Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime


Abstract
:1. Introduction
2. Ca2+ Signaling in Normal Endothelial Cells: A Brief Introduction
3. Enhanced Neovascularization
3.1. Vanilloid Transient Receptor Potential 4 (TRPV4)
3.2. Piezo Proteins
3.3. P2X7 Receptors
3.4. Stim1, Orai1 and Canonical Transient Receptor Potential 1 (TRPC1)
3.5. Neuronal Nicotinic Receptors (nAchRs)
3.6. Gasotransmitters-Activated Ca2+-Permeable Channels
3.7. Connexin 40 (Cx40)
3.8. Na+/H+ Exchanger-1 (NHE-1)
3.9. Two-Pore Channels (TPCs)
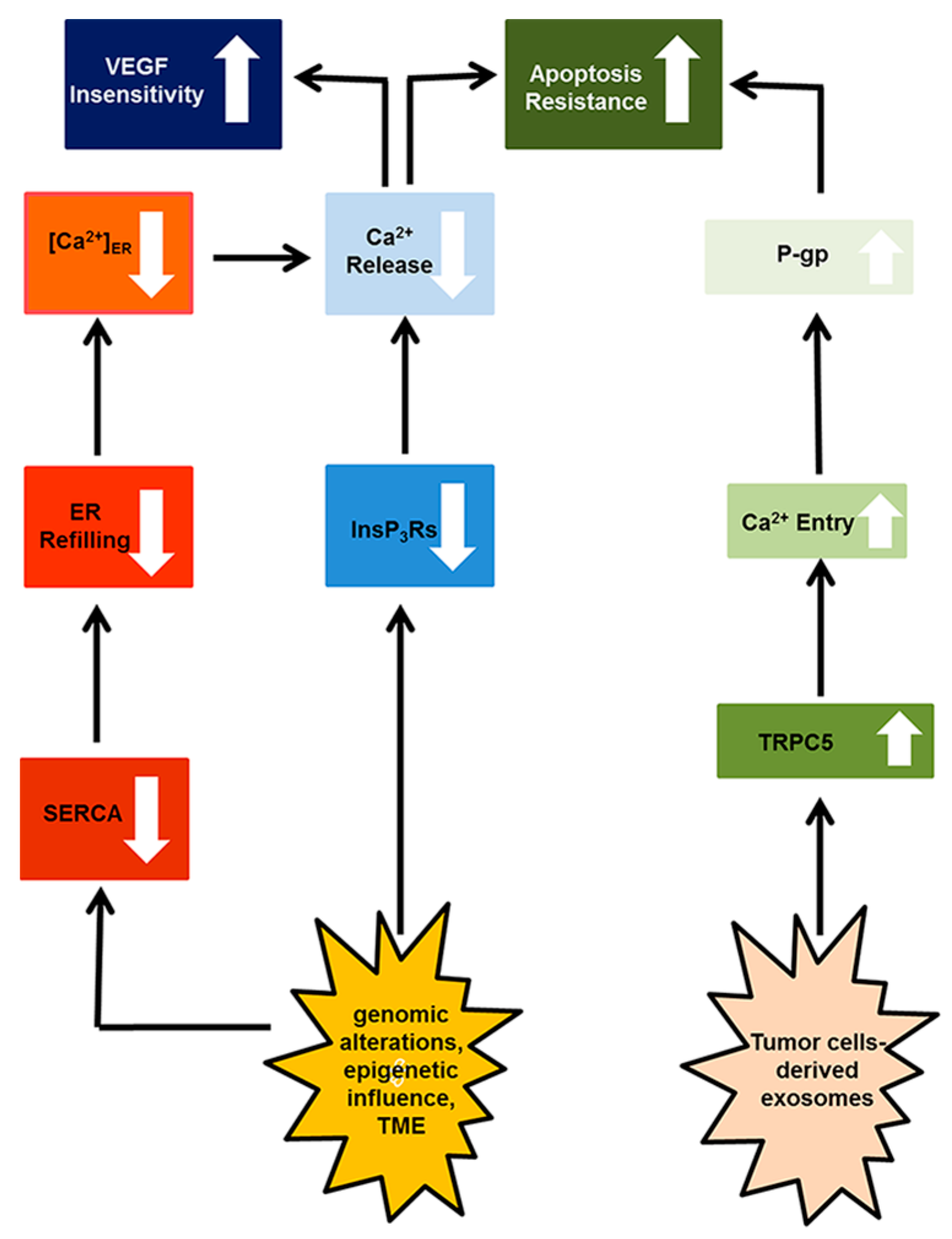
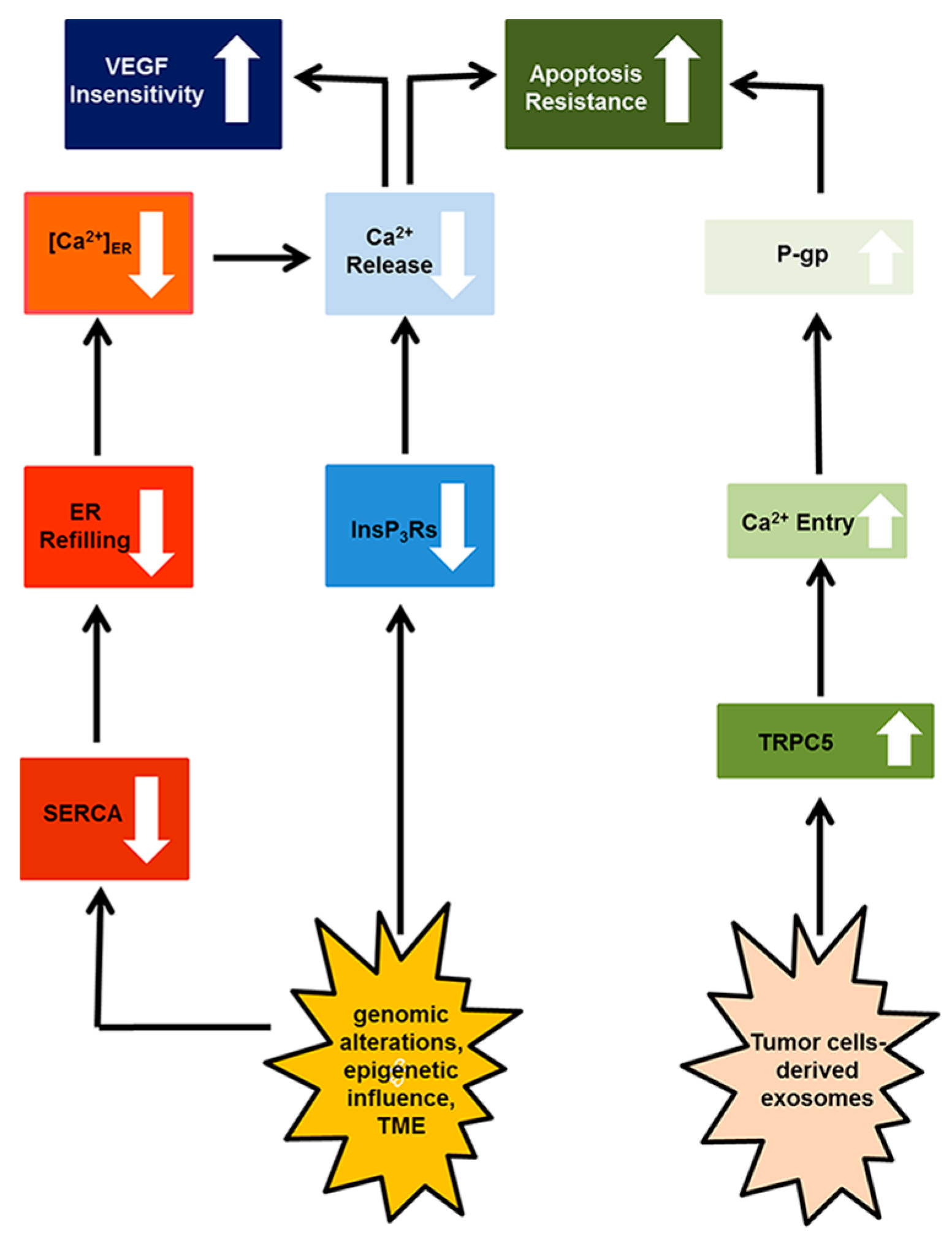
4. Resistance to Apoptosis
4.1. Canonical Transient Receptor Potential 5 (TRPC)
4.2. Inositol-1,4,5-Trisphosphate (InsP3) Receptors (InsP3Rs)
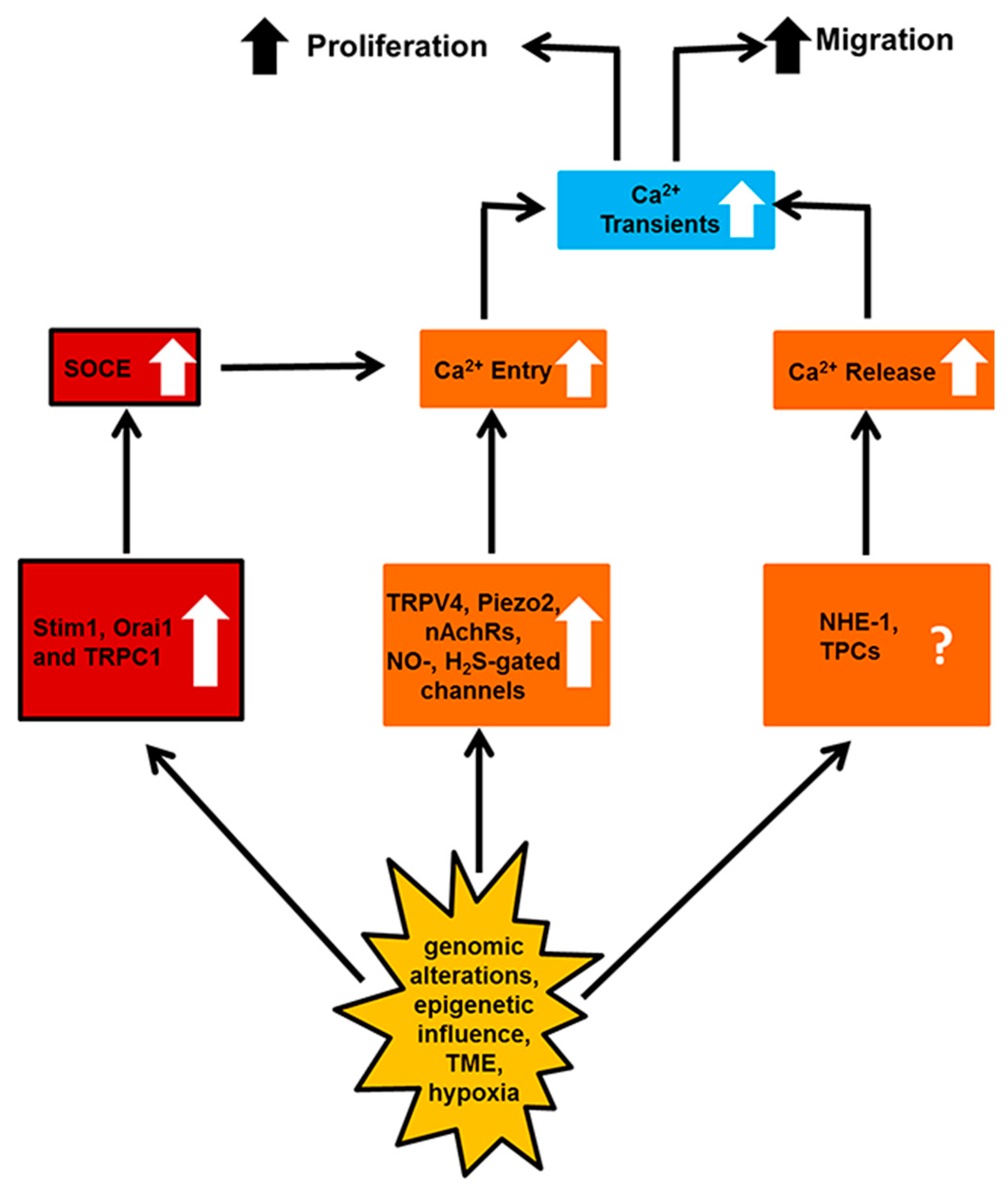
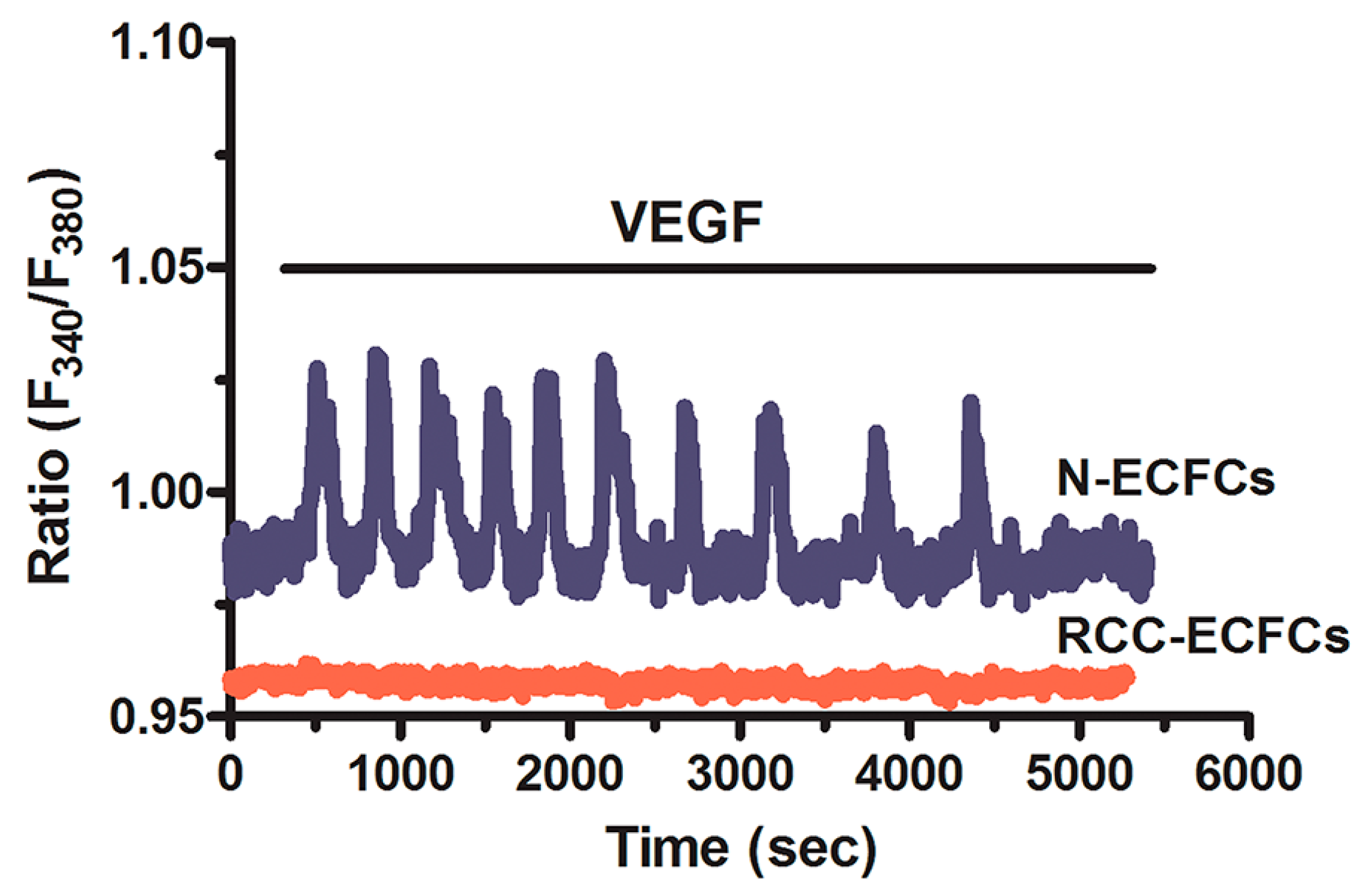
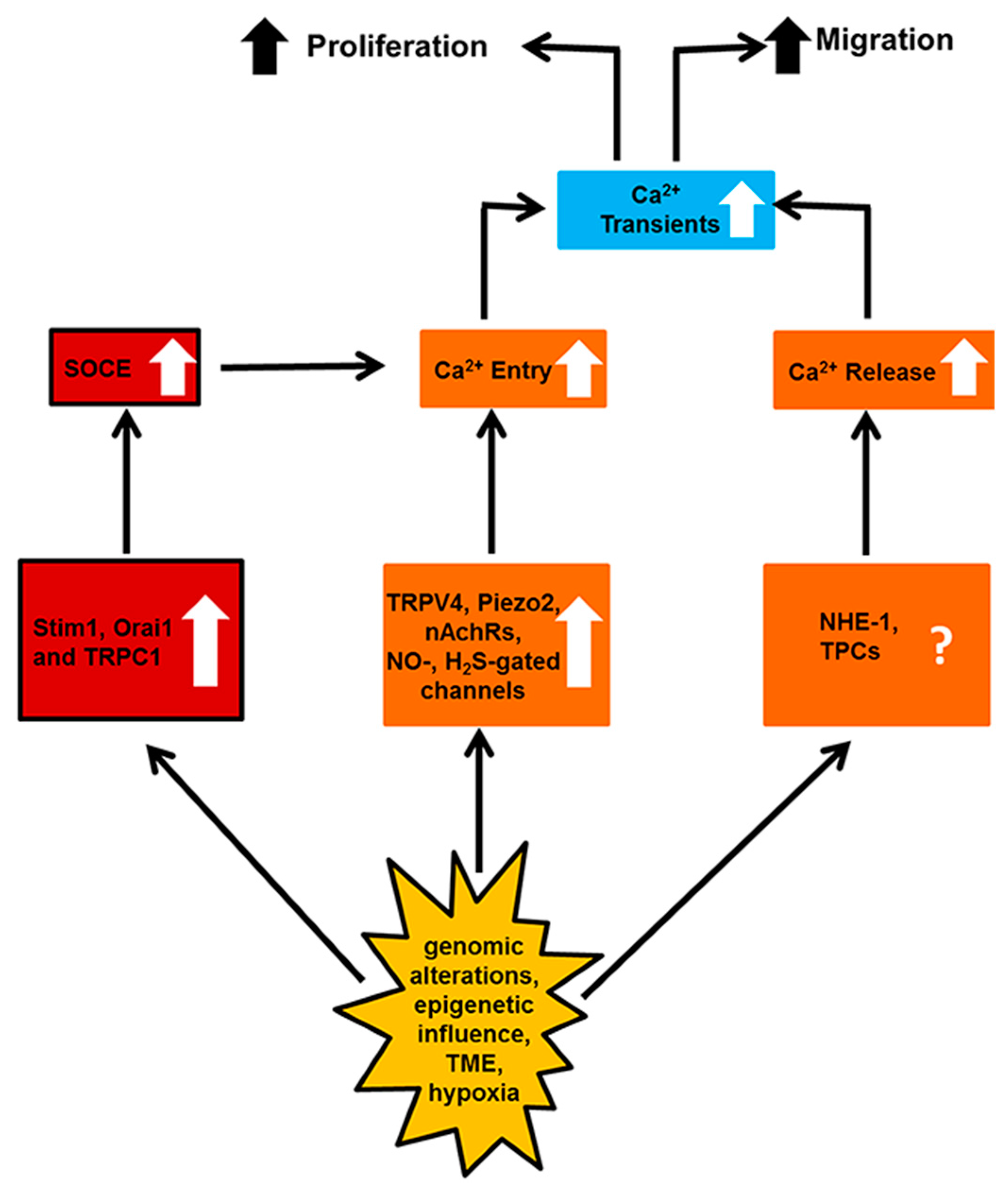
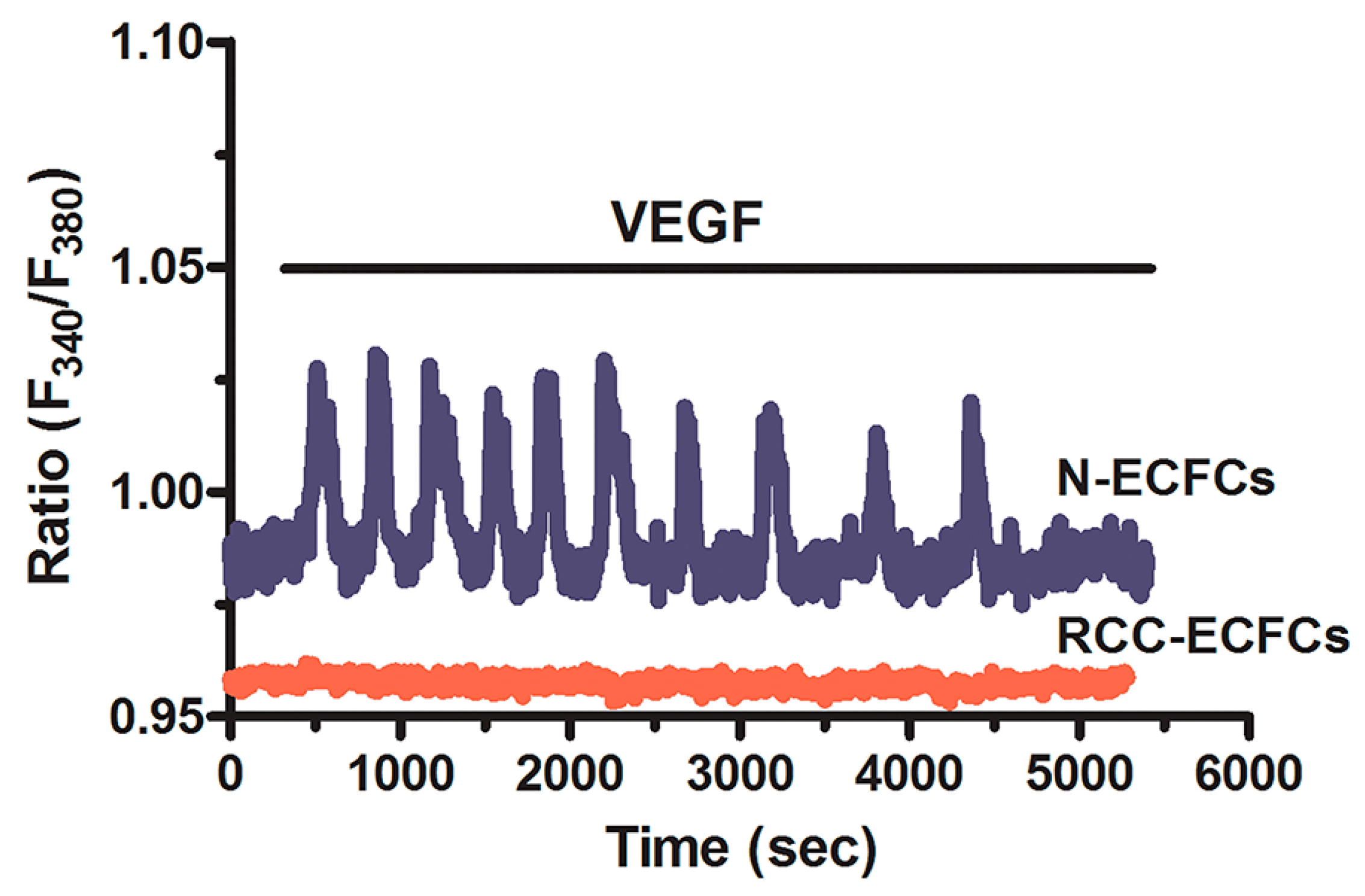
5. Targeting the Endothelial Ca2+ Toolkit to Circumvent the Resistance to Anticancer Treatments
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L.; Pla, A.F. Endothelial Calcium Machinery and Angiogenesis: Understanding Physiology to Interfere with Pathology. Curr. Med. Chem. 2009, 16, 4691–4703. [Google Scholar] [CrossRef] [PubMed]
- Troidl, C.; Nef, H.; Voss, S.; Schilp, A.; Kostin, S.; Troidl, K.; Szardien, S.; Rolf, A.; Schmitz-Rixen, T.; Schaper, W.; et al. Calcium-dependent signalling is essential during collateral growth in the pig hind limb-ischemia model. J. Mol. Cell. Cardiol. 2010, 49, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Troidl, C.; Troidl, K.; Schierling, W.; Cai, W.J.; Nef, H.; Mollmann, H.; Kostin, S.; Schimanski, S.; Hammer, L.; Elsasser, A.; et al. Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J. Cell. Mol. Med. 2009, 13, 2613–2621. [Google Scholar] [CrossRef] [PubMed]
- Potenza, D.M.; Guerra, G.; Avanzato, D.; Poletto, V.; Pareek, S.; Guido, D.; Gallanti, A.; Rosti, V.; Munaron, L.; Tanzi, F.; et al. Hydrogen sulphide triggers VEGF-induced intracellular Ca2+ signals in human endothelial cells but not in their immature progenitors. Cell Calcium 2014, 56, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Tritto, S.; Signorelli, S.; Taglietti, V.; Tanzi, F. Epidermal growth factor induces intracellular Ca2+ oscillations in microvascular endothelial cells. J. Cell. Physiol. 2003, 194, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Noren, D.P.; Chou, W.H.; Lee, S.H.; Qutub, A.A.; Warmflash, A.; Wagner, D.S.; Popel, A.S.; Levchenko, A. Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci. Signal. 2016, 9, ra20. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L.; Fiorio Pla, A. Calcium influx induced by activation of tyrosine kinase receptors in cultured bovine aortic endothelial cells. J. Cell. Physiol. 2000, 185, 454–463. [Google Scholar] [CrossRef]
- Gupta, S.K.; Lysko, P.G.; Pillarisetti, K.; Ohlstein, E.; Stadel, J.M. Chemokine receptors in human endothelial cells. Functional expression of CXCR4 and its transcriptional regulation by inflammatory cytokines. J. Biol. Chem. 1998, 273, 4282–4287. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Bonetti, E.; Dragoni, S.; Fontana, J.; Lodola, F.; Romani, R.B.; Laforenza, U.; Rosti, V.; Tanzi, F. Hematopoietic progenitor and stem cells circulate by surfing on intracellular Ca2+ waves: A novel target for cell-based therapy and anti-cancer treatment? Curr. Signal Trans. Ther. 2012, 7, 161–176. [Google Scholar] [CrossRef]
- Yang, C.; Ohk, J.; Lee, J.Y.; Kim, E.J.; Kim, J.; Han, S.; Park, D.; Jung, H.; Kim, C. Calmodulin Mediates Ca2+-Dependent Inhibition of Tie2 Signaling and Acts as a Developmental Brake During Embryonic Angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.P.; Luo, Y.G.; Chen, T.X.; Chen, F.R.; Wang, T.; Hu, Q. Ca2+ oscillation frequency regulates agonist-stimulated gene expression in vascular endothelial cells. J. Cell Sci. 2008, 121, 2511–2518. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhu, L.; Cai, L.; Zhang, J.; Zeng, X.; Li, J.; Su, Y.; Hu, Q. A stromal interaction molecule 1 variant up-regulates matrix metalloproteinase-2 expression by strengthening nucleoplasmic Ca2+ signaling. Biochim. Biophys. Acta 2016, 1863, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjo, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Avelino-Cruz, J.E.; Raqeeb, A.; Della Corte, A.; Cinelli, M.; Montagnani, S.; Guerra, G.; Moccia, F.; Tanzi, F. Ca2+-dependent nitric oxide release in the injured endothelium of excised rat aorta: A promising mechanism applying in vascular prosthetic devices in aging patients. BMC Surg. 2013, 13 (Suppl. 2), S40. [Google Scholar] [CrossRef] [PubMed]
- Charoensin, S.; Eroglu, E.; Opelt, M.; Bischof, H.; Madreiter-Sokolowski, C.T.; Kirsch, A.; Depaoli, M.R.; Frank, S.; Schrammel, A.; Mayer, B.; et al. Intact mitochondrial Ca2+ uniport is essential for agonist-induced activation of endothelial nitric oxide synthase (eNOS). Free Radic. Biol. Med. 2017, 102, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, P.; Eccles, S.A.; Yaqoob, M.M. Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell. Signal. 2017, 37, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Lyubchenko, T.; Woodward, H.; Veo, K.D.; Burns, N.; Nijmeh, H.; Liubchenko, G.A.; Stenmark, K.R.; Gerasimovskaya, E.V. P2Y1 and P2Y13 purinergic receptors mediate Ca2+ signaling and proliferative responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol. Cell Physiol. 2011, 300, C266–C275. [Google Scholar] [CrossRef] [PubMed]
- Sameermahmood, Z.; Balasubramanyam, M.; Saravanan, T.; Rema, M. Curcumin modulates SDF-1alpha/CXCR4-induced migration of human retinal endothelial cells (HRECs). Investig. Ophthalmol. Vis. Sci. 2008, 49, 3305–3311. [Google Scholar] [CrossRef] [PubMed]
- Pupo, E.; Pla, A.F.; Avanzato, D.; Moccia, F.; Cruz, J.E.; Tanzi, F.; Merlino, A.; Mancardi, D.; Munaron, L. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Radic. Biol. Med. 2011, 51, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Munaron, L. Functional properties of ion channels and transporters in tumour vascularization. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130103. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Remodelling of the Ca2+ Toolkit in Tumor Endothelium as a Crucial Responsible for the Resistance to Anticancer Therapies. Curr. Signal Trans. Ther. 2017, 12. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chiu, W.T.; Chen, Y.T.; Lin, P.Y.; Huang, H.J.; Chou, C.Y.; Chang, H.C.; Tang, M.J.; Shen, M.R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Carmeliet, P. SnapShot: Tumor angiogenesis. Cell 2012, 149, 1408.e1. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.C.; Nolan, D.; McDonnell, K.; Vahdat, L.; Benezra, R.; Altorki, N.; Mittal, V. Bone marrow-derived endothelial progenitor cells contribute to the angiogenic switch in tumor growth and metastatic progression. Biochim. Biophys. Acta 2009, 1796, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Cinelli, M.; Bonetti, E.; Guerra, G.; Rosti, V. Endothelial progenitor cells support tumour growth and metastatisation: Implications for the resistance to anti-angiogenic therapy. Tumour Biol. 2015, 36, 6603–6614. [Google Scholar] [CrossRef] [PubMed]
