Comparative Genome-Wide Survey of Single Nucleotide Variation Uncovers the Genetic Diversity and Potential Biomedical Applications among Six Macaca Species


Abstract
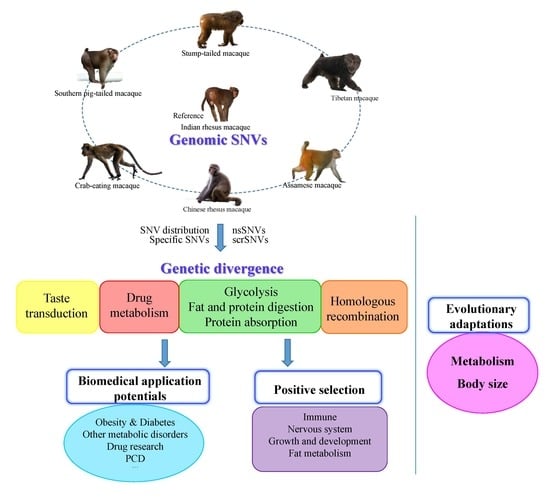
:
1. Introduction
2. Results
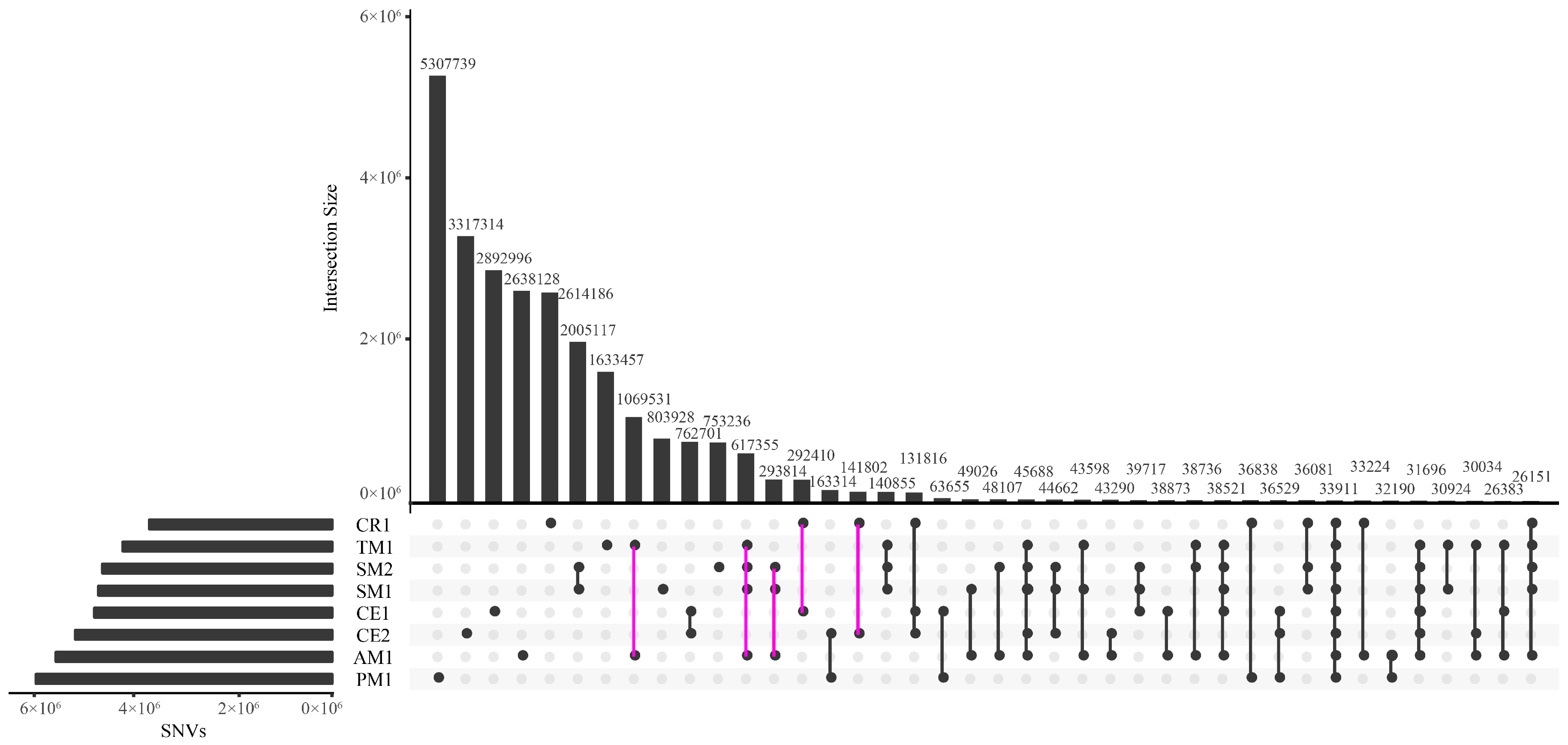
2.1. Genome-Wide Discovery of SNVs
2.2. Functional Annotation of SNVs
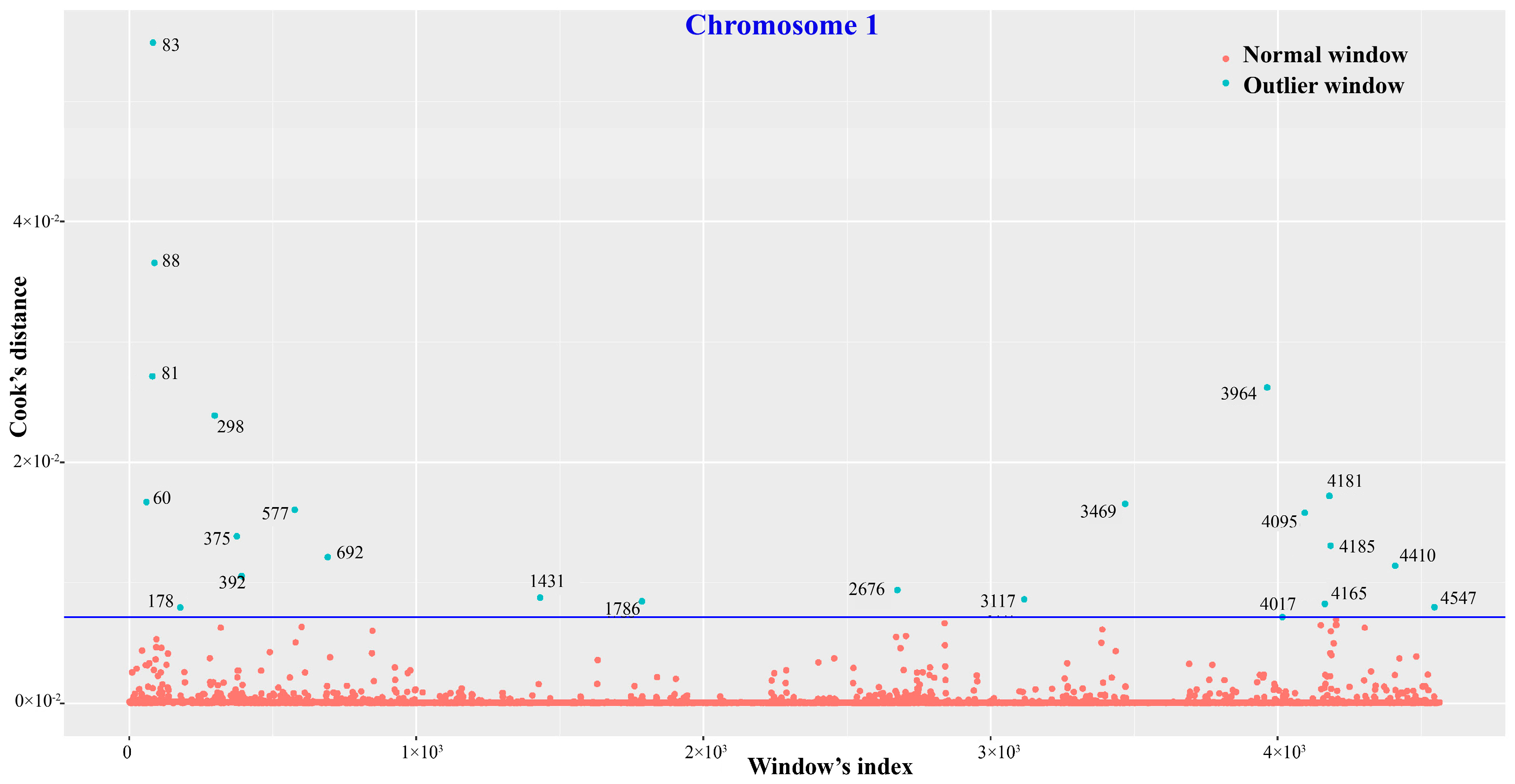
2.3. Characterization of SNV and nsSNV Distribution Patterns
2.4. Enrichment Analyses of Specific nsSNVs with Putative Functions
2.5. Putatively Damaging nsSNVs and Their Associated Diseases
2.6. SNVs Causing Stop Codon Changes
2.7. Positive Selection Based on SNVs
3. Discussion
3.1. Large Dataset of SNVs for Macaca
3.2. Suggestive Functional Divergence Inferred from SNV Distribution Patterns
3.3. Divergent Characteristics Inferred from Enrichment of Putative Functional SNVs
3.4. Application Potentials in Biomedical Research
3.5. Positive selections on Macaca Genomes
4. Materials and Methods
4.1. Genome Data and SNV Calling
4.2. Functional Annotation of SNVs
4.3. Determination of SNV Distribution Patterns and Outlier Detection
4.4. Functional Enrichment Analyses of SNVs
4.5. Identification of Positively Selected Genes Based on SNVs
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| SNV | Single nucleotide variation |
| NHP | Nonhuman primate |
| CNV | Copy number variation |
| A | Adenine |
| C | Cytosine |
| G | Guanine |
| T | Thymine |
| A | Alanine |
| T | Threonine |
| V | Valine |
| I | Isoleucine |
| P | Proline |
| L | Leucine |
| R | Arginine |
| H | Histidine |
| KEGG | Kyoto encyclopedia of genes and genomes |
| GO | Gene ontology |
| DSBR | Double strand break repair |
| HR | homologous recombination |
| PCD | Primary ciliary dyskinesia |
| ECM | Extracellular matrix |
| mTOR | Mammalian target of rapamycin |
| PPAR | Peroxisome proliferator-activated receptor |
| HDL | High-density lipoprotein |
| PSG | Positively selected gene |
| PAML | Phylogenetic analysis by maximum likelihood |
References
- Solari, S.; Baker, R.J. Mammal Species of the World, A Taxonomic and Geographic Reference. Mastozoología Neotrop. 2006, 13, 290–293. [Google Scholar] [CrossRef]
- Li, C.; Zhao, C.; Fan, P.F. White-cheeked macaque (Macaca leucogenys): A new macaque species from Medog, southeastern Tibet. Am. J. Primatol. 2015, 77, 753. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yu, J.; Li, J.; Li, P.; Fan, Z.; Niu, L.; Deng, J.; Yue, B. Mitochondrial Genome and Nuclear Markers Provide New Insight into the Evolutionary History of Macaques. PLoS ONE 2016, 11, e0154665. [Google Scholar] [CrossRef] [PubMed]
