Understanding the Progression of Bone Metastases to Identify Novel Therapeutic Targets

{kind=link}
{kind=link}
Abstract
1. Introduction
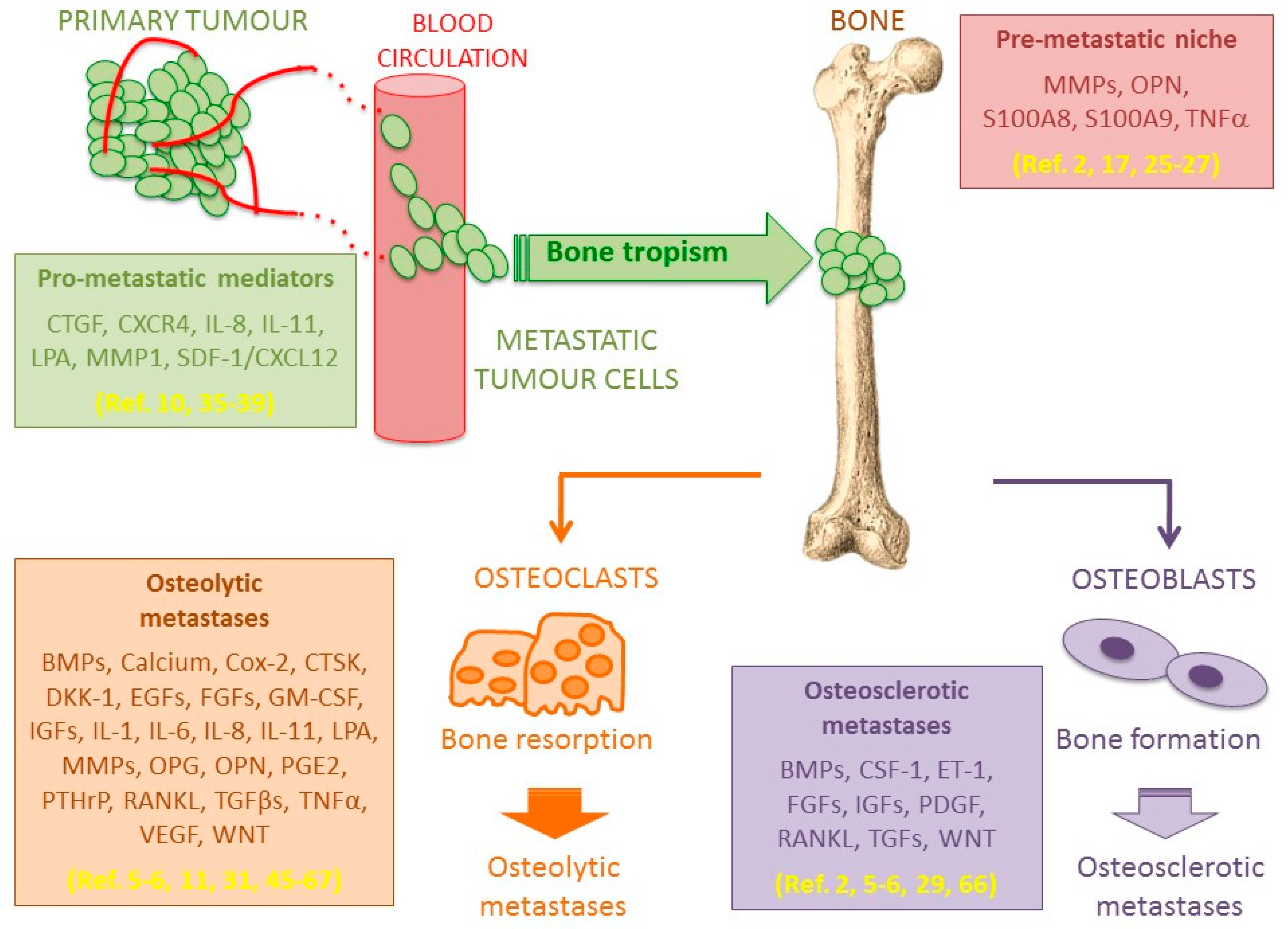
2. Seed and Soil Interactions
3. Theories of Metastatic Progression
4. Gene Expression Profiling of Bone Metastases
5. Bone Metastasis Microenvironment
5.1. Osteolytic Bone Metastases
5.2. Osteosclerotic Bone Metastases
6. Bone Metastasis Treatments
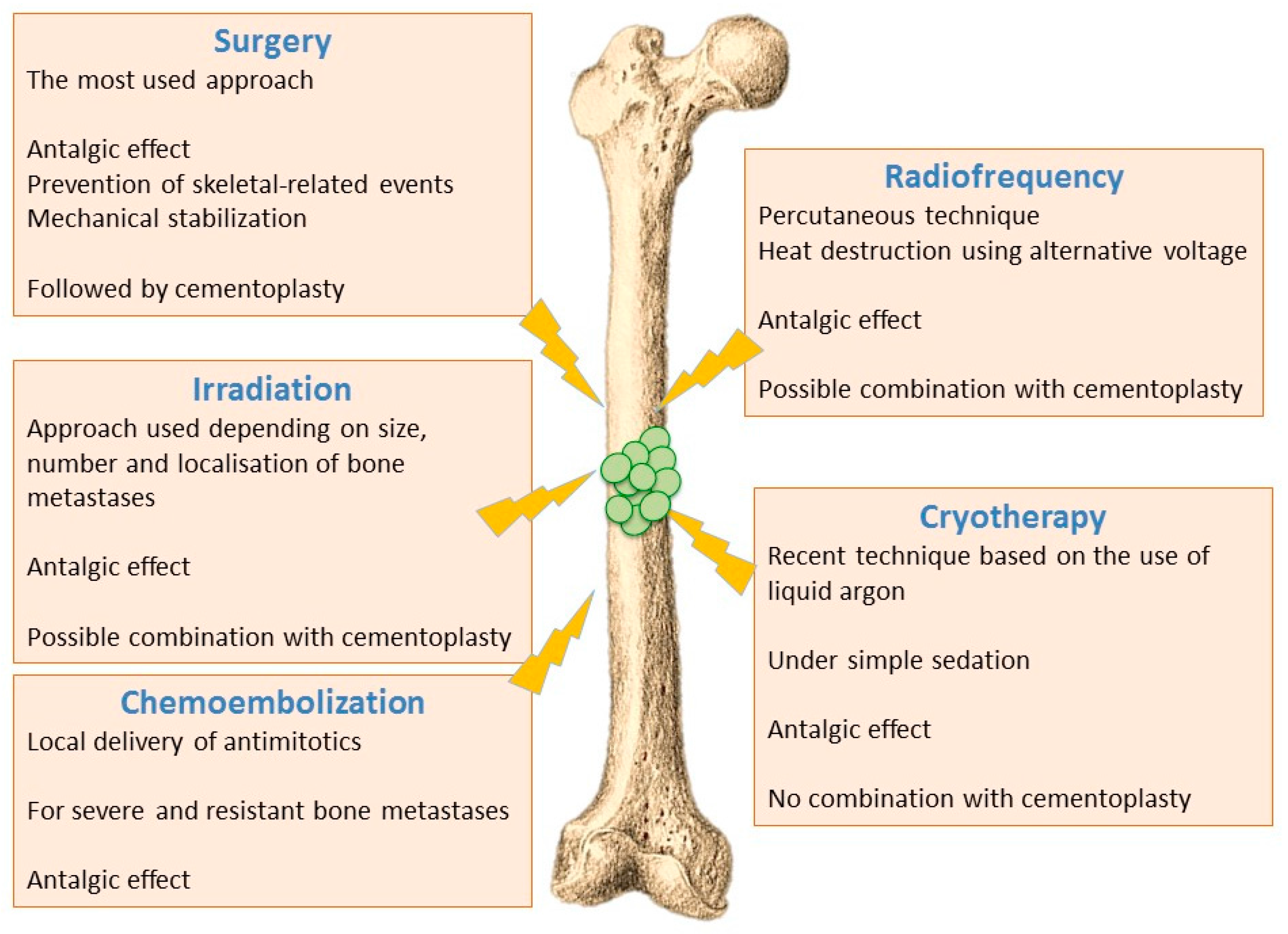
6.1. Ablative Procedures
6.1.1. Surgical Techniques
6.1.2. Irradiation Techniques
6.1.3. Thermal and Chemical Ablation
- Radiofrequency ablation - Radiofrequency ablation of bone metastases is a recent radiological percutaneous technique, using alternative current (450–600 kHz) applied through a needle entered in the metastasis under radioscopic control. Radiofrequency energy generates an overheating (60–70 °C), and cancer cells necrosis is obtained by heat destruction within a volume of 3 cm in diameter. This technique cannot be used in the close vicinity of nerve tissues, since they do not support a temperature above 45 °C. It is also difficult to implement this technique for metastases affecting flat bones (i.e., sternum, iliac wing). Radiofrequency can also be used in combination with cementoplasty, although this combination has not yet shown significant improvement compared to cementoplasty alone [72,74].
- Cryotherapy—Cryotherapy is the more recent technique that enables tumoral destruction, over areas up to 5 cm, by low temperature using a needle-like applicator and liquid argon. This technique involves generally little pain and it can be realized under simple sedation, contrary to radiotherapy that usually requires general anaesthesia. On the other hand, cryotherapy cannot be combined with cementoplasty since tissue cooling exerts an inhibitory effect on cement polymerization [72,74].
- Chemoembolization—Chemoembolization consists of delivering directly to tumours an antimitotic infusion or antimitotic-loaded microparticles (carboplatin and adriamycin). This technique is used to treat bone lesions that have been previously irradiated, or which are unresectable and resistant to other treatments. In 50% of cases, a partial or complete response is observed, and this method is often very effective in treating initial metastases (especially from breast cancer) [72,74].