- Yoder, M.C.; Ingram, D.A. The definition of EPCs and other bone marrow cells contributing to neoangiogenesis and tumor growth: Is there common ground for understanding the roles of numerous marrow-derived cells in the neoangiogenic process? Biochim. Biophys. Acta 2009, 1796, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Guerra, G. Ca2+ Signalling in Endothelial Progenitor Cells: Friend or Foe? J. Cell. Physiol. 2016, 231, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Maeng, Y.S.; Choi, H.J.; Kwon, J.Y.; Park, Y.W.; Choi, K.S.; Min, J.K.; Kim, Y.H.; Suh, P.G.; Kang, K.S.; Won, M.H.; et al. Endothelial progenitor cell homing: Prominent role of the IGF2-IGF2R-PLCbeta2 axis. Blood 2009, 113, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Ruffinatti, F.A.; Zuccolo, E. Intracellular Ca2+ Signals to Reconstruct A Broken Heart: Still A Theoretical Approach? Curr. Drug Targets 2015, 16, 793–815. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Poletto, V. May the remodeling of the Ca2+ toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochim. Biophys. Acta 2015, 1853, 1958–1973. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Lodola, F.; Dragoni, S.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Ca2+ signalling in endothelial progenitor cells: A novel means to improve cell-based therapy and impair tumour vascularisation. Curr. Vasc. Pharmacol. 2014, 12, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Fotia, V.; Tancredi, R.; Della Porta, M.G.; Rosti, V.; Bonetti, E.; Poletto, V.; Marchini, S.; Beltrame, L.; Gallizzi, G.; et al. Breast and renal cancer-Derived endothelial colony forming cells share a common gene signature. Eur. J. Cancer 2017, 77, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Baruffi, S.; Spaggiari, S.; Signorelli, S.; Castelli, L.; Magistretti, J.; Taglietti, V.; Tanzi, F. Ca2+ uptake by the endoplasmic reticulum Ca2+-ATPase in rat microvascular endothelial cells. Biochem. J. 2002, 364 Pt 1, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Guzman-Silva, A.; Torres-Jacome, J.; Tanzi, F.; Moccia, F. Na+-Ca2+ exchanger contributes to Ca2+ extrusion in ATP-stimulated endothelium of intact rat aorta. Biochem. Biophys. Res. Commun. 2010, 395, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Paszty, K.; Caride, A.J.; Bajzer, Z.; Offord, C.P.; Padanyi, R.; Hegedus, L.; Varga, K.; Strehler, E.E.; Enyedi, A. Plasma membrane Ca2+-ATPases can shape the pattern of Ca2+ transients induced by store-operated Ca2+ entry. Sci. Signal. 2015, 8, ra19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas, C.; Foskett, J.K. Mitochondrial Ca2+ signals in autophagy. Cell Calcium 2012, 52, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Chu, T.F.; Chen, H.I.; Jen, C.J. Heterogeneity of [Ca2+](i) signaling in intact rat aortic endothelium. FASEB J. 2000, 14, 797–804. [Google Scholar] [PubMed]
- Duza, T.; Sarelius, I.H. Localized transient increases in endothelial cell Ca2+ in arterioles in situ: Implications for coordination of vascular function. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2322–H2331. [Google Scholar] [CrossRef] [PubMed]
- Kansui, Y.; Garland, C.J.; Dora, K.A. Enhanced spontaneous Ca2+ events in endothelial cells reflect signalling through myoendothelial gap junctions in pressurized mesenteric arteries. Cell Calcium 2008, 44, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Pedriali, G.; Rimessi, A.; Sbano, L.; Giorgi, C.; Wieckowski, M.R.; Previati, M.; Pinton, P. Regulation of Endoplasmic Reticulum-Mitochondria Ca2+ Transfer and Its Importance for Anti-Cancer Therapies. Front. Oncol. 2017, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Marcu, R.; Wiczer, B.M.; Neeley, C.K.; Hawkins, B.J. Mitochondrial matrix Ca2+ accumulation regulates cytosolic NAD(+)/NADH metabolism, protein acetylation, and sirtuin expression. Mol. Cell. Biol. 2014, 34, 2890–2902. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ Uniporter Is a Mitochondrial Luminal Redox Sensor that Augments MCU Channel Activity. Mol. Cell 2017, 65, 1014.e7–1028.e7. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, M.; Parys, J.B.; Pinton, P.; Bultynck, G. ER functions of oncogenes and tumor suppressors: Modulators of intracellular Ca2+ signaling. Biochim. Biophys. Acta 2016, 1863 Pt B, 1364–1378. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Pandolfi, P.P. The role of PML in the control of apoptotic cell fate: A new key player at ER-mitochondria sites. Cell Death Differ. 2011, 18, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jin, Q.; Li, Y.; Ma, Q.; Wang, J.; Li, D.; Zhou, H.; Chen, Y. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca2+]c/VDAC-[Ca2+]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones 2018, 23, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Gottschalk, B.; Parichatikanond, W.; Eroglu, E.; Klec, C.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Resveratrol Specifically Kills Cancer Cells by a Devastating Increase in the Ca2+ Coupling Between the Greatly Tethered Endoplasmic Reticulum and Mitochondria. Cell. Physiol. Biochem. 2016, 39, 1404–1420. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-dependent Ca2+ entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr. Med. Chem. 2012, 19, 5802–5818. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Blatter, L.A. Tissue Specificity: SOCE: Implications for Ca2+ Handling in Endothelial Cells. Adv. Exp. Med. Biol. 2017, 993, 343–361. [Google Scholar] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [PubMed]
- Sundivakkam, P.C.; Freichel, M.; Singh, V.; Yuan, J.P.; Vogel, S.M.; Flockerzi, V.; Malik, A.B.; Tiruppathi, C. The Ca2+ sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca2+ entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol. Pharmacol. 2012, 81, 510–526. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, D.L.; Wu, S.; Chen, H.; Alexeyev, M.; St Croix, C.M.; Pitt, B.R.; Uhlig, S.; Stevens, T. Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ. Res. 2012, 110, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Cioffi, D.L.; Alexeyev, M.; Rich, T.C.; Stevens, T. Sodium entry through endothelial store-operated calcium entry channels: Regulation by Orai1. Am. J. Physiol. Cell Physiol. 2015, 308, C277–C288. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Jousset, H.; Konig, S.; Frieden, M. Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium 2011, 49, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Mederos y Schnitzler, M.; Brandes, R.P.; Grimminger, F.; Meissner, M.; Freichel, M.; et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 2012, 3, 649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeldt, D.S.; Grummer, M.A.; Magness, R.R.; Bird, I.M. Altered VEGF-stimulated Ca2+ signaling in part underlies pregnancy-adapted eNOS activity in UAEC. J. Endocrinol. 2014, 223, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hamdollah Zadeh, M.A.; Glass, C.A.; Magnussen, A.; Hancox, J.C.; Bates, D.O. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 2008, 15, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Antoniotti, S.; Lovisolo, D.; Fiorio Pla, A.; Munaron, L. Expression and functional role of bTRPC1 channels in native endothelial cells. FEBS Lett. 2002, 510, 189–195. [Google Scholar] [CrossRef]
- Ho, W.S.; Zheng, X.; Zhang, D.X. Role of endothelial TRPV4 channels in vascular actions of the endocannabinoid, 2-arachidonoylglycerol. Br. J. Pharmacol. 2015, 172, 5251–5264. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zinkevich, N.S.; Gebremedhin, D.; Gauthier, K.M.; Nishijima, Y.; Fang, J.; Wilcox, D.A.; Campbell, W.B.; Gutterman, D.D.; Zhang, D.X. Arachidonic acid-induced dilation in human coronary arterioles: Convergence of signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry. J. Am. Heart Assoc. 2013, 2, e000080. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Inoue, R.; Morii, T.; Takahashi, N.; Yamamoto, S.; Hara, Y.; Tominaga, M.; Shimizu, S.; Sato, Y.; Mori, Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2006, 2, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, A.; Gudermann, T. Another TRP to endothelial dysfunction: TRPM2 and endothelial permeability. Circ. Res. 2008, 102, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.Y.; Cheng, K.T.; Ma, Y.; Huang, Y.; Tang, N.L.; Yu, S.; Yao, X. CNGA2 contributes to ATP-induced noncapacitative Ca2+ influx in vascular endothelial cells. J. Vasc. Res. 2010, 47, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Frost, C.; Berra-Romani, R.; Tanzi, F.; Adams, D.J. Expression and function of neuronal nicotinic ACh receptors in rat microvascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H486–H491. [Google Scholar] [CrossRef] [PubMed]