- Vallender, E.J.; Miller, G.M. Nonhuman Primate Models in the Genomic Era: A Paradigm Shift. ILAR J. 2013, 54, 154–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, M.B.; Luciw, P.A. Macaque models of human infectious disease. ILAR J. 2008, 49, 220–255. [Google Scholar] [CrossRef] [PubMed]
- Hatziioannou, T.; Ambrose, Z.; Chung, N.P.Y.; Piatak, M.; Yuan, F.; Trubey, C.M.; Coalter, V.; Kiser, R.; Schneider, D.; Smedley, J. A macaque model of HIV-1 infection. Proc. Nat. Acad. Sci. USA 2009, 106, 4425–4429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pouladi, M.A.; Morton, A.J.; Hayden, M.R. Choosing an animal model for the study of Huntington’s disease. Nat. Rev. Neurosci. 2013, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kanthaswamy, S.; Trask, J.S.; Ng, J.; Oldt, R.F.; Mankowski, J.L.; Adams, R.J.; Smith, D.G. Genetic characterization of a captive colony of pigtailed macaques (Macaca nemestrina). J. Am. Assoc. Lab. Animal Sci. 2017, 56, 390–395. [Google Scholar]
- Norgren, R.B., Jr. Improving genome assemblies and annotations for nonhuman primates. ILAR J. 2013, 54, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Haus, T.; Ferguson, B.; Rogers, J.; Doxiadis, G.; Certa, U.; Rose, N.J.; Teepe, R.; Weinbauer, G.F.; Roos, C. Genome typing of nonhuman primate models: Implications for biomedical research. Trend. Gen. 2014, 30, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Seekatz, A.M.; Panda, A.; Rasko, D.A.; Toapanta, F.R.; Eloe-Fadrosh, E.A.; Khan, A.Q.; Liu, Z.; Shipley, S.T.; DeTolla, L.J.; Sztein, M.B. Differential response of the cynomolgus macaque gut microbiota to Shigella infection. PLoS ONE 2013, 8, e64212. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Q.; Na, L.; Lv, X.-L.; Liu, J.-D.; Liu, X.-M.; Ji, F.; Zheng, Y.-H.; Du, H.-L.; Kong, X.-G.; Zhou, J.-H. The TRIMCyp genotype in four species of macaques in China. Immunogenetics 2013, 65, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Trichel, A.; Rajakumar, P.; Murphey-Corb, M. Species-specific variation in SIV disease progression between Chinese and Indian subspecies of rhesus macaque. J. Med. Primatol. 2002, 31, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; Risch, N. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 2003, 33, 228. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zhang, Y.; Zhang, R.; Yang, L.; Li, M.; Ye, K.; Guo, X.; Wang, J.; Su, B. Genome sequence and global sequence variation map with 5.5 million SNPs in Chinese rhesus macaque. Genome Biol. 2011, 12, R63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.; Zhang, G.; Fang, X.; Zhang, Y.; Li, C.; Ling, F.; Cooper, D.N.; Li, Q.; Li, Y.; Van Gool, A.J. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat. Biotechnol. 