transition through MAPK/PLCγ/COX-2-mediated mechanisms. PLoS ONE 2012, 7, e38972. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Wientjes, M.G.; Au, J.L.-S. Expression of basic fibroblast growth factor correlates with resistance to paclitaxel in human patient tumors. Pharm. Res. 2006, 23, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, V.D.; Gangula, R.D.; Freeman, K.W.; Li, R.; Zhang, Y.; Wang, F.; Ayala, G.E.; Peterson, L.E.; Ittmann, M.; Spencer, D.M. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell 2007, 12, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Terai, H.; Soejima, K.; Yasuda, H.; Nakayama, S.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Ikemura, S.; Sato, T. Activation of the FGF2-FGFR1 autocrine pathway: A novel mechanism of acquired resistance to gefitinib in NSCLC. Mol. Cancer Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ji, W.; Yu, Y.; Li, Z.; Niu, X.; Xia, W.; Lu, S. FGFR1-ERK1/2-SOX2 axis promotes cell proliferation, epithelial–mesenchymal transition, and metastasis in FGFR1-amplified lung cancer. Oncogene 2018, 37, 5340–5354. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Wang, H.S.; Soong, Y.K. Paclitaxel-induced cell death: Where the cell cycle and apoptosis come together. Cancer 2000, 88, 2619–2628. [Google Scholar] [CrossRef]
- Milas, L.; Hunter, N.R.; Kurdoglu, B.; Mason, K.A.; Meyn, R.E.; Stephens, L.C.; Peters, L.J. Kinetics of mitotic arrest and apoptosis in murine mammary and ovarian tumors treated with taxol. Cancer Chemother. Pharmacol. 1995, 35, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.F.; Donaldson, K.L.; Fairchild, C.; Lee, F.Y.; Foster, S.A.; Demers, G.W.; Galloway, D.A. Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat. Med. 1996, 2, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Ivashkevich, A.; Redon, C.E.; Nakamura, A.J.; Martin, R.F.; Martin, O.A. Use of the gamma-H2AX assay to monitor DNA damage and repair in translational cancer research. Cancer Lett. 2012, 327, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Bijnsdorp, I.V.; Giovannetti, E.; Peters, G.J. Analysis of drug interactions. Methods Mol. Biol. 2011, 731, 421–434. [Google Scholar] [PubMed]
- Sharpe, R.; Pearson, A.; Herrera-Abreu, M.T.; Johnson, D.; Mackay, A.; Welti, J.C.; Natrajan, R.; Reynolds, A.R.; Reis-Filho, J.S.; Ashworth, A.; et al. FGFR signaling promotes the growth of triple-negative and basal-like breast cancer cell lines both in vitro and in vivo. Clin. Cancer Res. 2011, 17, 5275–5286. [Google Scholar] [CrossRef] [PubMed]
- Holz, C.; Niehr, F.; Boyko, M.; Hristozova, T.; Distel, L.; Budach, V.; Tinhofer, I. Epithelial-mesenchymal-transition induced by EGFR activation interferes with cell migration and response to irradiation and cetuximab in head and neck cancer cells. Radiother. Oncol. 2011, 101, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, K.S.; Rohatgi, N.; Halldorsson, S.; Briem, E.; Gudjonsson, T.; Gudmundsson, S.; Rolfsson, O. EGFR Signal-Network Reconstruction Demonstrates Metabolic Crosstalk in EMT. PLoS Comput. Biol. 2016, 12, e1004924. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.S.; Tan, L.; Smith, A.; Gray, N.S.; Wendt, M.K. Covalent Targeting of Fibroblast Growth Factor Receptor Inhibits Metastatic Breast Cancer. Mol. Cancer Ther. 2016, 15, 2096–2106. [Google Scholar] [CrossRef] [PubMed]
- Fumarola, C.; Cretella, D.; La Monica, S.; Bonelli, M.A.; Alfieri, R.; Caffarra, C.; Quaini, F.; Madeddu, D.; Falco, A.; Cavazzoni, A.; et al. Enhancement of the anti-tumor activity of FGFR1 inhibition in squamous cell lung cancer by targeting downstream signaling involved in glucose metabolism. Oncotarget 2017, 8, 91841–91859. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.H.; Lingohr, P.; Strasser, A.; Lehnen, N.C.; Braun, M.; Perner, S.; Holler, T.; Kristiansen, G.; Kalff, J.C.; Gutgemann, I. Fibroblast growth factor receptor 1 gene amplification in gastric adenocarcinoma. Hum. Pathol. 2015, 46, 1488–1495. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Al-Rohil, R.N.; Nazeer, T.; Sheehan, C.E.; Otto, G.A.; He, J.; Palmer, G.; Yelensky, R.; Lipson, D.; et al. Advanced urothelial carcinoma: Next-generation sequencing reveals diverse genomic alterations and targets of therapy. Mod. Pathol. 2014, 27, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Cortes, J. Rationale for targeting fibroblast growth factor receptor signaling in breast cancer. Breast Cancer Res. Treat. 