- LeMaistre, J.L.; Sanders, S.A.; Stobart, M.J.; Lu, L.; Knox, J.D.; Anderson, H.D.; Anderson, C.M. Coactivation of NMDA receptors by glutamate and D-serine induces dilation of isolated middle cerebral arteries. J. Cereb. Blood Flow Metab. 2012, 32, 537–547. [Google Scholar] [CrossRef] [PubMed]
- AbouAlaiwi, W.A.; Takahashi, M.; Mell, B.R.; Jones, T.J.; Ratnam, S.; Kolb, R.J.; Nauli, S.M. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ. Res. 2009, 104, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Berrout, J.; Jin, M.; O’Neil, R.G. Critical role of TRPP2 and TRPC1 channels in stretch-induced injury of blood-brain barrier endothelial cells. Brain Res. 2012, 1436, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Filosa, J.A.; Yao, X.; Rath, G. TRPV4 and the regulation of vascular tone. J. Cardiovasc. Pharmacol. 2013, 61, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014, 28, 4677–4685. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, B.; Tumova, S.; Muraki, K.; Bruns, A.; Ludlow, M.J.; Sedo, A.; Hyman, A.J.; McKeown, L.; Young, R.S.; et al. Piezo1 integration of vascular architecture with physiological force. Nature 2014, 515, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A Consensus Statement on Nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Hernandez, Y.; Laforenza, U.; Bonetti, E.; Fontana, J.; Dragoni, S.; Russo, M.; Avelino-Cruz, J.E.; Schinelli, S.; Testa, D.; Guerra, G.; et al. Store-operated Ca2+ entry is expressed in human endothelial progenitor cells. Stem Cells Dev. 2010, 19, 1967–1981. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Guerra, G.; Borghesi, A.; Stronati, M.; Rosti, V.; Tanzi, F.; et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013, 22, 2561–2580. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Poletto, V.; Dragoni, S.; Lim, D.; Biggiogera, M.; Aronica, A.; Cinelli, M.; De Luca, A.; Rosti, V.; Porta, C.; Guerra, G.; et al. Endoplasmic Reticulum Ca2+ Handling and Apoptotic Resistance in Tumor-Derived Endothelial Colony Forming Cells. J. Cell. Biochem. 2016, 117, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Pla, A.F.; Ong, H.L.; Cheng, K.T.; Brossa, A.; Bussolati, B.; Lockwich, T.; Paria, B.; Munaron, L.; Ambudkar, I.S. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 2012, 31, 200–212. [Google Scholar]
- Fiorio Pla, A.; Grange, C.; Antoniotti, S.; Tomatis, C.; Merlino, A.; Bussolati, B.; Munaron, L. Arachidonic acid-induced Ca2+ entry is involved in early steps of tumor angiogenesis. Mol. Cancer Res. 2008, 6, 535–545. [Google Scholar] [PubMed]
- Adapala, R.K.; Thoppil, R.J.; Ghosh, K.; Cappelli, H.C.; Dudley, A.C.; Paruchuri, S.; Keshamouni, V.; Klagsbrun, M.; Meszaros, J.G.; Chilian, W.M.; et al. Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene 2016, 35, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Thoppil, R.J.; Cappelli, H.C.; Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Thodeti, C.K. TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget 2016, 7, 25849–25861. [Google Scholar] [CrossRef] [PubMed]
- Thoppil, R.J.; Adapala, R.K.; Cappelli, H.C.; Kondeti, V.; Dudley, A.C.; Gary Meszaros, J.; Paruchuri, S.; Thodeti, C.K. TRPV4 channel activation selectively inhibits tumor endothelial cell proliferation. Sci. Rep. 2015, 5, 14257. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, C.; Zhou, R.M.; Yao, J.; Li, X.M.; Shen, Y.; Cheng, H.; Yuan, J.; Yan, B.; Jiang, Q. Piezo2 protein: A novel regulator of tumor angiogenesis and hyperpermeability. Oncotarget 2016, 7, 44630–44643. [Google Scholar] [CrossRef] [PubMed]
- Avanzato, D.; Genova, T.; Fiorio Pla, A.; Bernardini, M.; Bianco, S.; Bussolati, B.; Mancardi, D.; Giraudo, E.; Maione, F.; Cassoni, P.; et al. Activation of P2X7 and P2Y11 purinergic receptors inhibits migration and normalizes tumor-derived endothelial cells via cAMP signaling. Sci. Rep. 2016, 6, 32602. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Laforenza, U.; Bonetti, E.; Lim, D.; Dragoni, S.; Bottino, C.; Ong, H.L.; Guerra, G.; Ganini, C.; Massa, M.; et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS ONE 2012, 7, e42541. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, A.D.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [PubMed]
- Zuccolo, E.; Bottino, C.; Diofano, F.; Poletto, V.; Codazzi, A.C.; Mannarino, S.; Campanelli, R.; Fois, G.; Marseglia, G.L.; Guerra, G.; et al. Constitutive Store-Operated Ca2+ Entry Leads to Enhanced Nitric Oxide Production and Proliferation in Infantile Hemangioma-Derived Endothelial Colony-Forming Cells. Stem Cells Dev. 2016, 25, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Heeschen, C.; Weis, M.; Aicher, A.; Dimmeler, S.; Cooke, J.P. A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors. J. Clin. Investig. 2002, 110, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Heeschen, C.; Jang, J.J.; Weis, M.; Pathak, A.; Kaji, S.; Hu, R.S.; Tsao, P.S.; Johnson, F.L.; Cooke, J.P. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med. 2001, 7, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, Q.; Sun, J.; Yi, K.; Wu, L.; Tan, X. Nicotine improves the functional activity of late endothelial progenitor cells via nicotinic acetylcholine receptors. Biochem. Cell Biol. 2011, 89, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Natori, T.; Sata, M.; Washida, M.; Hirata, Y.; Nagai, R.; Makuuchi, M. Nicotine enhances neovascularization and promotes tumor growth. Mol. Cells 2003, 16, 143–146. [Google Scholar] [PubMed]
- Alonso, F.; Domingos-Pereira, S.; Le Gal, L.; Derre, L.; Meda, P.; Jichlinski, P.; Nardelli-Haefliger, D.; Haefliger, J.A. Targeting endothelial connexin40 inhibits tumor growth by reducing angiogenesis and improving vessel perfusion. Oncotarget 2016, 7, 14015–14028. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Scarpelli, A.; Lappano, R.; Pisano, A.; Santolla, M.F.; Avino, S.; De Marco, P.; Bussolati, B.; Maggiolini, M.; De Francesco, E.M. GPER is involved in the stimulatory effects of aldosterone in breast cancer cells and breast tumor-derived endothelial cells. Oncotarget 2016, 7, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Pan, Q.; Jiang, L.; Chen, Z.; Zhang, F.; Liu, Y.; Xing, H.; Shi, M.; Li, J.; Li, X.; et al. Tumor endothelial expression of P-glycoprotein upon microvesicular transfer of TrpC5 derived from adriamycin-resistant breast cancer cells. Biochem. Biophys. Res. Commun. 2014, 446, 85–90. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef] [PubMed]
