Transcriptional Regulation by CpG Sites Methylation in the Core Promoter Region of the Bovine SIX1 Gene: Roles of Histone H4 and E2F2

 , and
, and
Abstract
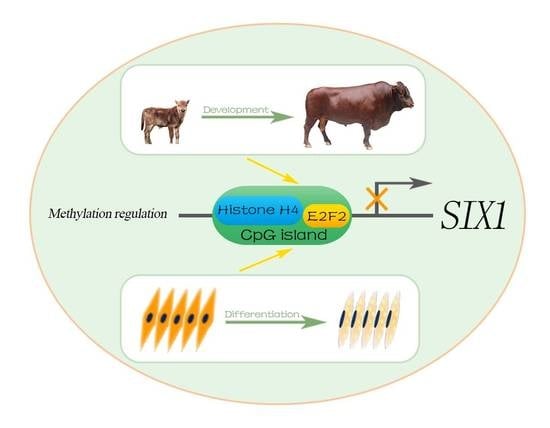
:
1. Introduction
2. Results
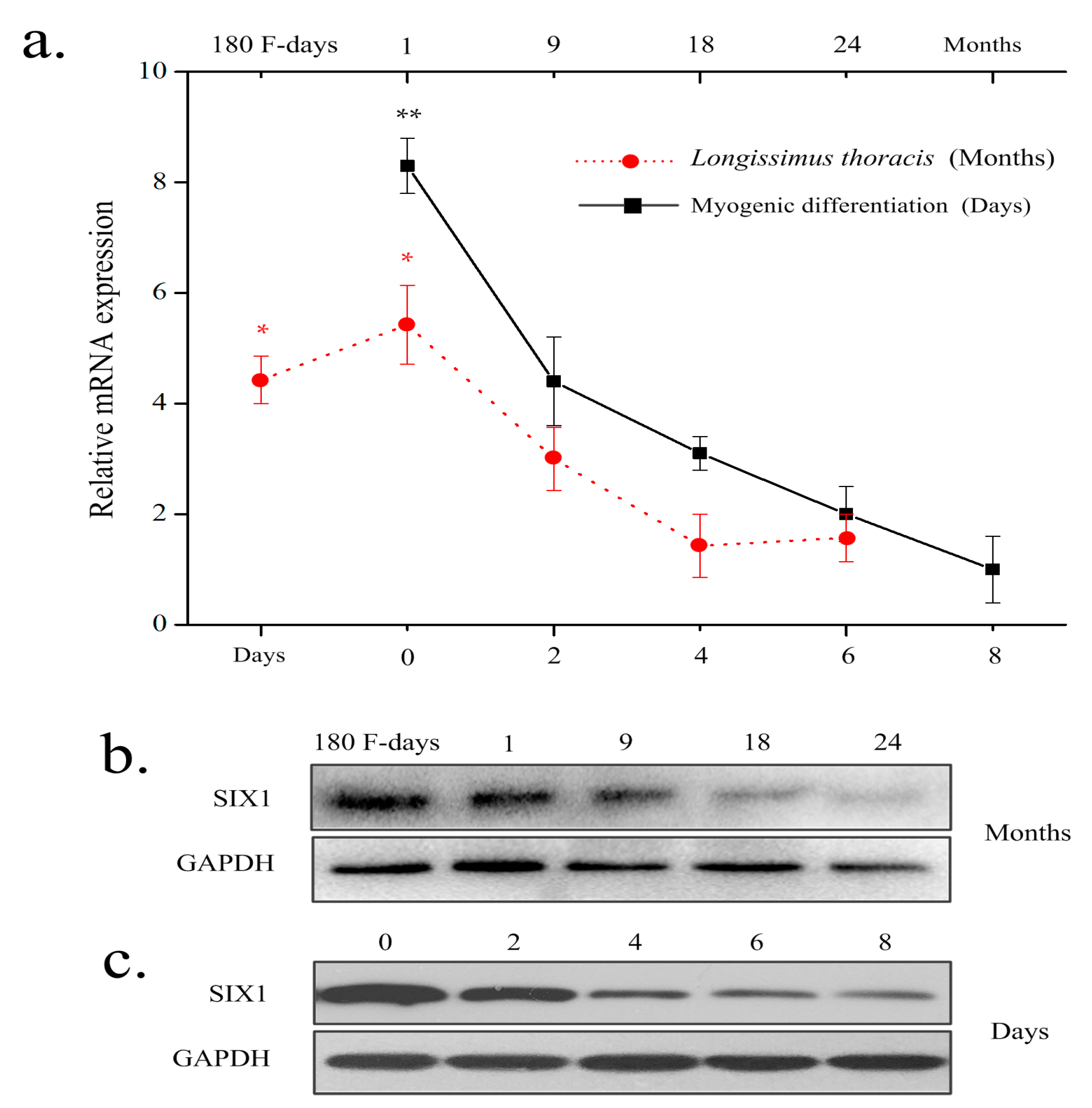
2.1. Expression Pattern of Bovine SIX1
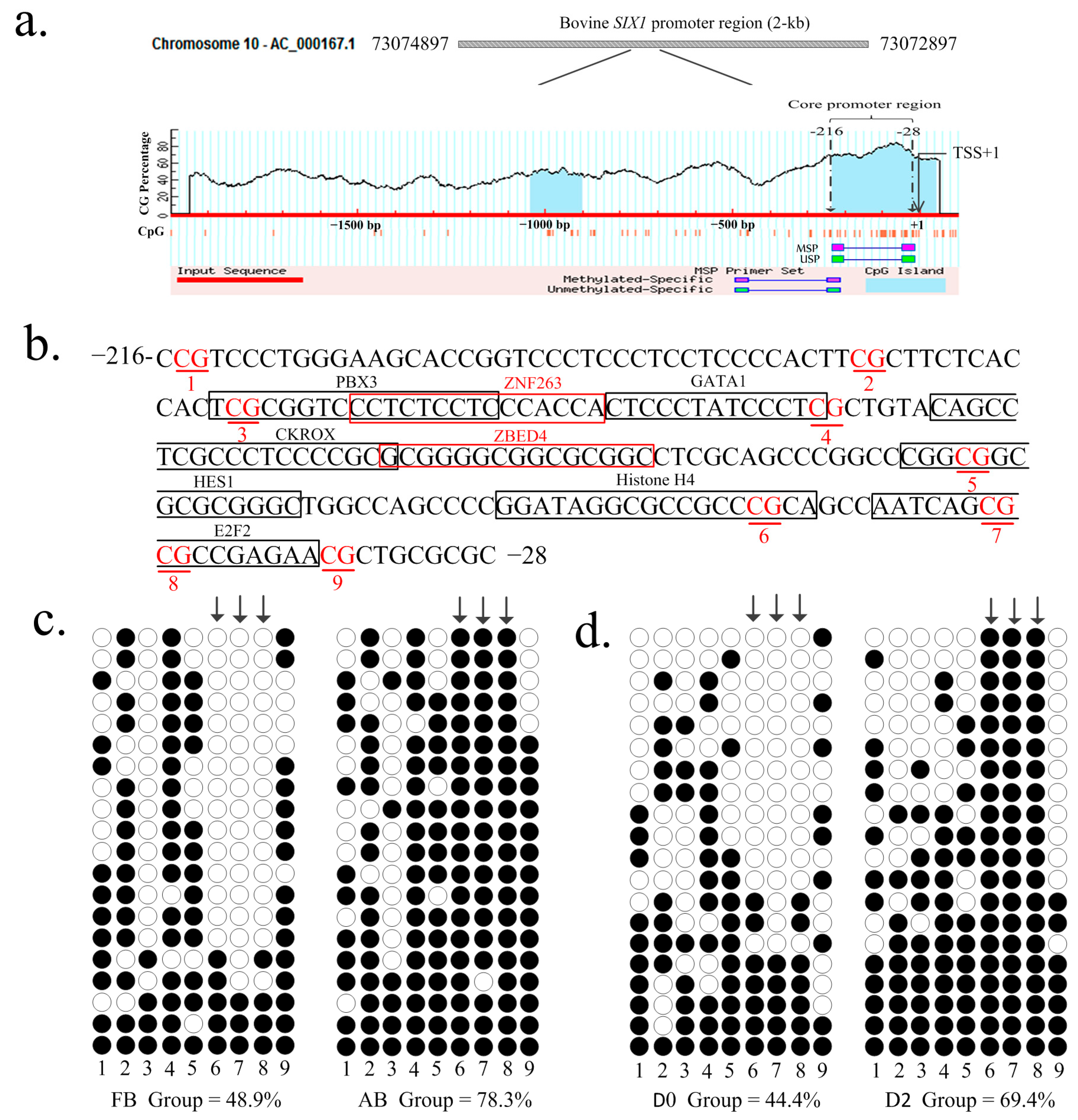
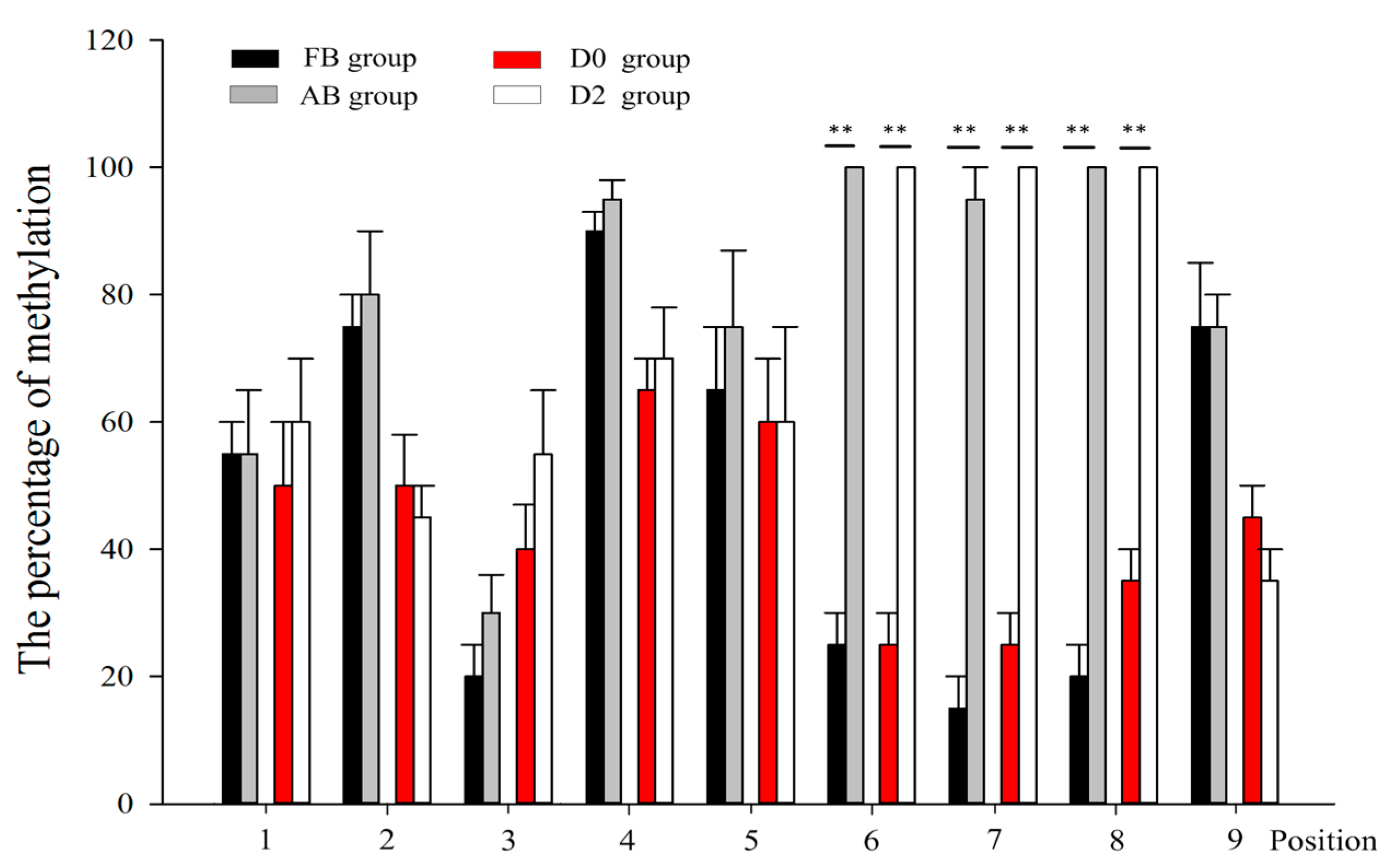
2.2. DNA Methylation Analysis by Bisulfite Sequencing Polymerase Chain Reaction (BSP)
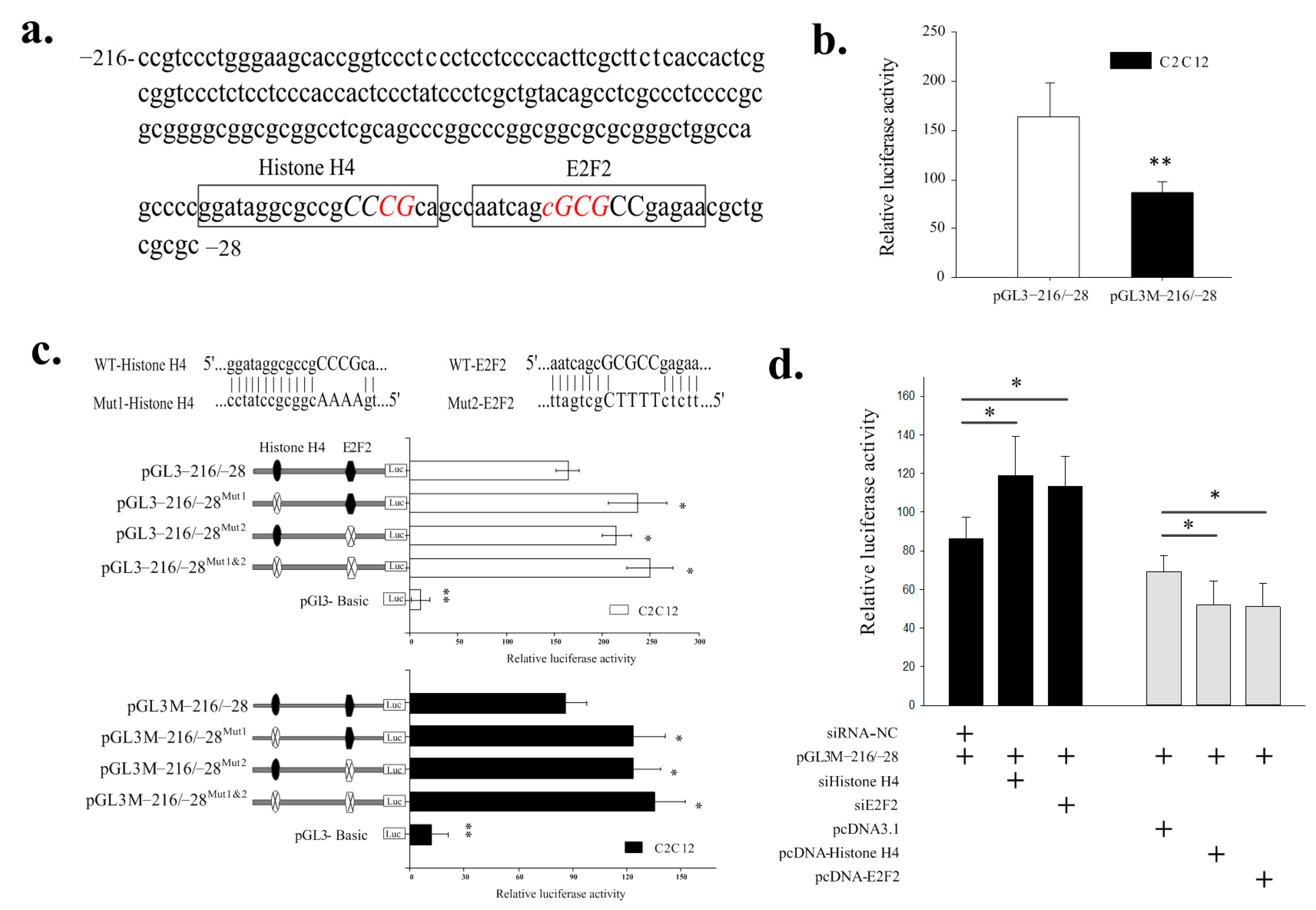
2.3. Identification of the Histone H4 and E2F2 Binding Sites as Transcriptional Repressors in the Core Promoter Region of the SIX1 Gene by DNA Methylation
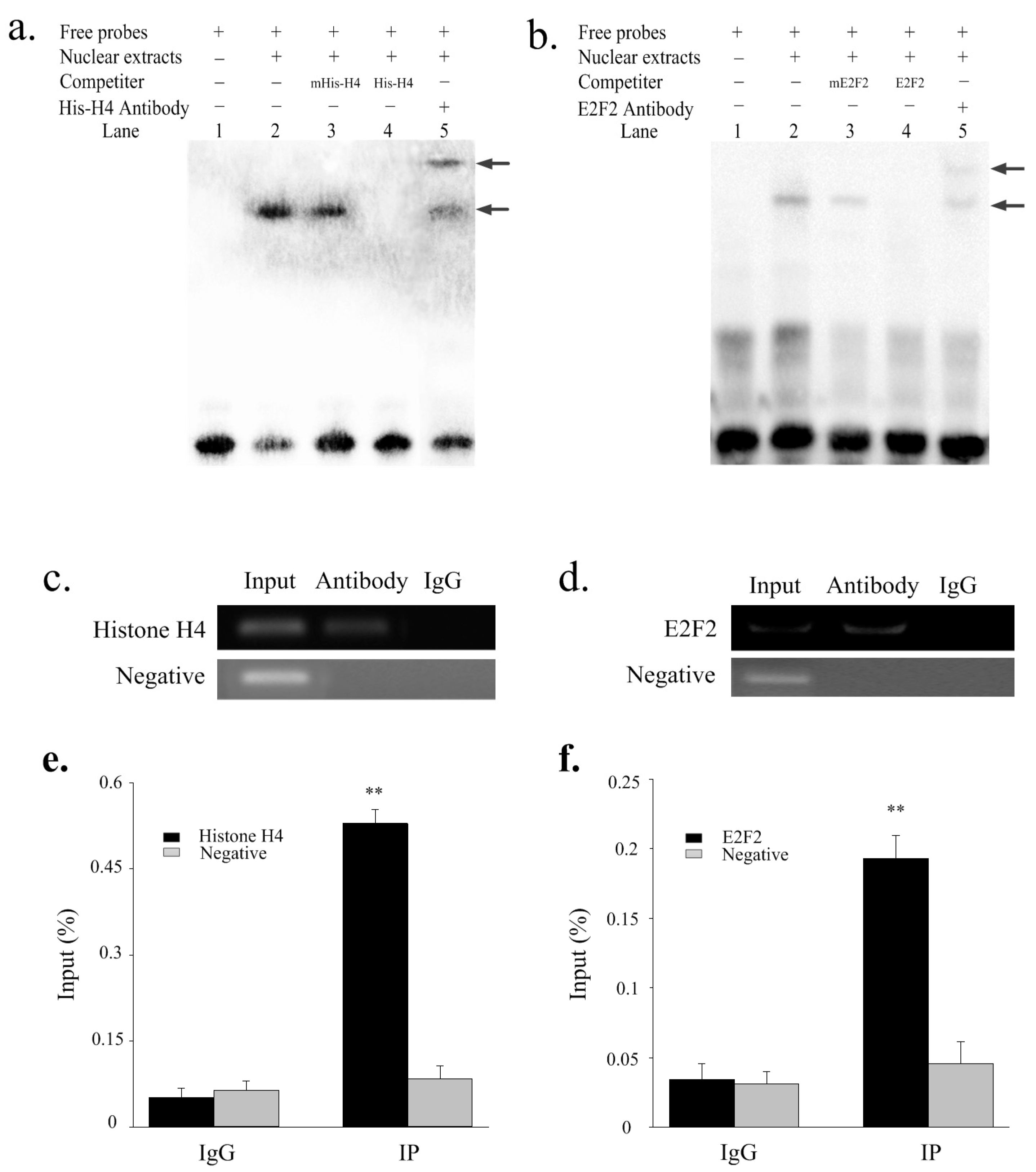
2.4. Histone H4 and E2F2 Bind to the SIX1 Promoter In Vitro and Vivo
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Quantitative PCR Analysis of Gene Expression Patterns
4.3. Western Blotting
4.4. DNA Preparation and Sodium Bisulfite Treatment and BSP
4.5. Potential TFs Luciferase Reporter Constructs and Substitution Mutation Constructs
4.6. In Vitro Methylation
4.7. Cell Transfection
4.8. Histone H4 and E2F2 Knockdown and Overexpression
4.9. Electrophoretic Mobility Shift Assays (EMSAs)
4.10. Chromatin Immunoprecipitation (ChIP) Assay
4.11. Statistical Analysis
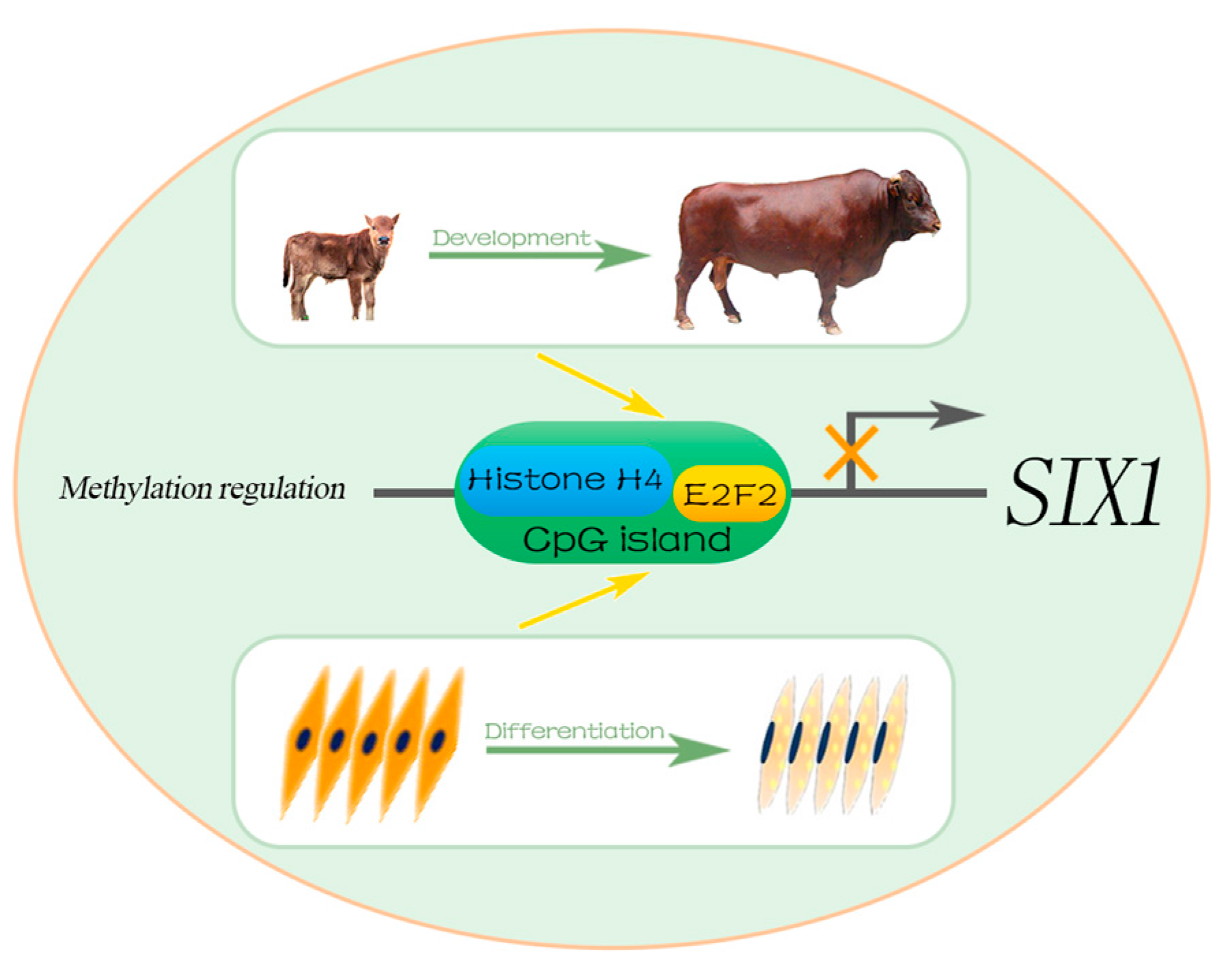
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TF | transcription factor |
| FB | fetal bovine |