2011, 29, 1019–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, C.; Raveendran, M.; Harris, R.A.; Fawcett, G.L.; Liu, X.; White, S.; Dahdouli, M.; Rio, D.D.; Below, J.E.; Salerno, W. The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res. 2016, 26, 1651. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.; Street, S.L.; Wright, H.; Pearson, C.; Jia, Y.; Thompson, S.L.; Allibone, P.; Dubay, C.J.; Spindel, E.; Norgren, R.B. Single nucleotide polymorphisms (SNPs) distinguish Indian-origin and Chinese-origin rhesus macaques (Macaca mulatta). BMC Genom. 2007, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Peng, J.; Shen, Q.S.; Chen, J.Y.; Han, G.; Luan, X.; Yan, S.; Xin, H.; Zhang, S.J.; Xu, L. RhesusBase PopGateway: Genome-Wide Population Genetics Atlas in Rhesus Macaque. Mol. Biol. Evol. 2016, 33, 1370. [Google Scholar] [CrossRef] [PubMed]
- Malhi, R.S.; Sickler, B.; Lin, D.; Satkoski, J.; Tito, R.Y.; George, D.; Kanthaswamy, S.; Smith, D.G. MamuSNP: A Resource for Rhesus Macaque (Macaca mulatta) Genomics. PLoS ONE 2007, 2, e438. [Google Scholar] [CrossRef] [PubMed]
- Ng, J. Comparisons of Genomic Variation in Rhesus Macaques, Cynomolgus Macaques, and Humans; University of California: Davis, CA, USA, 2015. [Google Scholar]
- Fan, Z.; Zhao, G.; Li, P.; Osada, N.; Xing, J.; Yi, Y.; Du, L.; Silva, P.; Wang, H.; Sakate, R. Whole-genome sequencing of tibetan macaque (Macaca thibetana) provides new insight into the macaque evolutionary history. Mol. Biol. Evol. 2014, 31, 1475. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Rogers, J.; Katze, M.G.; Bumgarner, R.; Weinstock, G.M.; Mardis, E.R.; Remington, K.A.; Strausberg, R.L.; Venter, J.C.; Wilson, R.K. Evolutionary and Biomedical Insights from the Rhesus Macaque Genome. Science 2007, 316, 222. [Google Scholar] [PubMed]
- Higashino, A.; Sakate, R.; Kameoka, Y.; Takahashi, I.; Hirata, M.; Tanuma, R.; Masui, T.; Yasutomi, Y.; Osada, N. Whole-genome sequencing and analysis of the Malaysian cynomolgus macaque (Macaca fascicularis) genome. Genome Biol. 2012, 13, R58. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Zhou, A.; Osada, N.; Yu, J.; Jiang, J.; Li, P.; Du, L.; Niu, L.; Deng, J.; Xu, H. Ancient hybridization and admixture in macaques (genus Macaca) inferred from whole genome sequences. Mol. Phylogenet. Evol. 2018, 127, 376. [Google Scholar] [CrossRef] [PubMed]
- Delson, E. Fossil macaques, phyletic relationships and a scenario of deployment. The Macaques: Studies in Ecology, Behavior and Evolution; Van Nostrand Reinhold Co.: New York, NY, USA, 1980; pp. 10–30. Available online: http://pages.nycep.org/ed/download/pdf/1980a%20Fossil%20macaques,%20phylogeny%20&%20deployment.pdf (accessed on 8 October 2018).