- Everaerts, W.; Nilius, B.; Owsianik, G. The vanilloid transient receptor potential channel TRPV4: From structure to disease. Prog. Biophys. Mol. Biol. 2010, 103, 2–17. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Pan, Q.; Chen, Z.; Sun, C.; Zhang, P.; Mao, A.; Zhu, Y.; Li, H.; Lu, C.; Xie, M.; et al. Treatment of hypertension by increasing impaired endothelial TRPV4-KCa2.3 interaction. EMBO Mol. Med. 2017, 9, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Hsu, P.Y.; Wang, T.M.; Miao, Z.F.; Lin, R.T.; Juo, S.H. TRPV4 Activation Contributes Functional Recovery from Ischemic Stroke via Angiogenesis and Neurogenesis. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hatano, N.; Suzuki, H.; Itoh, Y.; Muraki, K. TRPV4 partially participates in proliferation of human brain capillary endothelial cells. Life Sci. 2013, 92, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.D.; Thodeti, C.K.; Tytell, J.D.; Mammoto, A.; Overby, D.R.; Ingber, D.E. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr. Biol. (Camb.) 2010, 2, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Thodeti, C.K.; Matthews, B.; Ravi, A.; Mammoto, A.; Ghosh, K.; Bracha, A.L.; Ingber, D.E. TRPV4 Channels Mediate Cyclic Strain-Induced Endothelial Cell Reorientation Through Integrin-to-Integrin Signaling. Circ. Res. 2009, 104, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012, 336, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sullivan, M.N.; Chase, M.; Gonzales, A.L.; Earley, S. Calcineurin/nuclear factor of activated T cells-coupled vanilliod transient receptor potential channel 4 Ca2+ sparklets stimulate airway smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.H.; Su, Y.C.; Lai, P.L.; Zhang, Y.; Xu, Y.F.; Zhao, A.; Yao, G.Y.; Jia, C.H.; Lin, J.; Xu, S.; et al. Critical role of arachidonic acid-activated mTOR signaling in breast carcinogenesis and angiogenesis. Oncogene 2013, 32, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L. Shuffling the cards in signal transduction: Calcium, arachidonic acid and mechanosensitivity. World J. Biol. Chem. 2011, 2, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Tunset, H.M.; Cebulla, J.; Vettukattil, R.; Helgesen, H.; Feuerherm, A.J.; Engebraten, O.; Maelandsmo, G.M.; Johansen, B.; Moestue, S.A. Anti-vascular effects of the cytosolic phospholipase A2 inhibitor AVX235 in a patient-derived basal-like breast cancer model. BMC Cancer 2016, 16, 191. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef] [PubMed]
- Bagriantsev, S.N.; Gracheva, E.O.; Gallagher, P.G. Piezo proteins: Regulators of mechanosensation and other cellular processes. J. Biol. Chem. 2014, 289, 31673–31681. [Google Scholar] [CrossRef] [PubMed]
- Honore, E.; Martins, J.R.; Penton, D.; Patel, A.; Demolombe, S. The Piezo Mechanosensitive Ion Channels: May the Force Be with You! Rev. Physiol. Biochem. Pharmacol. 2015, 169, 25–41. [Google Scholar] [PubMed]
- Ranade, S.S.; Qiu, Z.; Woo, S.H.; Hur, S.S.; Murthy, S.E.; Cahalan, S.M.; Xu, J.; Mathur, J.; Bandell, M.; Coste, B.; et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10347–10352. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Beilby, H.; Davis, F.M.; Marcial, D.L.; Kenny, P.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Altered purinergic receptor-Ca2+ signaling associated with hypoxia-induced epithelial-mesenchymal transition in breast cancer cells. Mol. Oncol. 2016, 10, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Chen, X.; Wang, S.; Xie, T.; Du, X.; Liu, H.; Li, X.; Chen, J.; Zhang, B.; Liang, H.; et al. The expression of P2X(7) receptors in EPCs and their potential role in the targeting of EPCs to brain gliomas. Cancer Biol. Ther. 2015, 16, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Guerra, G.; Fiorio Pla, A.; Bertoni, G.; Rappa, A.; Poletto, V.; Bottino, C.; Aronica, A.; Lodola, F.; Cinelli, M.P.; et al. A functional Transient Receptor Potential Vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J. Cell. Physiol. 2015, 230, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, N.A.; Barth, S.; Waldeck-Weiermair, M.; Klec, C.; Strunk, D.; Malli, R.; Graier, W.F. TRPV1 mediates cellular uptake of anandamide and thus promotes endothelial cell proliferation and network-formation. Biol. Open 2014, 3, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; Jang, S.I.; Ambudkar, I.S. Distinct contributions of Orai1 and TRPC1 to agonist-induced [Ca2+](i) signals determine specificity of Ca2+-dependent gene expression. PLoS ONE 2012, 7, e47146. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Dragoni, S.; Poletto, V.; Rosti, V.; Tanzi, F.; Ganini, C.; Porta, C. Orai1 and Transient Receptor Potential Channels as novel molecular targets to impair tumor neovascularisation in renal cell carcinoma and other malignancies. Anticancer Agents Med. Chem. 2014, 14, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Giglione, P.; Paglino, C. Targeted therapy for renal cell carcinoma: Focus on 2nd and 3rd line. Expert Opin. Pharmacother. 2016, 17, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C457–C469. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, B.; Xie, Q.; Ye, D.; Zhang, D.; Zhu, Y.; Chen, H.; Zhu, B. STIM1 Mediates Hypoxia-Driven Hepatocarcinogenesis via Interaction with HIF-1. Cell Rep. 2015, 12, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J.; Fu, L.; Minton, D.R.; Mongan, N.P.; Nanus, D.M. The role of HIF1alpha in renal cell carcinoma tumorigenesis. J. Mol. Med. (Berl.) 2014, 92, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of the breast cancer stem cell phenotype by hypoxia-inducible factors. Clin. Sci. (Lond.) 2015, 129, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Weigand, L.; Lu, W.; Sylvester, J.T.; Semenza, G.L.; Shimoda, L.A. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ. Res. 2006, 98, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Leaute-Labreze, C.; Harper, J.I.; Hoeger, P.H. Infantile haemangioma. Lancet 2017, 390, 85–94. [Google Scholar] [CrossRef]
- Bischoff, J. Progenitor cells in infantile hemangioma. J. Craniofac. Surg. 2009, 20 (Suppl. 1), 695–697. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.A.; Melero-Martin, J.M.; Wu, X.; Paruchuri, S.; Boscolo, E.; Mulliken, J.B.; Bischoff, J. Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood 2006, 108, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, S.; Bischoff, J. Infantile hemangioma-mechanism(s) of drug action on a vascular tumor. Cold Spring Harb. Perspect. Med. 2011, 1, a006460. [Google Scholar] [CrossRef] [PubMed]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and extraneuronal nicotinic acetylcholine receptors. Curr. Neuropharmacol. 2017. [Google Scholar] [CrossRef]
- Yakel, J.L. Nicotinic ACh receptors in the hippocampal circuit; functional expression and role in synaptic plasticity. J. Physiol. 2014, 592, 4147–4153. [Google Scholar] [CrossRef] [PubMed]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Nicotinic acetylcholine receptors in cancer: Multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol. Sci. 2008, 29, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.P.; Ghebremariam, Y.T. Endothelial nicotinic acetylcholine receptors and angiogenesis. Trends Cardiovasc. Med. 2008, 18, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Angiogenic activity of nicotinic acetylcholine receptors: Implications in tobacco-related vascular diseases. Pharmacol. Ther. 2009, 121, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Dom, A.M.; Buckley, A.W.; Brown, K.C.; Egleton, R.D.; Marcelo, A.J.; Proper, N.A.; Weller, D.E.; Shah, Y.H.; Lau, J.K.; Dasgupta, P. The alpha7-nicotinic acetylcholine receptor and MMP-2/-9 pathway mediate the proangiogenic effect of nicotine in human retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4428–4438. [Google Scholar] [CrossRef] [PubMed]
- Smedlund, K.; Tano, J.Y.; Margiotta, J.; Vazquez, G. Evidence for operation of nicotinic and muscarinic acetylcholine receptor-dependent survival pathways in human coronary artery endothelial cells. J. Cell Biochem. 2011, 112, 1978–1984. [Google Scholar] [CrossRef] [PubMed]