| AB | adult bovine |
| QCMCs | Qinchuan cattle muscle cells |
| MRFs | myogenic regulatory factors |
| BSP | bisulfite sequencing polymerase |
| EMSA | electrophoretic mobility shift assay |
| ChIP | chromatin immunoprecipitation |
| TSS | transcriptional start sites |
| MSP | methylated-specific primer |
| USP | unmethylated-specific primer |
References
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Soloway, P.D. DNA methylation, imprinting and cancer. Eur. J. Hum. Genet. 2002, 10, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Takai, D.; Jones, P.A. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc. Natl. Acad. Sci. USA 2002, 99, 3740–3745. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.C.; Jones, P.A. Epigenetics and MicroRNAs. Pediatr. Res. 2007, 61, 24R–29R. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hu, X.; Li, J.; Du, Z.; Chen, L.; Yin, G.; Duan, J.; Zhang, H.; Zhao, Y.; Wang, J.; et al. Genome-wide mapping of DNA methylation in chicken. PLoS ONE 2011, 6, e19428. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Feng, S.; Joo, J.W.; Jacobsen, S.E.; Pellegrini, M. A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 2011, 12, R62. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Oghi, K.A.; Zhang, J.; Krones, A.; Bush, K.T.; Glass, C.K.; Nigam, S.K.; Aggarwal, A.K.; Maas, R.; Rose, D.W.; et al. Eya protein phosphatase activity regulates SIX1-dach-eya transcriptional effects in