- Li, J.; Han, K.; Xing, J.; Kim, H.-S.; Rogers, J.; Ryder, O.A.; Disotell, T.; Yue, B.; Batzer, M.A. Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene 2009, 448, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Depristo, M.A.; Banks, E.; Poplin, R.E.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Angel, G.D.; Rivas, M.A.; Hanna, M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491. [Google Scholar] [CrossRef] [PubMed]
- Lachance, J.; Vernot, B.; Elbers, C.C.; Ferwerda, B.; Froment, A.; Bodo, J.M.; Lema, G.; Fu, W.; Nyambo, T.B.; Rebbeck, T.R. Evolutionary History and Adaptation from High-Coverage Whole-Genome Sequences of Diverse African Hunter-Gatherers. Cell 2012, 150, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SATKOSKI TRASK, J.A.; Garnica, W.T.; Smith, D.G.; Houghton, P.; Lerche, N.; Kanthaswamy, S. Single-Nucleotide Polymorphisms Reveal Patterns of Allele Sharing Across the Species Boundary Between Rhesus (Macaca mulatta) and Cynomolgus (M. fascicularis) Macaques. Am. J. Primatol. 2013, 75, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Le, L.W.; Coon, M.; Nguyen, T.; Luan, W.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Cornish, A.S.; Maudhoo, M.D.; Gibbs, R.M.; Zhang, X.; Pandey, S.; Meehan, D.T.; Wipfler, K.; Bosinger, S.E.; Johnson, Z.P. A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol. Direct 2014, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, D.; Das, S.; Badoni, S.; Kumar, V.; Singh, M.; Bansal, K.C.; Tyagi, A.K.; Parida, S.K. Genome-wide high-throughput SNP discovery and genotyping for understanding natural (functional) allelic diversity and domestication patterns in wild chickpea. Sci. Rep. 2015, 5, 12468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.; Cho, Y.S.; Hu, H.; Kim, H.-M.; Jho, S.; Gadhvi, P.; Park, K.M.; Lim, J.; Paek, W.K.; Han, K. Whole genome sequence and analysis of the Marwari horse breed and its genetic origin. BMC Genom. 2014, 15, S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bimber, B.N.; Ramakrishnan, R.; Cervera-Juanes, R.; Madhira, R.; Peterson, S.M.; Norgren, R.B.; Ferguson, B. Whole genome sequencing predicts novel human disease models in rhesus macaques. Genomics 2017, 109, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Gokcumen, O.; Babb, P.L.; Iskow, R.C.; Zhu, Q.; Shi, X.; Mills, R.E.; Ionita-Laza, I.; Vallender, E.J.; Clark, A.G.; Johnson, W.E. Refinement of primate copy number variation hotspots identifies candidate genomic regions evolving under positive selection. Genome Biol. 2011, 12, R52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryukov, G.V.; Pennacchio, L.A.; Sunyaev, S.R. Most Rare Missense Alleles Are Deleterious in Humans: Implications for Complex Disease and Association Studies. Am. J. Hum. Gen. 2007, 80, 727. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Lamming, D.W. The mechanistic target of rapamycin: The grand conducTOR of metabolism and aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Linden, H.C.; Price, S.M. Cardiofaciocutaneous syndrome in a mother and two sons with a MEK2 mutation. Clin. Dysmorphol. 2011, 20, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Shi, X.; Wang, T.; Ying, C.; He, S.; Chen, Y. The prognostic and clinicopathological significance of IGF-1R in NSCLC: A meta-analysis. Cell. Physiol. Biochem. 2017, 43, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Liauchonak, I.; Dawoud, F.; Riat, Y.; Qorri, B.; Sambi, M.; Jain, J.; Kalaydina, R.-V.; Mendonza, N.; Bajwa, K.; Szewczuk, M.R. The Biased G-Protein-Coupled Receptor Agonism Bridges the Gap between the Insulin Receptor and the Metabolic Syndrome. Int. J. Mol. Sci. 2018, 19, 575. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nat. Protoc. 2016, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Dolphin, C.T.; Beckett, D.J.; Janmohamed, A.; Cullingford, T.E.; Smith, R.L.; Shephard, E.A.; Phillips, I.R. The flavin-containing monooxygenase 2 gene (FMO2) of humans, but not of other primates, encodes a truncated, nonfunctional protein. J. Biol. Chem. 1998, 273, 30599–30607. [Google Scholar] [CrossRef] [PubMed]