6.2. Reconstructive Procedures
6.3. Pharmacological Approaches
6.3.1. Bisphosphonates
6.3.2. Denosumab
6.3.3. Other Agents Targeting Bone Tissue
6.3.4. Agents Targeting Tumour Cells
6.3.5. Agents Targeting Angiogenesis
6.3.6. Agents Targeting Pain
7. Conclusions: Immunotherapy Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BMP | bone morphogenetic protein |
| BP | bisphosphonate |
| CTGF | connective tissue growth factor |
| Ctsk | cathepsin K |
| Coll | collagen |
| CXCL | C-X-C motif chemokine ligand |
| CXCR4 | C-X-C chemokine receptor type 4 |
| EMT | epithelial to mesenchymal transition |
| ET-1 | endothelin-1 |
| FGF | fibroblast growth factor |
| Ga | gallium |
| GM-CSF | granulocyte macrophage colony stimulating factor |
| HFG | halofuginone |
| HSCs | hematopoietic stem cells |
| IGF | insulin-like growth factor |
| IL | interleukin |
| LPA | lysophosphatidic acid |
| miRNAs | microRNAs |
| MMP | matrix metallo protein |
| MSC | mesenchymal stem cells |
| MSG | metastases suppressor gene |
| MTA1 | metastasis- associated protein 1 |
| NGF | nerve growth factor |
| OPG | osteoprotegerin |
| OPN | osteopontin |
| PDGF | platelet -derived growth factor |
| PgE2 | prostaglandin E2 |
| PMMA | poly[methyl methacrylate] |
| PPi | pyrophosphate |
| PTEN | phosphatase and tensin homolog |
| PTHrP | parathyroid hormone-related peptide |
| RANKL | receptor activator of nuclear factor—κB receptor ligand |
| SCP | single cell progeny |
| SDF-1 | stromal-derived growth factor-1 (/CXCL12) |
| siRNAs | small interfering RNAs |
| SRE | skeletal-related event |
| TCR | T cell receptor |
| TGFβ | transforming growth factor-beta |
| TNFα | tumor necrosis factor-alpha |
| VEGF | vascular endothelial growth factor |
| VLA4 | very late antigen 4 |
References
- Davila, D.; Antoniou, A.; Chaudhry, M.A. Evaluation of osseous metastasis in bone scintigraphy. Semin. Nucl. Med. 2015, 45, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Camparo, P.; Vieillefond, A. Molecular aspects of prostate cancer: Recent data from the literature. Bull. Cancer 2007, 94, F77–F88. [Google Scholar] [PubMed]
- Hamaoka, T.; Madewell, J.E.; Podoloff, D.A.; Hortobagyi, G.N.; Ueno, N.T. Bone imaging in metastatic breast cancer. J. Clin. Oncol. 2004, 22, 2942–2953. [Google Scholar] [CrossRef] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Cecchini, M.G.; Wetterwald, A.; van der Pluijm, G.; Thalmann, G.N. Molecular and Biological Mechanisms of Bone Metastasis. EAU Update Ser. 2005, 3, 214–226. [Google Scholar] [CrossRef]
- Buijs, J.T.; van der Pluijm, G. Osteotropic cancers: From primary tumor to bone. Cancer Lett. 2009, 273, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Hiraga, T. Crosstalk between cancer cells and bone microenvironment in bone metastasis. Biochem. Biophys. Res. Commun. 2005, 328, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Ata, R.; Antonescu, C.N. Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Gasparics, A.; Rosivall, L.; Krizbai, I.A.; Sebe, A. When the endothelium scores an own goal: Endothelial cells actively augment metastatic extravasation through endothelial-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1055–H1063. [Google Scholar] [CrossRef] [PubMed]
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Cote, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005, 435, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Iddon, J.; Byrne, G.; Bundred, N.J. Bone metastasis in breast cancer: The role of parathyroid hormone related protein. Surg. Oncol. 1999, 8, 13–25. [Google Scholar] [CrossRef]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Klein, G. Foulds’ dangerous idea revisited: The multistep development of tumors 40 years later. Adv. Cancer Res. 1998, 72, 1–23. [Google Scholar] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Cairns, J. Mutation selection and the natural history of cancer. Nature 1975, 255, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Langerod, A.; Carrieri, P.; Bergh, J.; Klaar, S.; Eyfjord, J.; Theillet, C.; Rodriguez, C.; Lidereau, R.; Bieche, I.; et al. The clinical value of somatic TP53 gene mutations in 1794 patients with breast cancer. Clin. Cancer Res. 2006, 12, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.S.; Gifford, A.M.; Greiner, A.L.; Kelleher, S.P.; Saelzler, M.P.; Ince, T.A.; Reinhardt, F.; Harris, L.N.; Hylander, B.L.; Repasky, E.A.; et al. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 2008, 133, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Koscielny, S.; Tubiana, M.; Le, M.G.; Valleron, A.J.; Mouriesse, H.; Contesso, G.; Sarrazin, D. Breast cancer: Relationship between the size of the primary tumour and the probability of metastatic dissemination. Br. J. Cancer 1984, 49, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Koscielny, S.; Tubiana, M. Parallel progression of tumour and metastases. Nat. Rev. Cancer 2010, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Collins, V.P.; Loeffler, R.K.; Tivey, H. Observations on growth rates of human tumors. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1956, 76, 988–1000. [Google Scholar] [PubMed]
- Husemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmuller, G.; et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Podsypanina, K.; Du, Y.C.; Jechlinger, M.; Beverly, L.J.; Hambardzumyan, D.; Varmus, H. Seeding and propagation of untransformed mouse mammary cells in the lung. Science 2008, 321, 1841–1844. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kittler, O.; Ragg, T.; Daskalakis, A.; Granzow, M.; Ahr, A.; Blankenstein, T.J.; Kaufmann, M.; Diebold, J.; Arnholdt, H.; Muller, P.; et al. From latent disseminated cells to overt metastasis: Genetic analysis of systemic breast cancer progression. Proc. Natl. Acad. Sci. USA 2003, 100, 7737–7742. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Zaharie, F.; Muresan, M.S.; Pop, L.; Fekete, Z.; Dima, D.; Frinc, I.; Trifa, A.; Berce, C.; Jurj, A.; et al. How to Diagnose and Treat a Cancer of Unknown Primary Site. J. Gastrointestin. Liver Dis. 2017, 26, 69–79. [Google Scholar] [PubMed]
- Kaplan, R.N.; Psaila, B.; Lyden, D. Bone marrow cells in the ‘pre-metastatic niche’: Within bone and beyond. Cancer Metastasis Rev. 2006, 25, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Hess, K.R.; Varadhachary, G.R.; Taylor, S.H.; Wei, W.; Raber, M.N.; Lenzi, R.; Abbruzzese, J.L. Metastatic patterns in adenocarcinoma. Cancer 2006, 106, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Minn, A.J.; Kang, Y.; Siegel, P.M.; Serganova, I.; Cordon-Cardo, C.; Olshen, A.B.; Gerald, W.L.; Massague, J. Identifying site-specific metastasis genes and functions. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Ara, T.; Declerck, Y.A. Interleukin-6 in bone metastasis and cancer progression. Eur. J. Cancer 2010, 46, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Ewing, J. Neoplastic Diseases: A Treatise on Tumors, 3rd ed.; WB Saunders: Philadelphia, PA, USA, 1928. [Google Scholar]
- Chiang, A.C.; Massague, J. Molecular basis of metastasis. N. Engl. J. Med. 2008, 359, 2814–2823. [Google Scholar] [CrossRef] [PubMed]
- Zarour, L.R.; Anand, S.; Billingsley, K.G.; Bisson, W.H.; Cercek, A.; Clarke, M.F.; Coussens, L.M.; Gast, C.E.; Geltzeiler, C.B.; Hansen, L.; et al. Colorectal Cancer Liver Metastasis: Evolving Paradigms and Future Directions. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Ruger, R. Molecular Mechanisms of Bone Metastasis. Cancer Genom. Proteom. 2016, 13, 1–12. [Google Scholar]
- Goerge, T.; Barg, A.; Schnaeker, E.M.; Poppelmann, B.; Shpacovitch, V.; Rattenholl, A.; Maaser, C.; Luger, T.A.; Steinhoff, M.; Schneider, S.W. Tumor-derived matrix metalloproteinase-1 targets endothelial proteinase-activated receptor 1 promoting endothelial cell activation. Cancer Res. 2006, 66, 7766–7774. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, A.; Cunningham, J.L. Connective tissue growth factor in tumor pathogenesis. Fibrogenesis Tissue Repair 2012, 5, S8. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Lacroix, M.; Lespagnard, L.; Larsimont, D.; Paesmans, M.; Body, J.J. Interleukins-6 and -11 expression in primary breast cancer and subsequent development of bone metastases. Cancer Lett. 2001, 169, 87–95. [Google Scholar] [CrossRef]
- Rose, A.A.; Pepin, F.; Russo, C.; Abou Khalil, J.E.; Hallett, M.; Siegel, P.M. Osteoactivin promotes breast cancer metastasis to bone. Mol. Cancer Res. 2007, 5, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Lyden, D.; Roberts, I. Megakaryocytes, malignancy and bone marrow vascular niches. J. Thromb. Haemost. 2012, 10, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A.; Mundy, G.R. Cancer and bone. Endocr. Rev. 1998, 19, 18–54. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Pissimissis, N.; Halapas, A.; Koutsilieris, M. Mechanisms of bone metastasis in prostate cancer: Clinical implications. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Buijs, J.T.; Stayrook, K.R.; Guise, T.A. The role of TGF-beta in bone metastasis: Novel therapeutic perspectives. Bonekey Rep. 2012, 1, 96. [Google Scholar] [CrossRef] [PubMed]
- Juarez, P.; Guise, T.A. TGF-beta in cancer and bone: Implications for treatment of bone metastases. Bone 2011, 48, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, T.; Gillespie, M.T.; Glatz, J.A.; Fukumoto, S.; Moseley, J.M.; Martin, T.J. Transforming growth factor beta stimulation of parathyroid hormone-related protein (PTHrP): A paracrine regulator? Mol. Cell. Endocrinol. 1993, 92, 55–62. [Google Scholar] [CrossRef]
- Kingsley, L.A.; Fournier, P.G.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Induction of epithelial-mesenchymal transition by transforming growth factor beta. Semin. Cancer Biol. 2012, 22, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Hata, K.; Nakanishi, M.; Nagae, M.; Nagayama, T.; Wakabayashi, H.; Nishisho, T.; Sakurai, T.; Hiraga, T. Involvement of acidic microenvironment in the pathophysiology of cancer-associated bone pain. Bone 2011, 48, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.T.; Xu, B.; Song, Q.B.; Zeng, H. The role of neural-related factors in the metastasis of the gastrointestinal cancer. J. Cancer Res. Ther. 2013, 9, S123–S128. [Google Scholar] [PubMed]