mammalian organogenesis. Nature 2003, 426, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Laclef, C.; Hamard, G.; Demignon, J.; Souil, E.; Houbron, C.; Maire, P. Altered myogenesis in SIX1-deficient mice. Development 2003, 130, 2239–2252. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Ookawara, S.; Sato, S.; Ando, Z.; Kageyama, R.; Kawakami, K. SIX1 is essential for early neurogenesis in the development of olfactory epithelium. Dev. Biol. 2007, 311, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Grand, F.L.; Grifone, R.; Mourikis, P.; Houbron, C.; Gigaud, C.; Pujol, J.; Marjorie, M.; Gilles, P.; Michael, R.; Shahragim, T.; et al. SIX1 regulates stem cell repair potential and self-renewal during skeletal muscle regeneration. J. Cell Biol. 2012, 198, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chu, A.; Chakroun, I.; Islam, U.; Blais, A. Cooperation between myogenic regulatory factors and SIX family transcription factors is important for myoblast differentiation. Nucleic Acids Res. 2010, 38, 6857–6871. [Google Scholar] [CrossRef] [PubMed]
- Yajima, H.; Motohashi, N.; Ono, Y.; Sato, S.; Ikeda, K.; Masuda, S.; Yada, E.; Kanesaki, H.; Miyagoe-Suzuki, Y.; Takeda, S.; et al. Six family genes control the proliferation and differentiation of muscle satellite cells. Exp. Cell Res. 2010, 316, 2932–2944. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Gui, L.; Raza, S.; Zhang, S.; Khan, R.; Wang, L.; Guo, H.; Zan, L. NRF1 and ZSCAN10 bind to the promoter region of the SIX1 gene and their effects body measurements in Qinchuan cattle. Sci. Rep. 2017, 7, 7867. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Ma, X.; Zhang, S.; Hong, J.; Gui, L.; Mei, C.; Guo, H.; Wang, L.; Ning, Y.; Zan, L. Characterization of the promoter region of the bovine SIX1 gene: Roles of MyoD, PAX7, CREB and MyoG. Sci. Rep. 2017, 77, 12599. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Ren, Z.; Liu, H.; Wang, L.; Huang, R.; Chen, J.; Zhang, L.; Li, P.; Xiong, Y. Core promoter analysis of porcine SIX1 gene and its regulation of the promoter activity by CpG methylation. Gene 2013, 529, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Takai, D. The role of DNA methylation in mammalian epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Borgel, J.; Guibert, S.; Li, Y.; Chiba, H.; Schübeler, D.; Sasaki, H.; Forné, T.; Weber, M. Targets and dynamics of promoter DNA methylation during early mouse development. Nat. Genet. 2010, 42, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tontifilippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kuick, R.; Hanash, S.; Richardson, B. DNA methylation inhibition increases T cell KIR expression through effects on both promoter methylation and transcription factors. Clin. Immunol. 2009, 130, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Osley, M.A. The regulation of histone synthesis in the cell cycle. Annu. Rev. Biochem. 