- Krueger, S.K.; Martin, S.R.; Yueh, M.-F.; Pereira, C.B.; Williams, D.E. Identification of active flavin-containing monooxygenase isoform 2 in human lung and characterization of expressed protein. Drug Metab. Dispos. 2002, 30, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Krueger, S.K.; Siddens, L.K.; Martin, S.R.; Yu, Z.; Pereira, C.B.; Cabacungan, E.T.; Hines, R.N.; Ardlie, K.G.; Raucy, J.L.; Williams, D.E. Differences in FMO2* 1 allelic frequency between Hispanics of Puerto Rican and Mexican descent. Drug Metab. Dispos. 2004, 32, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Uno, Y.; Uehara, S.; Kohara, S.; Murayama, N.; Yamazaki, H. Polymorphisms of CYP2D17 in cynomolgus and rhesus macaques: An evidence of the genetic basis for the variability of CYP2D-dependent drug metabolism. Drug Metab. Dispos. 2014, 42, 1407–1410. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, B.D.; Lee, E.-J.; Park, Y.; Jameson, J.L. A dominant negative peroxisome proliferator-activated receptor-γ knock-in mouse exhibits features of the metabolic syndrome. J. Biol. Chem. 2005, 280, 17118–17125. [Google Scholar] [CrossRef] [PubMed]
- Semple, R.K.; Chatterjee, V.K.K.; O’Rahilly, S. PPARγ and human metabolic disease. J. Clin. Investig. 2006, 116, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Petousi, N.; Glusman, G.; Yu, Y.; Bohlender, R.; Tashi, T.; Downie, J.M.; Roach, J.C.; Cole, A.M.; Lorenzo, F.R. Evolutionary history of Tibetans inferred from whole-genome sequencing. PLoS Genet. 2017, 13, e1006675. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Du, L.; Li, W.; Fan, Z.; Zeng, D.; Chen, H.; Zhou, L.; Yi, Y.; Yang, N.; Dou, K. Generation and characterization of the blood transcriptome of Macaca thibetana and comparative analysis with M. mulatta. Mol. BioSyst. 2017, 13, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair 2007, 6, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Raghavan, S.C. DNA double-strand break repair inhibitors as cancer therapeutics. Chem. Biol. 2015, 22, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Evers, B.; Helleday, T.; Jonkers, J. Targeting homologous recombination repair defects in cancer. Trend. Pharmacol. Sci. 2010, 31, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.O.; Alt, F.W. DNA double strand break repair and chromosomal translocation: Lessons from animal models. Oncogene 2001, 20, 5572–5579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Srihari, S.; Cao, K.-A.L.; Chenevix-Trench, G.; Simpson, P.T.; Ragan, M.A.; Khanna, K.K. A fine-scale dissection of the DNA double-strand break repair machinery and its implications for breast cancer therapy. Nucleic Acids Res. 2014, 42, 6106–6127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemberger, T.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: A nuclear receptor signaling pathway in lipid physiology. Ann. Rev. Cell Dev. Biol. 1996, 12, 335–363. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Nelson, M.C.; Ong, E.S.; Jones, Y.Z.; Ruiz-Lozano, P.; Chien, K.R.; Koder, A.; Evans, R.M. PPARγ is required for placental, cardiac, and adipose tissue development. Mol. Cell 1999, 4, 585–595. [Google Scholar] [CrossRef]
- Kubota, N.; Terauchi, Y.; Miki, H.; Tamemoto, H.; Yamauchi, T.; Komeda, K.; Satoh, S.; Nakano, R.; Ishii, C.; Sugiyama, T. PPARγ mediates high-fat diet–induced adipocyte hypertrophy and insulin resistance. Mol. Cell 1999, 4, 597–609. [Google Scholar] [CrossRef]
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Goto, T.; Kusudo, T.; Moriyama, T.; Kawada, T. The structures and functions of peroxisome proliferator-activated receptors (PPARs). Nihon rinsho. Jpn. J. Clin. Med. 2005, 63, 557–564. [Google Scholar]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Grüters, A. Severe early onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Kadekaro, A.L.; Kanto, H.; Kavanagh, R.; Abdel-Malek, Z.A. Significance of the melanocortin 1 receptor in regulating human melanocyte pigmentation, proliferation, and survival. Ann. N. Y. Acad. Sci. 2003, 994, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Qin, C. Medical Laboratory Animal Science; People’s Health Publishing House: Beijing, China, 2008. [Google Scholar]
- Schwartz, S.; Kemnitz, J.; Howard, C., Jr. Obesity in free-ranging rhesus macaques. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 1993, 17, 1–9. [Google Scholar]
- Takahashi, T.; Higashino, A.; Takagi, K.; Kamanaka, Y.; Abe, M.; Morimoto, M.; Kang, K.H.; Goto, S.; Suzuki, J.; Hamada, Y. Characterization of obesity in Japanese monkeys (Macaca fuscata) in a pedigreed colony. J. Med. Primatol. 2006, 35, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.A.; Arndt, T.P.; Leslie, K.E.; Pearl, D.L.; Turner, P.V. Obesity in rhesus and cynomolgus macaques: A comparative review of the condition and its implications for research. Comp. Med. 2011, 61, 514–526. [Google Scholar] [PubMed]