- Fucile, S. Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 2004, 35, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Guo, W.; Chen, W.; Fu, L.; Wang, J.; Tian, Y.; Xiao, X.; Kang, T.; Huang, W.; Deng, W. Nicotine promotes proliferation of human nasopharyngeal carcinoma cells by regulating alpha7AChR, ERK, HIF-1alpha and VEGF/PEDF signaling. PLoS ONE 2012, 7, e43898. [Google Scholar]
- Zhu, B.Q.; Heeschen, C.; Sievers, R.E.; Karliner, J.S.; Parmley, W.W.; Glantz, S.A.; Cooke, J.P. Second hand smoke stimulates tumor angiogenesis and growth. Cancer Cell 2003, 4, 191–196. [Google Scholar] [CrossRef]
- Altaany, Z.; Moccia, F.; Munaron, L.; Mancardi, D.; Wang, R. Hydrogen sulfide and endothelial dysfunction: Relationship with nitric oxide. Curr. Med. Chem. 2014, 21, 3646–3661. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.; Pla, A.F.; Moccia, F.; Tanzi, F.; Munaron, L. Old and new gasotransmitters in the cardiovascular system: Focus on the role of nitric oxide and hydrogen sulfide in endothelial cells and cardiomyocytes. Curr. Pharm. Biotechnol. 2011, 12, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Nussbaum, B.; Calzia, E.; Radermacher, P.; Wepler, M. Gaseous Mediators and Mitochondrial Function: The Future of Pharmacologically Induced Suspended Animation? Front. Physiol. 2017, 8, 691. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L. Intracellular calcium, endothelial cells and angiogenesis. Recent Pat. Anticancer Drug Discov. 2006, 1, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. Carbon monoxide: Pro- or anti-angiogenic agent? Comment on Ahmad et al. (Thromb Haemost 2015; 113: 329–337). Thromb. Haemost. 2015, 114, 432–433. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.N.; Boyd, N.H.; Walker, K.; Hjelmeland, A.B. NOS Expression and NO Function in Glioma and Implications for Patient Therapies. Antioxid. Redox Signal. 2017, 26, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Mocellin, S. Nitric oxide: Cancer target or anticancer agent? Curr. Cancer Drug Targets 2009, 9, 214–236. [Google Scholar] [CrossRef] [PubMed]
- Basudhar, D.; Somasundaram, V.; de Oliveira, G.A.; Kesarwala, A.; Heinecke, J.L.; Cheng, R.Y.; Glynn, S.A.; Ambs, S.; Wink, D.A.; Ridnour, L.A. Nitric Oxide Synthase-2-Derived Nitric Oxide Drives Multiple Pathways of Breast Cancer Progression. Antioxid. Redox Signal. 2017, 26, 1044–1058. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Genova, T.; Pupo, E.; Tomatis, C.; Genazzani, A.; Zaninetti, R.; Munaron, L. Multiple roles of protein kinase a in arachidonic acid-mediated Ca2+ entry and tumor-derived human endothelial cell migration. Mol. Cancer Res. 2010, 8, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Reforgiato, M.; Poletto, V.; Lodola, F.; Bottino, C.; Guido, D.; Rappa, A.; Pareek, S.; et al. Enhanced expression of Stim, Orai, and TRPC transcripts and proteins in endothelial progenitor cells isolated from patients with primary myelofibrosis. PLoS ONE 2014, 9, e91099. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L.; Avanzato, D.; Moccia, F.; Mancardi, D. Hydrogen sulfide as a regulator of calcium channels. Cell Calcium 2013, 53, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.C.; Boyle, J.P.; Porter, K.E.; Peers, C. Modulation of Ca2+ signalling in human vascular endothelial cells by hydrogen sulfide. Atherosclerosis 2010, 209, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Bertoni, G.; Pla, A.F.; Dragoni, S.; Pupo, E.; Merlino, A.; Mancardi, D.; Munaron, L.; Tanzi, F. Hydrogen sulfide regulates intracellular Ca2+ concentration in endothelial cells from excised rat aorta. Curr. Pharm. Biotechnol. 2011, 12, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Katsouda, A.; Bibli, S.I.; Pyriochou, A.; Szabo, C.; Papapetropoulos, A. Regulation and role of endogenously produced hydrogen sulfide in angiogenesis. Pharmacol. Res. 2016, 113 Pt A, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, R.; Liu, X.; Zhou, Y.; Qu, C.; Kikuiri, T.; Wang, S.; Zandi, E.; Du, J.; Ambudkar, I.S.; et al. Hydrogen Sulfide Maintains Mesenchymal Stem Cell Function and Bone Homeostasis via Regulation of Ca2+ Channel Sulfhydration. Cell Stem Cell 2014, 15, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Ujike, A.; Otsuguro, K.; Miyamoto, R.; Yamaguchi, S.; Ito, S. Bidirectional effects of hydrogen sulfide via ATP-sensitive K(+) channels and transient receptor potential A1 channels in RIN14B cells. Eur. J. Pharmacol. 2015, 764, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Nitric oxide inhibits capacitative Ca2+ entry and enhances endoplasmic reticulum Ca2+ uptake in bovine vascular endothelial cells. J. Physiol. 2002, 539 Pt 1, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Boeldt, D.S.; Krupp, J.; Yi, F.X.; Khurshid, N.; Shah, D.M.; Bird, I.M. Positive versus negative effects of VEGF165 on Ca2+ signaling and NO production in human endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H173–H181. [Google Scholar] [CrossRef] [PubMed]
- Boittin, F.X.; Alonso, F.; Le Gal, L.; Allagnat, F.; Beny, J.L.; Haefliger, J.A. Connexins and M3 muscarinic receptors contribute to heterogeneous Ca2+ signaling in mouse aortic endothelium. Cell. Physiol. Biochem. 2013, 31, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Culot, M.; Wang, N.; Bol, M.; Decrock, E.; De Vuyst, E.; da Costa, A.; Dauwe, I.; Vinken, M.; Simon, A.M.; et al. Connexin channels provide a target to manipulate brain endothelial calcium dynamics and blood-brain barrier permeability. J. Cereb. Blood Flow Metab. 2011, 31, 1942–1957. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Wang, N.; Decrock, E.; Bol, M.; Gadicherla, A.K.; Culot, M.; Cecchelli, R.; Bultynck, G.; Leybaert, L. Endothelial calcium dynamics, connexin channels and blood-brain barrier function. Prog. Neurobiol. 2013, 108, 1–20. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, E.; Shao, Q.; Wang, H.L.; Langlois, S.; Laird, D.W. Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res. 2006, 66, 9886–9894. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E.; Ruch, R.J. Gap junctions as targets for cancer chemoprevention and chemotherapy. Curr. Drug Targets 2002, 3, 465–482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; O’Carroll, S.J.; Henare, K.; Ching, L.M.; Ormonde, S.; Nicholson, L.F.; Danesh-Meyer, H.V.; Green, C.R. Connexin hemichannel induced vascular leak suggests a new paradigm for cancer therapy. FEBS Lett. 2014, 588, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Counillon, L.; Bouret, Y.; Marchiq, I.; Pouyssegur, J. Na(+)/H(+) antiporter (NHE1) and lactate/H(+) symporters (MCTs) in pH homeostasis and cancer metabolism. Biochim. Biophys. Acta 2016, 1863, 2465–2480. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.G.; Chen, Q.W.; Li, X.S.; Zheng, M.M.; Ke, D.Z.; Deng, W.; Li, G.Q.; Jiang, J.; Wu, Z.Q.; Wang, L.; et al. Suppression of NHE1 by small interfering RNA inhibits HIF-1alpha-induced angiogenesis in vitro via modulation of calpain activity. Microvasc. Res. 2011, 81, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Ayajiki, K.; Kindermann, M.; Hecker, M.; Fleming, I.; Busse, R. Intracellular pH and tyrosine phosphorylation but not calcium determine shear stress-induced nitric oxide production in native endothelial cells. Circ. Res. 1996, 78, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Yuen, N.; Lam, T.I.; Wallace, B.K.; Klug, N.R.; Anderson, S.E.; O’Donnell, M.E. Ischemic factor-induced increases in cerebral microvascular endothelial cell Na/H exchange activity and abundance: Evidence for involvement of ERK1/2 MAP kinase. Am. J. Physiol. Cell Physiol. 2014, 306, C931–C942. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, D.; Bussolino, F.; Garbarino, G.; Heller, R.; Turrini, F.; Pescarmona, G.; Cragoe, E.J., Jr.; Pegoraro, L.; Bosia, A. Role of Na+/H+ exchange in thrombin-induced platelet-activating factor production by human endothelial cells. J. Biol. Chem. 1988, 263, 19437–19446. [Google Scholar] [PubMed]