- Artacho-Cordon, F.; Rios-Arrabal, S.; Lara, P.C.; Artacho-Cordon, A.; Calvente, I.; Nunez, M.I. Matrix metalloproteinases: Potential therapy to prevent the development of second malignancies after breast radiotherapy. Surg. Oncol. 2012, 21, e143–e151. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.; Lee, S.C.; David, M.; Bordet, J.C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clezardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef] [PubMed]
- Peyruchaud, O.; Leblanc, R.; David, M. Pleiotropic activity of lysophosphatidic acid in bone metastasis. Biochim. Biophys. Acta 2013, 1831, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, S.; Castellan, P.; Manco, R.; Tenaglia, R.L. Reciprocal cross-talk between Prostaglandin E2 and bone in prostate cancer: A current review. Cent. Eur. J. Urol. 2011, 64, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Diament, M.J.; Stillitani, M.I.; Puricelli, L.; Bal de Kier-Joffe, E.; Klein, S. GM-CSF secreted by murine adenocarcinoma cells modulates tumor progression and immune activity. Oncol. Rep. 2003, 10, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Wakabayashi, H.; Naito, Y.; Kato, S.; Nakagawa, T.; Matsumine, A.; Sudo, A. Anti-tumor necrosis factor therapy inhibits lung metastasis in an osteosarcoma cell line. Oncology 2015, 88, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Verbovsek, U.; Van Noorden, C.J.; Lah, T.T. Complexity of cancer protease biology: Cathepsin K expression and function in cancer progression. Semin. Cancer Biol. 2015, 35, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Kruger, T.E.; Miller, A.H.; Godwin, A.K.; Wang, J. Bone sialoprotein and osteopontin in bone metastasis of osteotropic cancers. Crit. Rev. Oncol. Hematol. 2014, 89, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.; Raja, E.; Miyazono, K.; Tsubakihara, Y.; Moustakas, A. Mechanisms of action of bone morphogenetic proteins in cancer. Cytokine Growth Factor Rev. 2016, 27, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; Steensma, M.R.; Williams, B.O.; Zhong, Z. Skeletal metastasis: Treatments, mouse models, and the Wnt signaling. Chin. J. Cancer 2013, 32, 380–396. [Google Scholar] [CrossRef] [PubMed]
- Buijs, J.T.; Kuijpers, C.C.; van der Pluijm, G. Targeted therapy options for treatment of bone metastases; beyond bisphosphonates. Curr. Pharm. Des. 2010, 16, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Garrison, K.; Hahn, T.; Lee, W.C.; Ling, L.E.; Weinberg, A.D.; Akporiaye, E.T. The small molecule TGF-beta signaling inhibitor SM16 synergizes with agonistic OX40 antibody to suppress established mammary tumors and reduce spontaneous metastasis. Cancer Immunol. Immunother. 2012, 61, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Liepe, K.; Kotzerke, J. Internal radiotherapy of painful bone metastases. Methods 2011, 55, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Kurup, A.N.; Callstrom, M.R. Expanding role of percutaneous ablative and consolidative treatments for musculoskeletal tumours. Clin. Radiol. 2017, 72, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Szendroi, M.; Antal, I.; Szendroi, A.; Lazary, A.; Varga, P.P. Diagnostic algorithm, prognostic factors and surgical treatment of metastatic cancer diseases of the long bones and spine. EFORT Open Rev. 2017, 2, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Thariat, J.; Vignot, S.; Bensadoun, R.J.; Mornex, F. Improvements of ablative local treatments modify the management of the oligometastatic disease. Cancer Radiother. 2012, 16, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Kougioumtzopoulou, A.; Zygogianni, A.; Liakouli, Z.; Kypraiou, E.; Kouloulias, V. The role of radiotherapy in bone metastases: A critical review of current literature. Eur. J. Cancer Care 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Chiras, J.; Shotar, E.; Cormier, E.; Clarencon, F. Interventional radiology in bone metastases. Eur. J. Cancer Care 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Laredo, J.D.; Chiras, J.; Kemel, S.; Taihi, L.; Hamze, B. Vertebroplasty and interventional radiology procedures for bone metastases. Jt. Bone Spine 2017. [Google Scholar] [CrossRef] [PubMed]
