Extract of Sheng-Mai-San Ameliorates Myocardial Ischemia-Induced Heart Failure by Modulating Ca2+-Calcineurin-Mediated Drp1 Signaling Pathways

 ,
,
Abstract
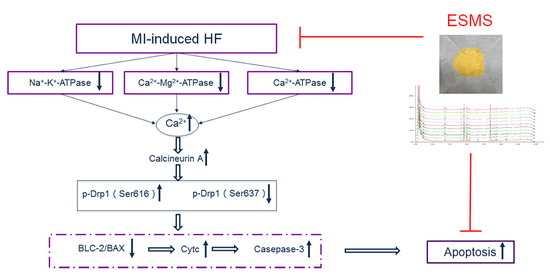
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
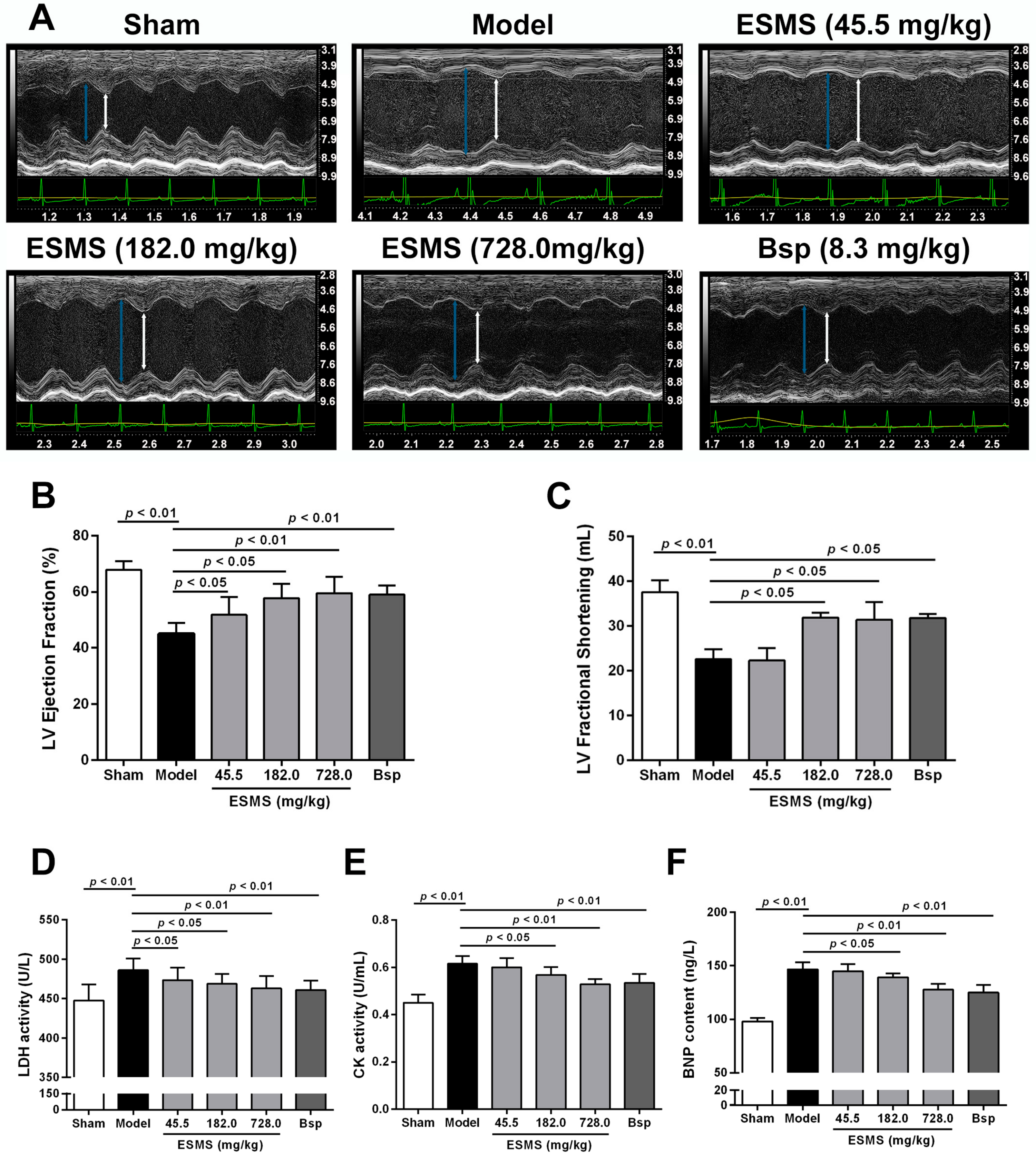
2.1. Extract of Sheng-Mai-San (ESMS) Ameliorated Cardiac Function and Decreased Myocardial Injury in Myocardial Ischemia (MI)-Induced Heart Failure (HF) Mice
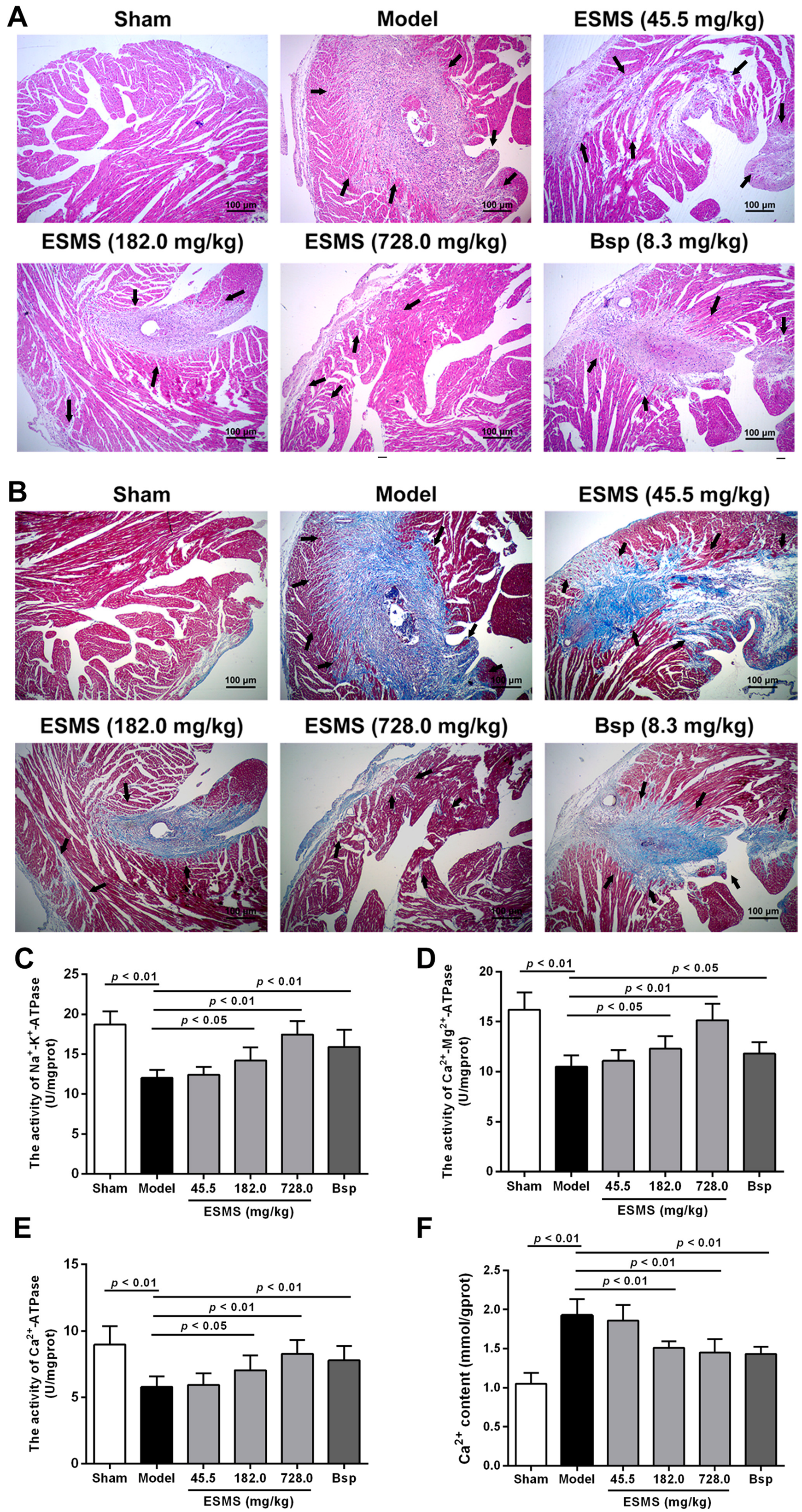