- Siffert, W.; Akkerman, J.W. Na+/H+ exchange and Ca2+ influx. FEBS Lett. 1989, 259, 1–4. [Google Scholar] [CrossRef]
- Danthuluri, N.R.; Kim, D.; Brock, T.A. Intracellular alkalinization leads to Ca2+ mobilization from agonist-sensitive pools in bovine aortic endothelial cells. J. Biol. Chem. 1990, 265, 19071–19076. [Google Scholar] [PubMed]
- Nishio, K.; Suzuki, Y.; Takeshita, K.; Aoki, T.; Kudo, H.; Sato, N.; Naoki, K.; Miyao, N.; Ishii, M.; Yamaguchi, K. Effects of hypercapnia and hypocapnia on [Ca2+]i mobilization in human pulmonary artery endothelial cells. J. Appl. Physiol. (1985) 2001, 90, 2094–2100. [Google Scholar] [CrossRef] [PubMed]
- Amith, S.R.; Fliegel, L. Regulation of the Na+/H+ Exchanger (NHE1) in Breast Cancer Metastasis. Cancer Res. 2013, 73, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Reshkin, S.J.; Greco, M.R.; Cardone, R.A. Role of pHi, and proton transporters in oncogene-driven neoplastic transformation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130100. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, A.K.; Mendes Lopes de Melo, J.; Morup, N.; Tritsaris, K.; Pedersen, S.F. Tumor microenvironment conditions alter Akt and Na+/H+ exchanger NHE1 expression in endothelial cells more than hypoxia alone: Implications for endothelial cell function in cancer. BMC Cancer 2017, 17, 542. [Google Scholar] [CrossRef] [PubMed]
- Orive, G.; Reshkin, S.J.; Harguindey, S.; Pedraz, J.L. Hydrogen ion dynamics and the Na+/H+ exchanger in cancer angiogenesis and antiangiogenesis. Br. J. Cancer 2003, 89, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Sonveaux, P.; Stock, C.; Perez-Sayans, M.; De Milito, A.; Avnet, S.; Garcia, A.G.; Harguindey, S.; Fais, S. Proton channels and exchangers in cancer. Biochim. Biophys. Acta 2015, 1848 Pt B, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.; Brailoiu, E.; Muallem, S. How does NAADP release lysosomal Ca2+? Channels 2014, 8, 174–175. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Cosker, F.; Cheviron, N.; Yamasaki, M.; Menteyne, A.; Lund, F.E.; Moutin, M.J.; Galione, A.; Cancela, J.M. The ecto-enzyme CD38 is a nicotinic acid adenine dinucleotide phosphate (NAADP) synthase that couples receptor activation to Ca2+ mobilization from lysosomes in pancreatic acinar cells. J. Biol. Chem. 2010, 285, 38251–38259. [Google Scholar] [CrossRef] [PubMed]
- Galione, A. A primer of NAADP-mediated Ca2+ signalling: From sea urchin eggs to mammalian cells. Cell Calcium 2015, 58, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Nusco, G.A.; Lim, D.; Kyozuka, K.; Santella, L. NAADP and InsP3 play distinct roles at fertilization in starfish oocytes. Dev. Biol. 2006, 294, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, G.C.; Gurzu, B.; Gao, X.; Parkesh, R.; Aley, P.K.; Trifa, D.I.; Galione, A.; Dun, N.J.; Madesh, M.; Patel, S.; et al. Acidic NAADP-sensitive calcium stores in the endothelium: Agonist-specific recruitment and role in regulating blood pressure. J. Biol. Chem. 2010, 285, 37133–37137. [Google Scholar] [CrossRef] [PubMed]
- Di Nezza, F.; Zuccolo, E.; Poletto, V.; Rosti, V.; De Luca, A.; Moccia, F.; Guerra, G.; Ambrosone, L. Liposomes as a Putative Tool to Investigate NAADP Signaling in Vasculogenesis. J. Cell. Biochem. 2017, 118, 3722–3729. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Pafumi, I.; Desideri, M.; Padula, F.; Montesano, C.; Passeri, D.; Nicoletti, C.; Orlandi, A.; Del Bufalo, D.; Sergi, M.; et al. NAADP-Dependent Ca2+ Signaling Controls Melanoma Progression, Metastatic Dissemination and Neoangiogenesis. Sci. Rep. 2016, 6, 18925. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.; Plant, T.D.; Obukhov, A.G.; Hofmann, T.; Gudermann, T.; Schultz, G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J. Biol. Chem. 2000, 275, 17517–17526. [Google Scholar] [CrossRef] [PubMed]
- Zholos, A.V. TRPC5. Handb. Exp. Pharmacol. 2014, 222, 129–156. [Google Scholar] [PubMed]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef] [PubMed]
- DeHaven, W.I.; Jones, B.F.; Petranka, J.G.; Smyth, J.T.; Tomita, T.; Bird, G.S.; Putney, J.W., Jr. TRPC channels function independently of STIM1 and Orai1. J. Physiol. 2009, 587 Pt 10, 2275–2298. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Lennerz, J.K.; Hein, A.; Link, A.S.; Kaczmarek, J.S.; Delling, M.; Uysal, S.; Pfeifer, J.D.; Riccio, A.; Clapham, D.E. Transient receptor potential cation channel, subfamily C, member 5 (TRPC5) is a cold-transducer in the peripheral nervous system. Proc. Natl. Acad. Sci. USA 2011, 108, 18114–18119. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Girardin, N.; Frieden, M. Transient Receptor Potential Canonical Channels Are Required for in Vitro Endothelial Tube Formation. J. Biol. Chem. 2012, 287, 5917–5927. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Colles, S.M.; Bhat, M.; Van Wagoner, D.R.; Birnbaumer, L.; Graham, L.M. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol. Biol. Cell 2008, 19, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Rosenbaum, M.A.; Birnbaumer, L.; Graham, L.M. Integration of TRPC6 and NADPH oxidase activation in lysophosphatidylcholine-induced TRPC5 externalization. Am. J. Physiol. Cell Physiol. 2017, 313, C541–C555. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cai, Y.; He, D.; Zou, C.; Zhang, P.; Lo, C.Y.; Xu, Z.; Chan, F.L.; Yu, S.; Chen, Y.; et al. Transient receptor potential channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16282–16287. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, Z.; Hua, D.; He, D.; Wang, L.; Zhang, P.; Wang, J.; Cai, Y.; Gao, C.; Zhang, X.; et al. Essential role for TrpC5-containing extracellular vesicles in breast cancer with chemotherapeutic resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 6389–6394. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chen, Z.; Zhu, Y.; Pan, Q.; Liu, Y.; Qi, X.; Jin, L.; Jin, J.; Ma, X.; Hua, D. Inhibition of transient receptor potential channel 5 reverses 5-Fluorouracil resistance in human colorectal cancer cells. J. Biol. Chem. 2015, 290, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, D.; Roseblade, A.; Oenarto, V.; Lu, J.F.; Bebawy, M. Proteins regulating the intercellular transfer and function of P-glycoprotein in multidrug-resistant cancer. Ecancermedicalscience 2017, 11, 768. [Google Scholar] [CrossRef] [PubMed]
- He, D.X.; Gu, X.T.; Jiang, L.; Jin, J.; Ma, X. A methylation-based regulatory network for microRNA 320a in chemoresistant breast cancer. Mol. Pharmacol. 2014, 86, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pan, Q.; Meng, H.; Jiang, Y.; Mao, A.; Wang, T.; Hua, D.; Yao, X.; Jin, J.; Ma, X. Enhancement of vascular endothelial growth factor release in long-term drug-treated breast cancer via transient receptor potential channel 5-Ca2+-hypoxia-inducible factor 1alpha pathway. Pharmacol. Res. 2015, 93, 36–42. [Google Scholar] [CrossRef] [PubMed]
- He, D.X.; Ma, X. Transient receptor potential channel C5 in cancer chemoresistance. Acta Pharmacol. Sin. 2016, 37, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Messai, Y.; Noman, M.Z.; Hasmim, M.; Janji, B.; Tittarelli, A.; Boutet, M.; Baud, V.; Viry, E.; Billot, K.; Nanbakhsh, A.; et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res. 2014, 74, 6820–6832. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Bertoli, A.; Sorgato, M.C.; Moccia, F. Generation and usage of aequorin lentiviral vectors for Ca2+ measurement in sub-cellular compartments of hard-to-transfect cells. Cell Calcium 2016, 59, 228–239. [Google Scholar] [CrossRef] [PubMed]