- Pikis, S.; Goldstein, J.; Spektor, S. Potential neurotoxic effects of polymethylmethacrylate during cranioplasty. J. Clin. Neurosci. 2015, 22, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Ratasvuori, M.; Wedin, R.; Keller, J.; Nottrott, M.; Zaikova, O.; Bergh, P.; Kalen, A.; Nilsson, J.; Jonsson, H.; Laitinen, M. Insight opinion to surgically treated metastatic bone disease: Scandinavian Sarcoma Group Skeletal Metastasis Registry report of 1195 operated skeletal metastasis. Surg. Oncol. 2013, 22, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Van Driel, M.; van Leeuwen, J.P. Cancer and bone: A complex complex. Arch. Biochem. Biophys. 2014, 561, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zou, L.; He, Y.; Bunger, C. Molecular treatment strategies and surgical reconstruction for metastatic bone diseases. Cancer Treat. Rev. 2008, 34, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Verron, E.; Bouler, J.M. Is bisphosphonate therapy compromised by the emergence of adverse bone disorders? Drug Discov. Today 2014, 19, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.; Gismondi, A.; Santoni, A. Chemokines and NK cells: Regulators of development, trafficking and functions. Immunol. Lett. 2012, 145, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Gronich, N.; Rennert, G. Beyond aspirin-cancer prevention with statins, metformin and bisphosphonates. Nat. Rev. Clin. Oncol. 2013, 10, 625–642. [Google Scholar] [CrossRef] [PubMed]
- Odri, G.; Kim, P.P.; Lamoureux, F.; Charrier, C.; Battaglia, S.; Amiaud, J.; Heymann, D.; Gouin, F.; Redini, F. Zoledronic acid inhibits pulmonary metastasis dissemination in a preclinical model of Ewing’s sarcoma via inhibition of cell migration. BMC Cancer 2014, 14, 169. [Google Scholar] [CrossRef] [PubMed]
- Clezardin, P.; Ebetino, F.H.; Fournier, P.G. Bisphosphonates and cancer-induced bone disease: Beyond their antiresorptive activity. Cancer Res. 2005, 65, 4971–4974. [Google Scholar] [CrossRef] [PubMed]
- Guise, T.A. Antitumor effects of bisphosphonates: Promising preclinical evidence. Cancer Treat. Rev. 2008, 34 (Suppl. S1), S19–S24. [Google Scholar] [CrossRef] [PubMed]
- Clezardin, P. Mechanisms of action of bisphosphonates in oncology: A scientific concept evolving from antiresorptive to anticancer activities. Bonekey Rep. 2013, 2, 267. [Google Scholar] [CrossRef] [PubMed]
- Benzaid, I.; Monkkonen, H.; Bonnelye, E.; Monkkonen, J.; Clezardin, P. In vivo phosphoantigen levels in bisphosphonate-treated human breast tumors trigger Vgamma9Vdelta2 T-cell antitumor cytotoxicity through ICAM-1 engagement. Clin. Cancer Res. 2012, 18, 6249–6259. [Google Scholar] [CrossRef] [PubMed]
- Benzaid, I.; Monkkonen, H.; Stresing, V.; Bonnelye, E.; Green, J.; Monkkonen, J.; Touraine, J.L.; Clezardin, P. High phosphoantigen levels in bisphosphonate-treated human breast tumors promote Vgamma9Vdelta2 T-cell chemotaxis and cytotoxicity in vivo. Cancer Res. 2011, 71, 4562–4572. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, L.J.; Di Bella, G.; Belvedere, M.; Barbagallo, M. Physiology of the aging bone and mechanisms of action of bisphosphonates. Biogerontology 2011, 12, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, V.; Tchekmedyian, N.S.; Rosen, L.S.; Zheng, M.; Hei, Y.J. Clinical benefit of zoledronic acid in patients with lung cancer and other solid tumors: Analysis based on history of skeletal complications. Clin. Lung Cancer 2004, 6, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Gleason, D.M.; Murray, R.; Tchekmedyian, S.; Venner, P.; Lacombe, L.; Chin, J.L.; Vinholes, J.J.; Goas, J.A.; Zheng, M.; et al. Long-term efficacy of zoledronic acid for the prevention of skeletal complications in patients with metastatic hormone-refractory prostate cancer. J. Natl. Cancer Inst. 2004, 96, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Confavreux, C.B.; Clezardin, P. Bone antiresorptive agents in the treatment of bone metastases associated with solid tumours or multiple myeloma. Bonekey Rep. 2015, 4, 744. [Google Scholar] [CrossRef] [PubMed]
- Van Poznak, C.H.; Von Roenn, J.H.; Temin, S. American society of clinical oncology clinical practice guideline update: Recommendations on the role of bone-modifying agents in metastatic breast cancer. J. Oncol. Pract. 2011, 7, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.; Gnant, M.; Morgan, G.; Clezardin, P. Effects of bone-targeted agents on cancer progression and mortality. J. Natl. Cancer Inst. 2012, 104, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Khosla, S.; Burr, D.; Cauley, J.; Dempster, D.W.; Ebeling, P.R.; Felsenberg, D.; Gagel, R.F.; Gilsanz, V.; Guise, T.; Koka, S.; et al. Bisphosphonate-associated osteonecrosis of the jaw: Report of a task force of the American Society for Bone and Mineral Research. J. Bone Miner. Res. 2007, 22, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Lezot, F.; Chesneau, J.; Battaglia, S.; Brion, R.; Castaneda, B.; Farges, J.C.; Heymann, D.; Redini, F. Preclinical evidence of potential craniofacial adverse effect of zoledronic acid in pediatric patients with bone malignancies. Bone 2014, 68, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kopp, L.; Anderson, N.; Cornelius, K.; Herzog, C.; Hughes, D.; Huh, W. Novel bone cancer drugs: Investigational agents and control paradigms for primary bone sarcomas (Ewing’s sarcoma and osteosarcoma). Expert Opin. Investig. Drugs 2008, 17, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, C.; Passaro, A.; Gori, B.; Del Signore, E.; Migliorino, M.R.; Ricciardi, S.; Fulvi, A.; de Marinis, F. Bone and brain metastasis in lung cancer: Recent advances in therapeutic strategies. Ther. Adv. Med. Oncol. 