1991, 60, 827–861. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, Z.Q.; Xia, L.; Feng, Q.; Erdjument-Bromage, H.; Strahl, B.D.; Briggs, S.D.; Allis, C.D.; Wong, J.; Tempst, P.; et al. Methylation of Histone H4 at Arginine 3 Facilitating Transcriptional Activation by Nuclear Hormone Receptor. Science 2001, 293, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.S.; Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Montecino, M.; Zaidi, S.K.; Braastad, C. An architectural perspective of cell-cycle control at the G1/S phase cell-cycle transition. J. Cell. Physiol. 2006, 209, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.J.; Xie, R.; Hussain, S.; Lian, J.B.; Riveraperez, J.; Jones, S.N.; Stein, J.L.; Stein, G.S.; Wijnen, A.J. Functional coupling of transcription factor HiNF-P and Histone H4 gene expression during pre- and post-natal mouse development. Gene 2011, 483, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA methylation and Histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Dimova, D.K.; Dyson, N.J. The E2F transcriptional network: Old acquaintances with new faces. Oncogene 2005, 24, 2810–2826. [Google Scholar] [CrossRef] [PubMed]
- Degregori, J.; Johnson, D. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Curr. Mol. Med. 2006, 6, 739–748. [Google Scholar] [PubMed]