- De la Puente, P.; Muz, B.; Azab, F.; Azab, A.K. Cell trafficking of endothelial progenitor cells in tumor progression. Clin. Cancer. Res. 2013, 19, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Szczylik, C.; Porta, C.; Gore, M. Treatment selection in metastatic renal cell carcinoma: Expert consensus. Nat. Rev. Clin. Oncol. 2012, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Imarisio, I.; Canipari, C.; Chen, K.; Neary, M.; Duh, M.S. Safety and treatment patterns of multikinase inhibitors in patients with metastatic renal cell carcinoma at a tertiary oncology center in Italy. BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Imarisio, I.; Ganini, C.; Sacchi, L.; Quaglini, S.; Giunta, V.; De Amici, M. Changes in circulating pro-angiogenic cytokines, other than VEGF, before progression to sunitinib therapy in advanced renal cell carcinoma patients. Oncology 2013, 84, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. Tumor refractoriness to anti-VEGF therapy. Oncotarget 2016, 7, 46668–46677. [Google Scholar] [CrossRef] [PubMed]
- Hiles, J.J.; Kolesar, J.M. Role of sunitinib and sorafenib in the treatment of metastatic renal cell carcinoma. Am. J. Health Syst. Pharm. 2008, 65, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Wakabayashi, T.; Kidoya, H.; Muramatsu, F.; Takara, K.; Eino, D.; Yamane, K.; Iba, T.; Takakura, N. Endothelial Side Population Cells Contribute to Tumor Angiogenesis and Antiangiogenic Drug Resistance. Cancer Res. 2016, 76, 3200–3210. [Google Scholar] [CrossRef] [PubMed]
- Loges, S.; Schmidt, T.; Carmeliet, P. Mechanisms of resistance to anti-angiogenic therapy and development of third-generation anti-angiogenic drug candidates. Genes Cancer 2010, 1, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Sunryd, J.C.; Cheon, B.; Graham, J.B.; Giorda, K.M.; Fissore, R.A.; Hebert, D.N. TMTC1 and TMTC2 are novel endoplasmic reticulum tetratricopeptide repeat-containing adapter proteins involved in calcium homeostasis. J. Biol. Chem. 2014, 289, 16085–16099. [Google Scholar] [CrossRef] [PubMed]
- Sammels, E.; Parys, J.B.; Missiaen, L.; De Smedt, H.; Bultynck, G. Intracellular Ca2+ storage in health and disease: A dynamic equilibrium. Cell Calcium 2010, 47, 297–314. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Prevarskaya, N. Targeting apoptosis by the remodelling of calcium-transporting proteins in cancerogenesis. FEBS J. 2013, 280, 5500–5510. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Targeting Ca2+ transport in cancer: Close reality or long perspective? Expert Opin. Ther. Targets 2013, 17, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Vanoverberghe, K.; Vanden Abeele, F.; Mariot, P.; Lepage, G.; Roudbaraki, M.; Bonnal, J.L.; Mauroy, B.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004, 11, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Giorgi, C.; Pinton, P. Novel frontiers in calcium signaling: A possible target for chemotherapy. Pharmacol. Res. 2015, 99, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Manago, A.; Zoratti, M.; Gulbins, E.; Szabo, I. Pharmacological targeting of ion channels for cancer therapy: In vivo evidences. Biochim. Biophys. Acta 2016, 1863 Pt B, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, N.; Lindemann, O.; Schwab, A. TRP channels and STIM/ORAI proteins: Sensors and effectors of cancer and stroma cell migration. Br. J. Pharmacol. 2014, 171, 5524–5540. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L. Systems biology of ion channels and transporters in tumor angiogenesis: An omics view. Biochim. Biophys. Acta 2015, 1848 Pt B, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Kito, H.; Yamamura, H.; Suzuki, Y.; Yamamura, H.; Ohya, S.; Asai, K.; Imaizumi, Y. Regulation of store-operated Ca2+ entry activity by cell cycle dependent up-regulation of Orai2 in brain capillary endothelial cells. Biochem. Biophys. Res. Commun. 2015, 459, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Park, E.; Jung, E.; Seong, Y.J.; Hong, M.; Lee, S.; Burford, J.; Gyarmati, G.; Peti-Peterdi, J.; Srikanth, S.; et al. ORAI1 Activates Proliferation of Lymphatic Endothelial Cells in Response to Laminar Flow Through Kruppel-Like Factors 2 and 4. Circ. Res. 2017, 120, 1426–1439. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Du, L.; Zhou, Y.; Li, M. Store-operated CRAC channel inhibitors: Opportunities and challenges. Future Med. Chem. 2016, 8, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Luzzi, K.J.; Varghese, H.J.; MacDonald, I.C.; Schmidt, E.E.; Kohn, E.C.; Morris, V.L.; Marshall, K.E.; Chambers, A.F.; Groom, A.C. Inhibition of angiogenesis in liver metastases by carboxyamidotriazole (CAI). Angiogenesis 1998, 2, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Oliver, V.K.; Patton, A.M.; Desai, S.; Lorang, D.; Libutti, S.K.; Kohn, E.C. Regulation of the pro-angiogenic microenvironment by carboxyamido-triazole. J. Cell. Physiol. 2003, 197, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Patton, A.M.; Kassis, J.; Doong, H.; Kohn, E.C. Calcium as a molecular target in angiogenesis. Curr. Pharm. Des. 2003, 9, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Mignen, O.; Brink, C.; Enfissi, A.; Nadkarni, A.; Shuttleworth, T.J.; Giovannucci, D.R.; Capiod, T. Carboxyamidotriazole-induced inhibition of mitochondrial calcium import blocks capacitative calcium entry and cell proliferation in HEK-293 cells. J. Cell Sci. 2005, 118 Pt 23, 5615–5623. [Google Scholar] [CrossRef] [PubMed]
- Enfissi, A.; Prigent, S.; Colosetti, P.; Capiod, T. The blocking of capacitative calcium entry by 2-aminoethyl diphenylborate (2-APB) and carboxyamidotriazole (CAI) inhibits proliferation in Hep G2 and Huh-7 human hepatoma cells. Cell Calcium 2004, 36, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Faehling, M.; Kroll, J.; Fohr, K.J.; Fellbrich, G.; Mayr, U.; Trischler, G.; Waltenberger, J. Essential role of calcium in vascular endothelial growth factor A-induced signaling: Mechanism of the antiangiogenic effect of carboxyamidotriazole. FASEB J. 2002, 16, 1805–1807. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Palad, A.J.; Wasilenko, W.J.; Blackmore, P.F.; Pincus, W.A.; Schechter, G.L.; Spoonster, J.R.; Kohn, E.C.; Somers, K.D. Inhibition of head and neck squamous cell carcinoma growth and invasion by the calcium influx inhibitor carboxyamido-triazole. Clin. Cancer. Res. 1997, 3, 1915–1921. [Google Scholar] [PubMed]
- Moody, T.W.; Chiles, J.; Moody, E.; Sieczkiewicz, G.J.; Kohn, E.C. CAI inhibits the growth of small cell lung cancer cells. Lung Cancer 2003, 39, 279–288. [Google Scholar] [CrossRef]
- Hussain, M.M.; Kotz, H.; Minasian, L.; Premkumar, A.; Sarosy, G.; Reed, E.; Zhai, S.; Steinberg, S.M.; Raggio, M.; Oliver, V.K.; et al. Phase II trial of carboxyamidotriazole in patients with relapsed epithelial ovarian cancer. J. Clin. Oncol. 2003, 21, 4356–4363. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Molema, G. Angiogenesis: Potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol. Rev. 2000, 52, 237–268. [Google Scholar] [PubMed]
- Stadler, W.M.; Rosner, G.; Small, E.; Hollis, D.; Rini, B.; Zaentz, S.D.; Mahoney, J.; Ratain, M.J. Successful implementation of the randomized discontinuation trial design: An application to the study of the putative antiangiogenic agent carboxyaminoimidazole in renal cell carcinoma—CALGB 69901. J. Clin. Oncol. 2005, 23, 3726–3732. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Rahman, T. Unveiling some FDA-approved drugs as inhibitors of the store-operated Ca2+ entry pathway. Sci. Rep. 2017, 7, 12881. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.; Duncton, M.A. TRPV4 agonists and antagonists. Curr. Top. Med. Chem. 2011, 11, 2216–2226. [Google Scholar] [CrossRef] [PubMed]