2014, 6, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Clemons, M.; Gelmon, K.A.; Pritchard, K.I.; Paterson, A.H. Bone-targeted agents and skeletal-related events in breast cancer patients with bone metastases: The state of the art. Curr. Oncol. 2012, 19, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Peddi, P.; Lopez-Olivo, M.A.; Pratt, G.F.; Suarez-Almazor, M.E. Denosumab in patients with cancer and skeletal metastases: A systematic review and meta-analysis. Cancer Treat. Rev. 2013, 39, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.D.; Bolognese, M.A.; Lewiecki, E.M.; McClung, M.R.; Ding, B.; Austin, M.; Liu, Y.; San Martin, J.; Amg Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: A randomized blinded phase 2 clinical trial. Bone 2008, 43, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, S.; Guise, T.A.; Chirgwin, J. The critical role of the bone microenvironment in cancer metastases. Mol. Cell. Endocrinol. 2009, 310, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Heymann, M.F.; Stresing, V.; Mori, K.; Redini, F.; Heymann, D. Current therapeutic strategies and novel approaches in osteosarcoma. Cancers 2013, 5, 591–616. [Google Scholar] [CrossRef] [PubMed]
- Fili, S.; Karalaki, M.; Schaller, B. Mechanism of bone metastasis: The role of osteoprotegerin and of the host-tissue microenvironment-related survival factors. Cancer Lett. 2009, 283, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Wittrant, Y.; Theoleyre, S.; Chipoy, C.; Padrines, M.; Blanchard, F.; Heymann, D.; Redini, F. RANKL/RANK/OPG: New therapeutic targets in bone tumours and associated osteolysis. Biochim. Biophys. Acta 2004, 1704, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Verron, E.; Loubat, A.; Carle, G.F.; Vignes-Colombeix, C.; Strazic, I.; Guicheux, J.; Rochet, N.; Bouler, J.M.; Scimeca, J.C. Molecular effects of gallium on osteoclastic differentiation of mouse and human monocytes. Biochem. Pharmacol. 2012, 83, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Strazic Geljic, I.; Melis, N.; Boukhechba, F.; Schaub, S.; Mellier, C.; Janvier, P.; Laugier, J.P.; Bouler, J.M.; Verron, E.; Scimeca, J.C. Gallium enhances reconstructive properties of a calcium phosphate bone biomaterial. J. Tissue Eng. Regen. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Strazic-Geljic, I.; Guberovic, I.; Didak, B.; Schmid-Antomarchi, H.; Schmid-Alliana, A.; Boukhechba, F.; Bouler, J.M.; Scimeca, J.C.; Verron, E. Gallium, a promising candidate to disrupt the vicious cycle driving osteolytic metastases. Biochem. Pharmacol. 2016, 116, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, J.C.; Verron, E. The multiple therapeutic applications of miRNAs for bone regenerative medicine. Drug Discov. Today 2017, 22, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Green, D.; Mohorianu, I.; McNamara, I.; Dalmay, T.; Fraser, W.D. miR-16 is highly expressed in Paget’s associated osteosarcoma. Endocr. Relat. Cancer 2017, 24, L27–L31. [Google Scholar] [CrossRef] [PubMed]
- Dostalova Merkerova, M.; Krejcik, Z.; Votavova, H.; Belickova, M.; Vasikova, A.; Cermak, J. Distinctive microRNA expression profiles in CD34+ bone marrow cells from patients with myelodysplastic syndrome. Eur. J. Hum. Genet. 2011, 19, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Qin, X.J.; Cao, D.L.; Zhu, Y.; Yao, X.D.; Zhang, S.L.; Dai, B.; Ye, D.W. An elevated serum miR-141 level in patients with bone-metastatic prostate cancer is correlated with more bone lesions. Asian J. Androl. 2013, 15, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Krzeszinski, J.Y.; Wei, W.; Huynh, H.; Jin, Z.; Wang, X.; Chang, T.C.; Xie, X.J.; He, L.; Mangala, L.S.; Lopez-Berestein, G.; et al. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature 2014, 512, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar]
- Guise, T.A.; Mohammad, K.S. Endothelins in bone cancer metastases. Cancer Treat. Res. 2004, 118, 197–212. [Google Scholar] [PubMed]
- Qiao, L.; Liang, Y.; Li, N.; Hu, X.; Luo, D.; Gu, J.; Lu, Y.; Zheng, Q. Endothelin-A receptor antagonists in prostate cancer treatment-a meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 3465–3473. [Google Scholar] [PubMed]
- Bae, M.H.; Oh, S.H.; Park, C.J.; Lee, B.R.; Kim, Y.J.; Cho, Y.U.; Jang, S.; Lee, J.H.; Kim, N.; Park, S.H.; et al. VLA-4 and CXCR4 expression levels show contrasting prognostic impact (favorable and unfavorable, respectively) in acute myeloid leukemia. Ann. Hematol. 2015, 94, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Yoon, Y.; Votaw, J.; Goodman, M.M.; Williams, L.; Shim, H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005, 65, 967–971. [Google Scholar] [PubMed]