- Murga, M.; Fernández-Capetillo, O.; Field, S.; Moreno, B.; Borlado, L.; Fujiwara, Y.; Balomenos, D.; Vicario, A.; Carrera, A.C.; Orkin, S.H.; et al. Mutation of E2F2 in Mice Causes Enhanced T Lymphocyte Proliferation, Leading to the Development of Autoimmunity. Immunity 2001, 15, 959–970. [Google Scholar] [CrossRef]
- DeGregori, J.; Johnson, D.G. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr. Mol. Med. 2006, 6, 739–748. [Google Scholar] [PubMed]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W., 3rd.; et al. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, H.; Schulz, R. Protection of CpG islands against de novo DNA methylation during oogenesis is associated with the recognition site of E2f1 and E2f2. Epigenet. Chromatin 2014, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Jones, P.; Barnard, V.; Hebert, C.; Terlecki, S.; Wijeratne, W. Estimation of the developmental age of the bovine fetus and newborn calf. Vet. Rec. 1990, 126, 279–284. [Google Scholar] [PubMed]
- Huang, Y.; Zhan, Z.; Sun, Y.; Cao, X.; Li, M.; Wang, J.; Lan, X.; Lei, C.; Zhang, C.; Chen, H. Intragenic DNA methylation status down-regulates bovine IGF2 gene expression in different developmental stages. Gene 2014, 534, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Kumaki, Y.; Oda, M.; Okano, M. QUMA: Quantification tool for methylation analysis. Nucleic Acids Res. 2008, 36, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.K.; James, J.C.; Mirmira, R.G. Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J. Biol. Chem. 2002, 277, 13286–13293. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Name | Primer Sequence (5′ to 3′) | Temperature (°C) | Product Length (bp) | Amplified Region |