- Coddou, C.; Yan, Z.; Obsil, T.; Huidobro-Toro, J.P.; Stojilkovic, S.S. Activation and regulation of purinergic P2X receptor channels. Pharmacol. Rev. 2011, 63, 641–683. [Google Scholar] [CrossRef] [PubMed]
- Pafumi, I.; Favia, A.; Gambara, G.; Papacci, F.; Ziparo, E.; Palombi, F.; Filippini, A. Regulation of Angiogenic Functions by Angiopoietins through Calcium-Dependent Signaling Pathways. BioMed Res. Int. 2015, 2015, 965271. [Google Scholar] [CrossRef] [PubMed]
- Rubaiy, H.N.; Ludlow, M.J.; Bon, R.S.; Beech, D.J. Pico145—Powerful new tool for TRPC1/4/5 channels. Channels 2017, 11, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Naylor, J.; Minard, A.; Gaunt, H.J.; Amer, M.S.; Wilson, L.A.; Migliore, M.; Cheung, S.Y.; Rubaiy, H.N.; Blythe, N.M.; Musialowski, K.E.; et al. Natural and synthetic flavonoid modulation of TRPC5 channels. Br. J. Pharmacol. 2016, 173, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lu, Y.; Qu, C.; Miller, M.; Tian, J.; Thakur, D.P.; Zhu, J.; Deng, Z.; Hu, X.; Wu, M.; et al. Identification and optimization of 2-aminobenzimidazole derivatives as novel inhibitors of TRPC4 and TRPC5 channels. Br. J. Pharmacol. 2015, 172, 3495–3509. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Shi, J.; Zhu, Y.; Kustov, M.; Tian, J.B.; Stevens, A.; Wu, M.; Xu, J.; Long, S.; Yang, P.; et al. Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J. Biol. Chem. 2011, 286, 33436–33446. [Google Scholar] [CrossRef] [PubMed]
- Majeed, Y.; Amer, M.S.; Agarwal, A.K.; McKeown, L.; Porter, K.E.; O’Regan, D.J.; Naylor, J.; Fishwick, C.W.; Muraki, K.; Beech, D.J. Stereo-selective inhibition of transient receptor potential TRPC5 cation channels by neuroactive steroids. Br. J. Pharmacol. 2011, 162, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Bonora, M.; Missiroli, S.; Poletti, F.; Ramirez, F.G.; Morciano, G.; Morganti, C.; Pandolfi, P.P.; Mammano, F.; Pinton, P. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget 2015, 6, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.T.; Paulsen, E.S.; Sehgal, P.; Moller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Dionne, C.A.; Christensen, S.B. Targeting thapsigargin towards tumors. Steroids 2015, 97, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra86. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Moy, P.; Kim, S.; Xia, Y.; Rajasekaran, A.; Navarro, V.; Knudsen, B.; Bander, N.H. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997, 57, 3629–3634. [Google Scholar] [PubMed]
- Quynh Doan, N.T.; Christensen, S.B. Thapsigargin, Origin, Chemistry, Structure-Activity Relationships and Prodrug Development. Curr. Pharm. Des. 2015, 21, 5501–5517. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Gkika, D. Emerging role of TRP channels in cell migration: From tumor vascularization to metastasis. Front. Physiol. 2013, 4, 311. [Google Scholar] [CrossRef] [PubMed]
- Munaron, L.; Genova, T.; Avanzato, D.; Antoniotti, S.; Fiorio Pla, A. Targeting calcium channels to block tumor vascularization. Recent Pat. Anticancer Drug Discov. 2013, 8, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, P.; Baba, A.; Matsuda, T.; Djamgoz, M.B.; Yaqoob, M.M.; Eccles, S.A. Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells. J. Biol. Chem. 2011, 286, 37919–37931. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharmacol. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef] [PubMed]
- Stobart, J.L.; Lu, L.; Anderson, H.D.; Mori, H.; Anderson, C.M. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2013, 110, 3149–3154. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Lin, J.H.; Arcuino, G.; Gao, Q.; Yang, J.; Nedergaard, M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001, 7, 1010–1015. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Channel/Transporter | Tumor and Cell Type (T-EC, T-ECFC T-EPC) | Expression Levels (Transcripts and/or Proteins) | Effect on Tumor Vascularization | Strategy to Target Tumor Vascularization | Ref. |
|---|---|---|---|---|---|
| TRPV4 | Breast Cancer: T-ECs | ↑ | Stimulates B-TEC proliferation, migration and in vitro tubulogenesis | Channel blockade with shTRPV4 or with CAI (0.1–10 µM) | [88,89] |
| TRPV4 | Lewis Lung Carcinoma: T-ECs (isolated from prostate adenocarcinoma) | ↓ | Inhibits T-EC mechanosensation, proliferation and migration in vitro and promotes the formation of a malfunctioning, leaky and exceedingly expanded vascular network in vivo | Injection of TRPV4 agonist GSK (10 μg/kg) to normalize tumor vasculature and favor cisplatin-induced tumor regression | [90,91,92] |
| Piezo2 proteins | Glioma: T-ECs | ↑ | Regulates tumor angiogenesis, vascular leakage and permeability | Blockade with siPiezo2 | [93] |
| P2X7Rs | Breast cancer: T-ECs | ↑ | Inhibits B-TEC migration and normalizes B-TECs-derived vessels in vitro | Activated by BzATP (50 µM) | [94] |
| Stim1, Orai1, TRPC1 | Renal cellular carcinoma: T-ECFCs | ↑ | Stimulate T-EPC proliferation and in vitro tubulogenesis | Blockade with siStim1 and siOrai1 and with YM-58483/BTP2 (20 µM), La3+ (10 µM), Gd3+ (10 µM), CAI (2–10 µM), 2-APB (50 µM), and genistein (50 µM) | [95] |
| Stim1, Orai1, TRPC1 | Breast cancer: T-ECFCs | = | Control T-ECFC proliferation and in vitro tubulogenesis | Blockade with YM-58483/BTP2 (20 µM), La3+ (10 µM), and CAI (10 µM) | [96] |
| Stim1, Orai1, TRPC1 | Infantile hemangioma: T-ECFCs | ↑ | Control T-ECFCs proliferation in vitro | Blockade with with YM-58483/BTP2 (20 µM), La3+ (10 µM), and Pyr6 (10 µM) | [97] |
| α7-nAchRs | Lewis lung carcinoma: T-ECs and T-EPCs | Not determined | Controls tumor growth and angiogenesis in vivo | Blockade with mecamylamine (1.0 μg/kg) or hexamethonium (1.0 μg/kg) | [98,99] |
| Stimulates EPC proliferation, migration and tubulogenesis in vitro and EPC recruitment in vivo | Blockade in vitro with mecamylamine (1 µM) and α-bungarotoxin (10 nM) and in vivo with mecamylamine (0.24 mg/kg per day) | [100,101] | |||
| Connexin40 | Melanoma and urogenital cancers: T-EC | ↑ | Stimulates tumor angiogenesis and growth in vivo | Blockade in vivo with 40Gap27 peptide (100 μg) | [102] |
| NHE-1 | Breast cancer: TECs | Not determined | Stimulates B-TEC migration in vitro | Blocked with siNHE-1 and with cariporide (50 µM) | [103] |
| Channel/Transporter | Tumor and Cell Type (T-EC and T-EPC) | Expression Levels | Effect on Tumor Vascularization | Strategy to Target Tumor Vascularization | Ref. |
|---|---|---|---|---|---|
| TRPC5 | Breast Cancer: T-ECs | ↑ | Stimulates endothelial resistance to adriamycin | Channel blockade with the specific blocking antibody T5E3 (concentration not reported) | [104] |
| InsP3Rs | RCC: T-ECFCs | ↓ | Favor T-ECFC resistance to rapamycin | Preventing InsP3-dependent ER–mitochondria Ca2+ shuttle with selective InsP3R inhibitors or cytosolic Ca2+ buffers (e.g., BAPTA) | [87] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. https://doi.org/10.3390/ijms19010217
Moccia F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. International Journal of Molecular Sciences. 2018; 19(1):217. https://doi.org/10.3390/ijms19010217
Chicago/Turabian StyleMoccia, Francesco. 2018. "Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime" International Journal of Molecular Sciences 19, no. 1: 217. https://doi.org/10.3390/ijms19010217
APA StyleMoccia, F. (2018). Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. International Journal of Molecular Sciences, 19(1), 217. https://doi.org/10.3390/ijms19010217