- Wu, Z.; McRoberts, K.S.; Theodorescu, D. The role of PTEN in prostate cancer cell tropism to the bone micro-environment. Carcinogenesis 2007, 28, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Peyruchaud, O.; Serre, C.M.; NicAmhlaoibh, R.; Fournier, P.; Clezardin, P. Angiostatin inhibits bone metastasis formation in nude mice through a direct anti-osteoclastic activity. J. Biol. Chem. 2003, 278, 45826–45832. [Google Scholar] [CrossRef] [PubMed]
- Krzeszinski, J.Y.; Wan, Y. New therapeutic targets for cancer bone metastasis. Trends Pharmacol. Sci. 2015, 36, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Malisetty, V.L.; Penugurti, V.; Panta, P.; Chitta, S.K.; Manavathi, B. MTA1 expression in human cancers—Clinical and pharmacological significance. Biomed. Pharmacother. 2017, 95, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Lozano-Ondoua, A.N.; Symons-Liguori, A.M.; Vanderah, T.W. Cancer-induced bone pain: Mechanisms and models. Neurosci. Lett. 2013, 557 Pt A, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Honore, P.; Luger, N.M.; Sabino, M.A.; Schwei, M.J.; Rogers, S.D.; Mach, D.B.; O’Keefe, P.F.; Ramnaraine, M.L.; Clohisy, D.R.; Mantyh, P.W. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nat. Med. 2000, 6, 521–528. [Google Scholar] [PubMed]
- Chwistek, M. Recent advances in understanding and managing cancer pain. F1000Research 2017, 6, 945. [Google Scholar] [CrossRef] [PubMed]
- Itatani, Y.; Kawada, K.; Inamoto, S.; Yamamoto, T.; Ogawa, R.; Taketo, M.M.; Sakai, Y. The Role of Chemokines in Promoting Colorectal Cancer Invasion/Metastasis. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Salanga, C.L.; Handel, T.M.; Allen, S.J. Chemokines and cancer: Migration, intracellular signalling and intercellular communication in the microenvironment. Biochem. J. 2008, 409, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Rollins, B.J. Inflammatory chemokines in cancer growth and progression. Eur. J. Cancer 2006, 42, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Sarvaiya, P.J.; Guo, D.; Ulasov, I.; Gabikian, P.; Lesniak, M.S. Chemokines in tumor progression and metastasis. Oncotarget 2013, 4, 2171–2185. [Google Scholar] [CrossRef] [PubMed]
- Vicari, A.P.; Caux, C. Chemokines in cancer. Cytokine Growth Factor Rev. 2002, 13, 143–154. [Google Scholar] [CrossRef]
- Santoni, M.; Bracarda, S.; Nabissi, M.; Massari, F.; Conti, A.; Bria, E.; Tortora, G.; Santoni, G.; Cascinu, S. CXC and CC chemokines as angiogenic modulators in nonhaematological tumors. Biomed. Res. Int. 2014, 2014, 768758. [Google Scholar] [CrossRef] [PubMed]
- Stockmann, C.; Schadendorf, D.; Klose, R.; Helfrich, I. The impact of the immune system on tumor: Angiogenesis and vascular remodeling. Front. Oncol. 2014, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, Y.; Oupicky, D. Potential of CXCR4/CXCL12 Chemokine Axis in Cancer Drug Delivery. Curr. Pharmacol. Rep. 2016, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D. Regulatory T cells: How do they find their space in the immunological arena? Semin. Cancer Biol. 2006, 16, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Franciszkiewicz, K.; Boissonnas, A.; Boutet, M.; Combadiere, C.; Mami-Chouaib, F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res. 2012, 72, 6325–6332. [Google Scholar] [CrossRef] [PubMed]
- Hemdan, N.Y. Anti-cancer versus cancer-promoting effects of the interleukin-17-producing T helper cells. Immunol. Lett. 2013, 149, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A.; Mackay, C.R. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today 1998, 19, 568–574. [Google Scholar] [CrossRef]
- Viola, A.; Sarukhan, A.; Bronte, V.; Molon, B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012, 33, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Savino, B.; Locati, M.; Zammataro, L.; Allavena, P.; Bonecchi, R. The chemokine system in cancer biology and therapy. Cytokine Growth Factor Rev. 2010, 21, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmid-Alliana, A.; Schmid-Antomarchi, H.; Al-Sahlanee, R.; Lagadec, P.; Scimeca, J.-C.; Verron, E. Understanding the Progression of Bone Metastases to Identify Novel Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 148. https://doi.org/10.3390/ijms19010148
Schmid-Alliana A, Schmid-Antomarchi H, Al-Sahlanee R, Lagadec P, Scimeca J-C, Verron E. Understanding the Progression of Bone Metastases to Identify Novel Therapeutic Targets. International Journal of Molecular Sciences. 2018; 19(1):148. https://doi.org/10.3390/ijms19010148
Chicago/Turabian StyleSchmid-Alliana, Annie, Heidy Schmid-Antomarchi, Rasha Al-Sahlanee, Patricia Lagadec, Jean-Claude Scimeca, and Elise Verron. 2018. "Understanding the Progression of Bone Metastases to Identify Novel Therapeutic Targets" International Journal of Molecular Sciences 19, no. 1: 148. https://doi.org/10.3390/ijms19010148
APA StyleSchmid-Alliana, A., Schmid-Antomarchi, H., Al-Sahlanee, R., Lagadec, P., Scimeca, J.-C., & Verron, E. (2018). Understanding the Progression of Bone Metastases to Identify Novel Therapeutic Targets. International Journal of Molecular Sciences, 19(1), 148. https://doi.org/10.3390/ijms19010148