2015, 150, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sideris, S.; Aoun, F.; Zanaty, M.; Martinez, N.C.; Latifyan, S.; Awada, A.; Gil, T. Efficacy of weekly paclitaxel treatment as a single agent chemotherapy following first-line cisplatin treatment in urothelial bladder cancer. Mol. Clin. Oncol. 2016, 4, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Takahi, Y.; Ichimaru, N.; Okumi, M.; Takahara, S.; Nonomura, N. Successful treatment of metastatic urothelial carcinoma arising in a transplanted renal allograft with paclitaxel, cisplatin, and gemcitabine combination therapy: A case report. BMC Res. Notes 2015, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Vaishampayan, U.; Du, W.; Redman, B.; Smith, D.C. Combination paclitaxel, carboplatin, and gemcitabine is an active treatment for advanced urothelial cancer. J. Clin. Oncol. 2001, 19, 2527–2533. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.-L. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Xian, W.; Schwertfeger, K.L.; Vargo-Gogola, T.; Rosen, J.M. Pleiotropic effects of FGFR1 on cell proliferation, survival, and migration in a 3D mammary epithelial cell model. J. Cell Biol. 2005, 171, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar] [CrossRef] [PubMed]
- Jirawatnotai, S.; Hu, Y.; Michowski, W.; Elias, J.E.; Becks, L.; Bienvenu, F.; Zagozdzon, A.; Goswami, T.; Wang, Y.E.; Clark, A.B.; et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 2011, 474, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.P.; Qiang, L.; Zhang, J.L. Transcription activated p73-modulated cyclin D1 expression leads to doxorubicin resistance in gastric cancer. Exp. Ther. Med. 2018, 15, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Byron, S.A.; Loch, D.C.; Pollock, P.M. Fibroblast growth factor receptor inhibition synergizes with Paclitaxel and Doxorubicin in endometrial cancer cells. Int. J. Gynecol. Cancer 2012, 22, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, Y.; Ressler, S.J.; Ittmann, M.M.; Ayala, G.E.; Dang, T.D.; Wang, F.; Rowley, D.R. FGFR1 is essential for prostate cancer progression and metastasis. Cancer Res. 2013, 73, 3716–3724. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Berghoff, A.S.; Berger, W.; Ilhan-Mutlu, A.; Dinhof, C.; Widhalm, G.; Dieckmann, K.; Wohrer, A.; Hackl, M.; von Deimling, A.; et al. High rate of FGFR1 amplifications in brain metastases of squamous and non-squamous lung cancer. Lung Cancer 2014, 83, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Wang, X.; Zheng, C.; Ma, W. Downregulation of microRNA-214 and overexpression of FGFR-1 contribute to hepatocellular carcinoma metastasis. Biochem. Biophys. Res. Commun. 2013, 439, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Koh, M.J.; Jeong, H.J.; Cho, N.H.; Choi, Y.D.; Cho, D.Y.; Lee, H.Y.; Rha, S.Y. Fibroblast Growth Factor Receptor 1 Overexpression Is Associated with Poor Survival in Patients with Resected Muscle Invasive Urothelial Carcinoma. Yonsei Med. J. 2016, 57, 831–839. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.H.; Ryu, H.; Ock, C.-Y.; Suh, K.J.; Lee, J.Y.; Kim, J.-W.; Lee, J.-O.; Kim, J.W.; Kim, Y.J.; Lee, K.-W.; et al. BGJ398, A Pan-FGFR Inhibitor, Overcomes Paclitaxel Resistance in Urothelial Carcinoma with FGFR1 Overexpression. Int. J. Mol. Sci. 2018, 19, 3164. https://doi.org/10.3390/ijms19103164
Kim SH, Ryu H, Ock C-Y, Suh KJ, Lee JY, Kim J-W, Lee J-O, Kim JW, Kim YJ, Lee K-W, et al. BGJ398, A Pan-FGFR Inhibitor, Overcomes Paclitaxel Resistance in Urothelial Carcinoma with FGFR1 Overexpression. International Journal of Molecular Sciences. 2018; 19(10):3164. https://doi.org/10.3390/ijms19103164
Chicago/Turabian StyleKim, Se Hyun, Haram Ryu, Chan-Young Ock, Koung Jin Suh, Ji Yun Lee, Ji-Won Kim, Jeong-Ok Lee, Jin Won Kim, Yu Jung Kim, Keun-Wook Lee, and et al. 2018. "BGJ398, A Pan-FGFR Inhibitor, Overcomes Paclitaxel Resistance in Urothelial Carcinoma with FGFR1 Overexpression" International Journal of Molecular Sciences 19, no. 10: 3164. https://doi.org/10.3390/ijms19103164
APA StyleKim, S. H., Ryu, H., Ock, C.-Y., Suh, K. J., Lee, J. Y., Kim, J.-W., Lee, J.-O., Kim, J. W., Kim, Y. J., Lee, K.-W., Bang, S.-M., Kim, J. H., Lee, J. S., Ahn, J. B., Kim, K.-J., & Rha, S. Y. (2018). BGJ398, A Pan-FGFR Inhibitor, Overcomes Paclitaxel Resistance in Urothelial Carcinoma with FGFR1 Overexpression. International Journal of Molecular Sciences, 19(10), 3164. https://doi.org/10.3390/ijms19103164