- Kemnitz, J.W. Obesity in macaques: Spontaneous and induced. Adv. Vet. Sci. Comp. Med. 1984, 28, 81–114. [Google Scholar] [PubMed]
- Bremer, A.A.; Stanhope, K.L.; Graham, J.L.; Cummings, B.P.; Wang, W.; Saville, B.R.; Havel, P.J. Fructose-fed rhesus monkeys: A nonhuman primate model of insulin resistance, metabolic syndrome, and type 2 diabetes. Clin. Transl. Sci. 2011, 4, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.D.; Kavanagh, K.; Ward, G.M.; Auerbach, B.J.; Harwood, H.J., Jr.; Kaplan, J.R. Old world nonhuman primate models of type 2 diabetes mellitus. ILAR J. 2006, 47, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, S.; Ghosh, B.; Al-Dhubiab, B.E.; Nair, A.B. Understanding type 1 diabetes: Etiology and models. Can. J. Diabetes 2013, 37, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yu, L.; He, Y.; Wang, B. Nonhuman primate models of type 1 diabetes mellitus for islet transplantation. J. Diabetes Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, L.E.; Yin, W.T.; Busalacchi, K.B.; Chua, M.; O’Neal, W.K.; Grubb, B.R. Conditional deletion of dnaic1 in a murine model of primary ciliary dyskinesia causes chronic rhinosinusitis. Am. J. Resp. Cell Mol. Biol. 2010, 43, 55. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Lu, H.; Sun, J.; Pu, X.; Tan, R.; Ren, J. Spontaneous lesions of target tissue in Macaca fascicularis used in toxicity studies. Chin. J. Pharmacol. Toxicol. 2013, 27, 548. [Google Scholar]
- Cornish, A.S.; Gibbs, R.M.; Norgren, R.B. Exome screening to identify loss-of-function mutations in the rhesus macaque for development of preclinical models of human disease. BMC Genom. 2016, 17, 170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, J.; Tian, L.; Lu, D.; Yuan, K.; Yuan, Y.; Xu, S. Differential natural selection of human zinc transporter genes between African and Non-African populations. Sci. Rep. 2015, 5, 9658. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Tian, L.; Zhang, C.; Yuan, K.; Xu, S. Genetic diversity and natural selection footprints of the glycine amidinotransferase gene in various human populations. Sci. Rep. 2016, 6, 18755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schillaci, M.A. Sexual selection and the evolution of brain size in primates. PLoS ONE 2006, 1, e62. [Google Scholar] [CrossRef] [PubMed]
- Sequencing, T.M.G.; Consortium, A. The common marmoset genome provides insight into primate biology and evolution. Nat. Genet. 2014, 46, 850. [Google Scholar]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duforet-Frebourg, N.; Luu, K.; Laval, G.; Bazin, E.; Blum, M.G. Detecting genomic signatures of natural selection with principal component analysis: Application to the 1000 Genomes data. Mol. Biol. Evol. 2015, 33, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Vallender, E.J.; Lahn, B.T. Positive selection on the human genome. Human Mol. Genet. 2004, 13, R245–R254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009, 41, 393. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chen, L.-L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.-B.; Hao, B.-H.; Lyon, M.P. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013, 45, 59. [Google Scholar] [CrossRef] [PubMed]
- Team, R. RStudio: Integrated Development Environment for R; RStudio Inc.: Boston, MA, USA, 2016. [Google Scholar]
- Reimand, J.; Arak, T.; Vilo, J. g:Profiler—A web server for functional interpretation of gene lists (2011 update). Nucleic Acids Res. 2016, 44, W83. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107. [Google Scholar] [CrossRef] [PubMed]
- Löytynoja, A.; Goldman, N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 2008, 320, 1632–1635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Liu, C.J.; Shi, M.; Kong, L.; Chen, J.Y.; Zhou, W.Z.; Zhu, X.; Yu, P.; Wang, J.; Yang, X. RhesusBase: A knowledgebase for the monkey research community. Nucleic Acids Res. 2013, 41, 892–905. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scientific Names | Species Symbol | Sample Identifier(s) | GenBank Accession(s) | Sequencing Platform(s) | #Reads | Depth | Total Usable Sites | Sex | Sample Origin(s) | Source(s) |