|---|---|---|---|---|---|
| qRT-PCR | GAPDH | F: CCAACGTGTCTGTTGTGGAT
R: CTGCTTCACCACCTTCTTGA | 60.0 | 80 | 778/857 |
| SIX1 | F: GCCAAGGAAAGGGAGAACA
R: GACTCTGGGGAGGTGAGAACT | 60.0 | 127 | 866–992 | |
| Promoter cloning | SIX1-F/R | F:
CGGGGTACCCTCACGTTGCAAGGTCCTGAC R: GGAAGATCTGCGTTCTCGGCGCGCTGATTG | 61.5 | 189 | −216/−28 |
| Methylation analysis | SIX1-MSF/MSR | F: TCGTTTTTGGGAAGTATTGGT
R: ACACACAACGTTCTCAACG F: TTGTTTTTGGGAAGTATTGGT R: ACACACAACATTCTCAACA | 50.4 | 189 | −216/−28 |
| SIX1-USF/USR | 51.5 | 189 | −216/−28 | ||
| Site-mut and EMSA | Histone H4 forward | CCCGGATAGGCGCCG CCCGCAGCCAATCAGCGCG | −51/−17 | ||
| Histone H4 reverse | CGCGCTGATTGGCTGC GGGCGGCGCCTATCCGGG | ||||
| mHistone H4 forward | CCCGGATAGGCGCCG AAAACAGCCAATCAGCGCG | −51/−17 | |||
| mHistone H4 reverse | CGCGCTGATTGGCTGC TTTTGGCGCCTATCCGGG | ||||
| E2F2 forward | CCGCAGCCAATCAGC GCGCCGAGAACGCTGCGCG | −35/−2 | |||
| E2F2 reverse | CGCGCAGCGTTCTC GGCGCGCTGATTGGCTGCGG | ||||
| mE2F2 forward | CCGCAGCCAATCAGC GAAAAGAGAACGCTGCGCG | −35/−2 | |||
| mE2F2 reverse | CGCGCAGCGTTCTC TTTTCGCTGATTGGCTGCGG | ||||
| ChIP | ChIP-Histone H4/E2F2 | F: CCACCACTCCCTATCCCTCGCTGTA | 60 | 196 | −145/50 |
| R: TGCCGGGGAACTTGGTTTCTGTT | |||||