|---|---|---|---|---|---|---|---|---|---|---|
| M. mulatta mulatta | IR | IR | -- | Illumina | 20,100,000 | 5.1X | --- | Female | Washington National Primate Research Center | Gibbs et al. 2007 [23] |
| M. Mulatta lasiota | CR | CR1 | SRA023856 | Illumina | 3,299,851,568 | 45.65X | 2,264,143,011 | Female | Yunnan, China | Yan et al. 2011 [16] |
| M. fascicularis | CE | CE1 | SRA023855 | Illumina | 3,299,851,568 | 43.96X | 2,245,482,535 | Female | Vietnam | Yan et al. 2011 [16] |
| CE2 | -- | SOLiD 3+ | 3,692,987,634 | 24.69X | 2,261,105,771 | Female | Malaysia | Higashino et al. 2012 [24] | ||
| M. arctoides | SM | SM1 | SRX1470574 | Illumina | 1,001,034,260 | 34.55X | 2,280,352,231 | Female | Southwestern China | Fan et al. 2018 [25] |
| SM2 | SRX1470575 | Illumina | 471,805,366 | 20.51X | 2,079,812,789 | Female | Southwestern China | Fan et al. 2018 [25] | ||
| M. thibetana | TM | TM1 | SRP032525 | Illumina | 1,275,012,390 | 36.92X | 2,281,638,762 | Female | Sichuan, China | Fan et al. 2014 [22] |
| M. assamensis | AM | AM1 | SRX1470561 | Illumina | 1,231,654,664 | 54.04X | 2,011,347,545 | Male | Yunnan, China | Fan et al. 2018 [25] |
| M. nemestrina | PM | PM1 | SRX1022644 | Illumina | 770,413,198 | 25.59X | 2,246,079,419 | Female | Washington National Primate Research Center | Baylor College of Medicine |
| Species Symbol | Sample Identifier(s) | #SNVs | %SNVs | #Homo. | #Het. | %Het. | Ti/Tv | #Specific | %Specific | #Specific Het. |
|---|---|---|---|---|---|---|---|---|---|---|
| CR | CR1 | 9,384,359 | 3.47/kb | 3,458,482 | 5,925,877 | 63.15 | 2.23 | 2,614,186 | 27.86 | 2,297,127 |
| CE | CE1 | 11,751,302 | 4.35/kb | 5,004,945 | 6,746,357 | 57.41 | 2.21 | 2,892,996 | 24.62 | 2,464,551 |
| CE2 | 12,000,848 | 4.44/kb | 4,812,493 | 7,188,355 | 59.90 | 2.23 | 3,317,314 | 27.64 | 2,900,657 | |
| 5,089,889 † | -- | 2,712,160 | 2,377,729 | 46.71 | 2.25 | 762,701 | 14.98 | 369,245 | ||
| SM | SM1 | 12,712,801 | 4.69/kb | 8,985,648 | 3,727,153 | 29.32 | 2.21 | 803,928 | 6.32 | 740,450 |
| SM2 | 11,035,407 | 4.08/kb | 7,861,537 | 3,173,870 | 28.76 | 2.17 | 753,236 | 6.83 | 696,062 | |
| 9,353,661 † | -- | 6,931,659 | 2,422,002 | 25.89 | 2.19 | 2,005,117 | 21.43 | 661,821 | ||
| TM | TM1 | 11,937,445 | 4.42/kb | 9,889,106 | 2,048,339 | 17.16 | 2.21 | 1,633,457 | 13.68 | 701,115 |
| AM | AM1 | 12,249,208 | 4.52/kb | 6,770,425 | 5,478,783 | 44.73 | 2.17 | 2,638,128 | 21.54 | 2,300,208 |
| PM | PM1 | 13,914,612 | 5.15/kb | 7,613,888 | 6,300,724 | 45.28 | 2.18 | 5,307,739 | 38.15 | 3,860,264 |
| (a) | ||||||||||||
| Total SNVs | Intergenic | Genic | Up/Down Stream | Exonic | Splicing | Intronic | UTR | ncRNA | ||||
| CR1 | 9,384,359 | 6,172,058 | 65.77% | 3,212,301 | 34.23% | 144,929 | 56,515 | 0.60% | 426 | 2,976,764 | 28,536 | 5131 |
| CE1 | 11,751,302 | 7,595,956 | 64.64% | 4,155,346 | 35.36% | 181,392 | 71,807 | 0.61% | 524 | 3,858,404 | 37,045 | 6174 |
| CE2 | 12,000,848 | 7,836,824 | 65.30% | 4,164,024 | 34.70% | 189,573 | 82,821 | 0.69% | 545 | 3,844,219 | 39,977 | 6889 |
| SM | 9,353,661 | 5,956,902 | 63.69% | 3,396,759 | 36.31% | 149,351 | 63,179 | 0.68% | 411 | 3,145,443 | 33,775 | 4600 |
| TM1 | 11,937,445 | 7,642,434 | 64.02% | 4,295,011 | 35.98% | 188,920 | 77,819 | 0.65% | 519 | 3,980,620 | 40,541 | 6592 |
| AM1 | 12,249,208 | 7,880,410 | 64.33% | 4,368,798 | 35.67% | 184,730 | 73,890 | 0.60% | 505 | 4,063,127 | 40,412 | 6134 |
| PM1 | 13,914,612 | 8,944,532 | 64.28% | 4,970,080 | 35.72% | 214,348 | 87,789 | 0.63% | 612 | 4,613,059 | 46,798 | 7474 |
| (b) | ||||||||||||
| Synonymous | Nonsynonymous | Stop Codon Gain | Stop Codon Lost | |||||||||
| CR1 | All | 33,295 | 58.91% | 22,839 | 40.41% | 328 | 0.58% | 53 | 0.09% | |||
| Specific | 9576 | 60.79% | 6077 | 38.58% | 92 | 0.58% | 8 | 0.05% | ||||
| CE1 | All | 42,590 | 59.31% | 28,772 | 40.07% | 392 | 0.55% | 53 | 0.07% | |||
| Specific | 10,392 | 59.50% | 6963 | 39.87% | 101 | 0.58% | 10 | 0.06% | ||||
| CE2 | All | 48,535 | 58.60% | 33,769 | 40.77% | 464 | 0.56% | 53 | 0.06% | |||
| Specific | 12,259 | 59.93% | 8091 | 39.55% | 100 | 0.49% | 7 | 0.03% | ||||
| SM | All | 38,424 | 60.82% | 24,478 | 38.74% | 237 | 0.38% | 40 | 0.06% | |||
| Specific | 7753 | 61.25% | 4839 | 38.23% | 61 | 0.48% | 5 | 0.04% | ||||
| TM1 | All | 45,939 | 59.03% | 31,441 | 40.40% | 375 | 0.48% | 64 | 0.08% | |||
| Specific | 6057 | 58.76% | 4195 | 40.70% | 52 | 0.50% | 4 | 0.04% | ||||
| AM1 | All | 43,945 | 59.47% | 29,497 | 39.92% | 400 | 0.54% | 48 | 0.06% | |||
| Specific | 10,381 | 60.70% | 6599 | 38.58% | 115 | 0.67% | 8 | 0.05% | ||||
| PM1 | All | 51,763 | 58.96% | 35,503 | 40.44% | 466 | 0.53% | 57 | 0.06% | |||
| Specific | 20,034 | 61.25% | 12,507 | 38.24% | 154 | 0.47% | 15 | 0.05% | ||||
| (a) | |||||
| GO Terms | ID | Outlier Gene Counts | Genome-wide Gene Counts | pValue | Gene ID |
| Proteolysis | GO:0006508 | 16 | 151 | 0.0198 | ENSMMUG00000008649, ENSMMUG00000013071, ENSMMUG00000012849, ENSMMUG00000005527, ENSMMUG00000007838, ENSMMUG00000008264, ENSMMUG00000003339, ENSMMUG00000004413, ENSMMUG00000015029, ENSMMUG00000007209, ENSMMUG00000006734, ENSMMUG00000016370, ENSMMUG00000007785, ENSMMUG00000001344, ENSMMUG00000019294, ENSMMUG00000005120 |
| Regulation of proteolysis | GO:0030162 | 6 | 40 | 0.0374 | ENSMMUG00000001344, ENSMMUG00000012849, ENSMMUG00000005527, ENSMMUG00000007209, ENSMMUG00000006734, ENSMMUG00000005120 |
| Positive regulation of innate immune response | GO:0045089 | 3 | 12 | 0.0448 | ENSMMUG00000019932, ENSMMUG00000003373, ENSMMUG00000008854 |
| Cation transmembrane transporter activity | GO:0008324 | 12 | 116 | 0.0463 | ENSMMUG00000031030, ENSMMUG00000015607, ENSMMUG00000006442, ENSMMUG00000018390, ENSMMUG00000013626, ENSMMUG00000030358, ENSMMUG00000007087, ENSMMUG00000032213, ENSMMUG00000004969, ENSMMUG00000007062, ENSMMUG00000007061, ENSMMUG00000010257 |
| (b) | |||||
| Pathways | ID | Outlier Gene Counts | Genome-wide Gene Counts | pvalue | Gene ID |
| Taste transduction | mcc04742 | 10 | 70 | 0.0113 | ENSMMUG00000007062, ENSMMUG00000020698, ENSMMUG00000021005, ENSMMUG00000015717, ENSMMUG00000016272, ENSMMUG00000011771, ENSMMUG00000022440, ENSMMUG00000022439, ENSMMUG00000032291, ENSMMUG00000004773 |
| Homologous recombination | mcc03440 | 5 | 23 | 0.0163 | ENSMMUG00000007197, ENSMMUG00000003130, ENSMMUG00000022442, ENSMMUG00000014487, ENSMMUG00000019014 |
| Fat digestion and absorption | mcc04975 | 5 | 26 | 0.0247 | ENSMMUG00000007692, ENSMMUG00000000825, ENSMMUG00000031036, ENSMMUG00000000148, ENSMMUG00000002724 |
| Pancreatic secretion | mcc04972 | 9 | 73 | 0.0339 | ENSMMUG00000020698, ENSMMUG00000031036, ENSMMUG00000018390, ENSMMUG00000015298, ENSMMUG00000010306, ENSMMUG00000000148, ENSMMUG00000021397, ENSMMUG00000032208, ENSMMUG00000002724 |
| Species/Population Symbol | CR | CE_Viet | CE_Mal | SM | TM | AM | PM |
|---|---|---|---|---|---|---|---|
| Gene Counts | 13 | 14 | 17 | 18 | 12 | 18 | 33 |
| Gene Symbol | BRI3 † KSR1 ZMYND10 PGGT1B SHARPIN EVI2B EP400 RBP3 TBX4 MYRF CHST1 NCKAP5 ‡ SEC16B ‡ | KSR1 † PIN1 ZNF787DDT IGFL1 FCAMR CCDC33 ‡ EVC2 ‡ FRMPD2‡ SF3B1 FAM53A HAP1 ‡ KIAA0825 ZNF474 ‡ | CMYA5 †‡ BAHD1 EVI2B HTT ‡ ZNF646 KIAA1671 ‡ KCNMB2 NHLRC1 BAP1 RNF222 SH2D2A DROSHA | ETFB † ACE SKIV2L BAHD1 EP400 URB2 C9ORF131 PRR14L LMNB1 DIS3L2 KLF13 APOBR ‡ ASXL1 ‡ DACT2 AIM1 SPAG5 THEM6 RSL24D1 | CSRP1 † VSTM2L LYST ‡ DIS3L2 BAHD1 ACAN DDIAS KIAA1549 EXOSC6 THEM6 LYRM5 RSL24D 1 | KCNK1 † AFAP1L1 JSRP1 OGFR INPP5E MYD88 BAHD1 ASPM ‡ EVC2 LMNB1 KIF26B‡ KIAA1671 ‡ GAMT SRRM 2 AQP1 ‡ LYST ‡ ZNF330 THEM6 | HTT †‡, SRRM2 ‡ CCDC17 ‡, WHAMM BDP1 ‡, ALPK3 ‡ NDNF, ACAN ‡ TRIM28, LYST ‡ LRRC10B, RARRES1 ‡ CMIP, RCSD1 ERCC6 ‡, TFG GAMT, KIF26B ‡ SLC9C2, NAT6 DHRS9, EXO1 ‡ HAVCR1, TNK2 ‡ DDX31, XIRP2 ‡ WDR73, HVCN1 ‡ TMEM126B ‡, APOBR, NDUFV3 PRDM1, YWHAE |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Fan, Z.; Sun, T.; Peng, C.; Yue, B.; Li, J. Comparative Genome-Wide Survey of Single Nucleotide Variation Uncovers the Genetic Diversity and Potential Biomedical Applications among Six Macaca Species. Int. J. Mol. Sci. 2018, 19, 3123. https://doi.org/10.3390/ijms19103123
Li J, Fan Z, Sun T, Peng C, Yue B, Li J. Comparative Genome-Wide Survey of Single Nucleotide Variation Uncovers the Genetic Diversity and Potential Biomedical Applications among Six Macaca Species. International Journal of Molecular Sciences. 2018; 19(10):3123. https://doi.org/10.3390/ijms19103123
Chicago/Turabian StyleLi, Jing, Zhenxin Fan, Tianlin Sun, Changjun Peng, Bisong Yue, and Jing Li. 2018. "Comparative Genome-Wide Survey of Single Nucleotide Variation Uncovers the Genetic Diversity and Potential Biomedical Applications among Six Macaca Species" International Journal of Molecular Sciences 19, no. 10: 3123. https://doi.org/10.3390/ijms19103123