| ChIP-control | F: GTTTTGTTTACCACTAGCTTTTC | 60 | 134 | Exon 2 | |
| R: ATCCTTGTAGGAGTTCCCTTT | |||||
| siRNA | siHistone H4 | UGUGACUGUUGUGACUACAGCUGUATT | |||
| siE2F2 | GAUAUCCGUGCCGUAGGCAACUUCATT | ||||
| siRNA-NC | UUCUCCGAACGUGUCACGUTT | ||||
| Overexpression | Histone H4-CDSF/R | F: CCGATATCATGCCGCCACCTGGGAAAGTTC | 62 | 1551 | NM_001038531 |
| R: GCTCTAGATCACACCATCTGGACTTCTGG | |||||
| E2F2-CDSF/R | F: CCGATATCATGTGGGGTAGGCCCCACCCA | 63.5 | 1299 | XM_869196 | |
| R: GCTCTAGATCAGTTAATCAGCAGGTCCC | |||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, D.; Li, A.; Zhao, C.; Wang, H.; Mei, C.; Khan, R.; Zan, L. Transcriptional Regulation by CpG Sites Methylation in the Core Promoter Region of the Bovine SIX1 Gene: Roles of Histone H4 and E2F2. Int. J. Mol. Sci. 2018, 19, 213. https://doi.org/10.3390/ijms19010213
Wei D, Li A, Zhao C, Wang H, Mei C, Khan R, Zan L. Transcriptional Regulation by CpG Sites Methylation in the Core Promoter Region of the Bovine SIX1 Gene: Roles of Histone H4 and E2F2. International Journal of Molecular Sciences. 2018; 19(1):213. https://doi.org/10.3390/ijms19010213
Chicago/Turabian StyleWei, Dawei, Anning Li, Chunping Zhao, Hongbao Wang, Chugang Mei, Rajwali Khan, and Linsen Zan. 2018. "Transcriptional Regulation by CpG Sites Methylation in the Core Promoter Region of the Bovine SIX1 Gene: Roles of Histone H4 and E2F2" International Journal of Molecular Sciences 19, no. 1: 213. https://doi.org/10.3390/ijms19010213