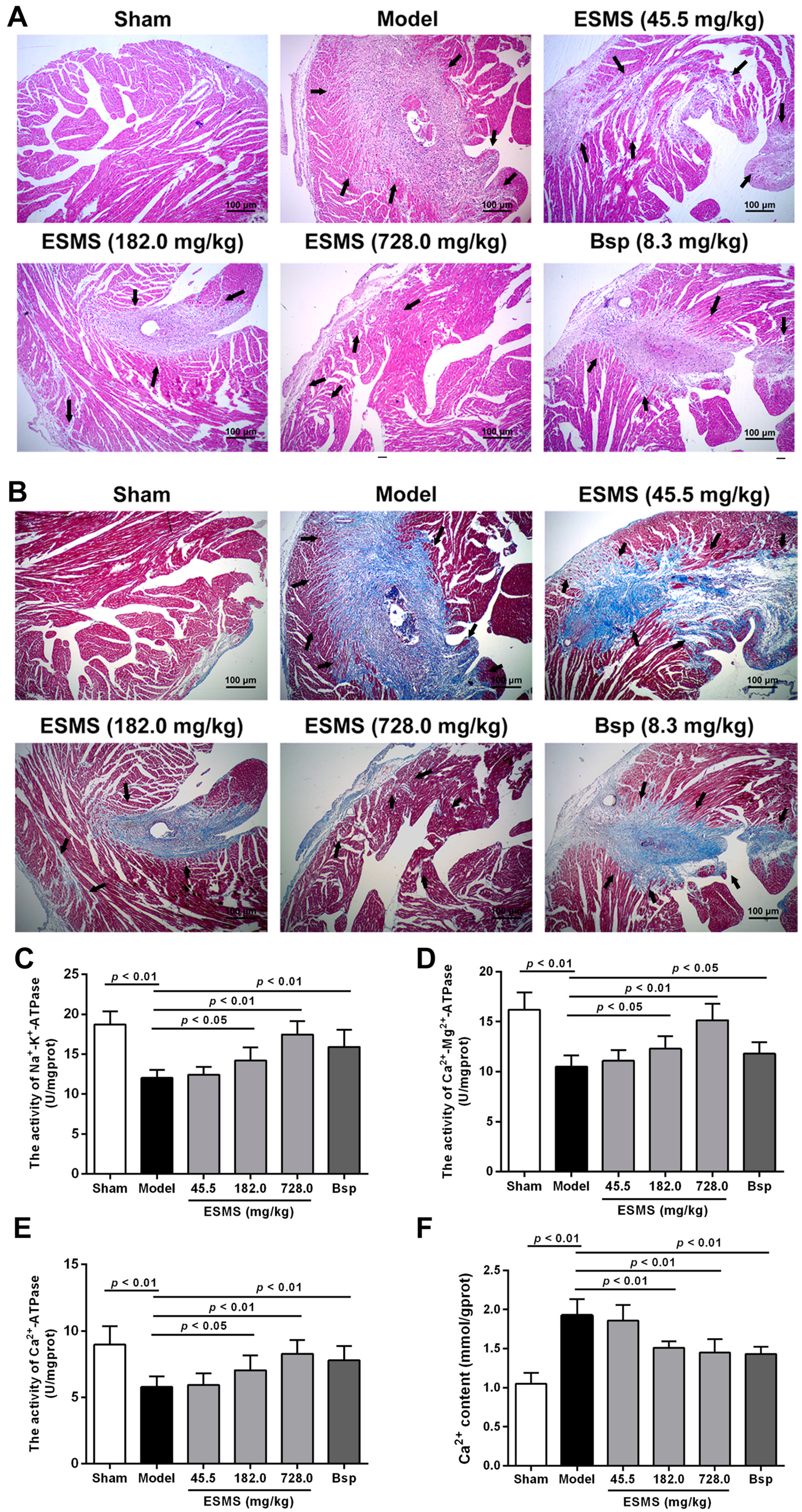
2.2. ESMS Ameliorated Cardiac Pathological Changes and Fibrosis in MI-Induced HF Mice
2.3. ESMS Increased ATPase Activity and Reduced Intracellular Ca2+ Concentration in MI-Induced HF Mice
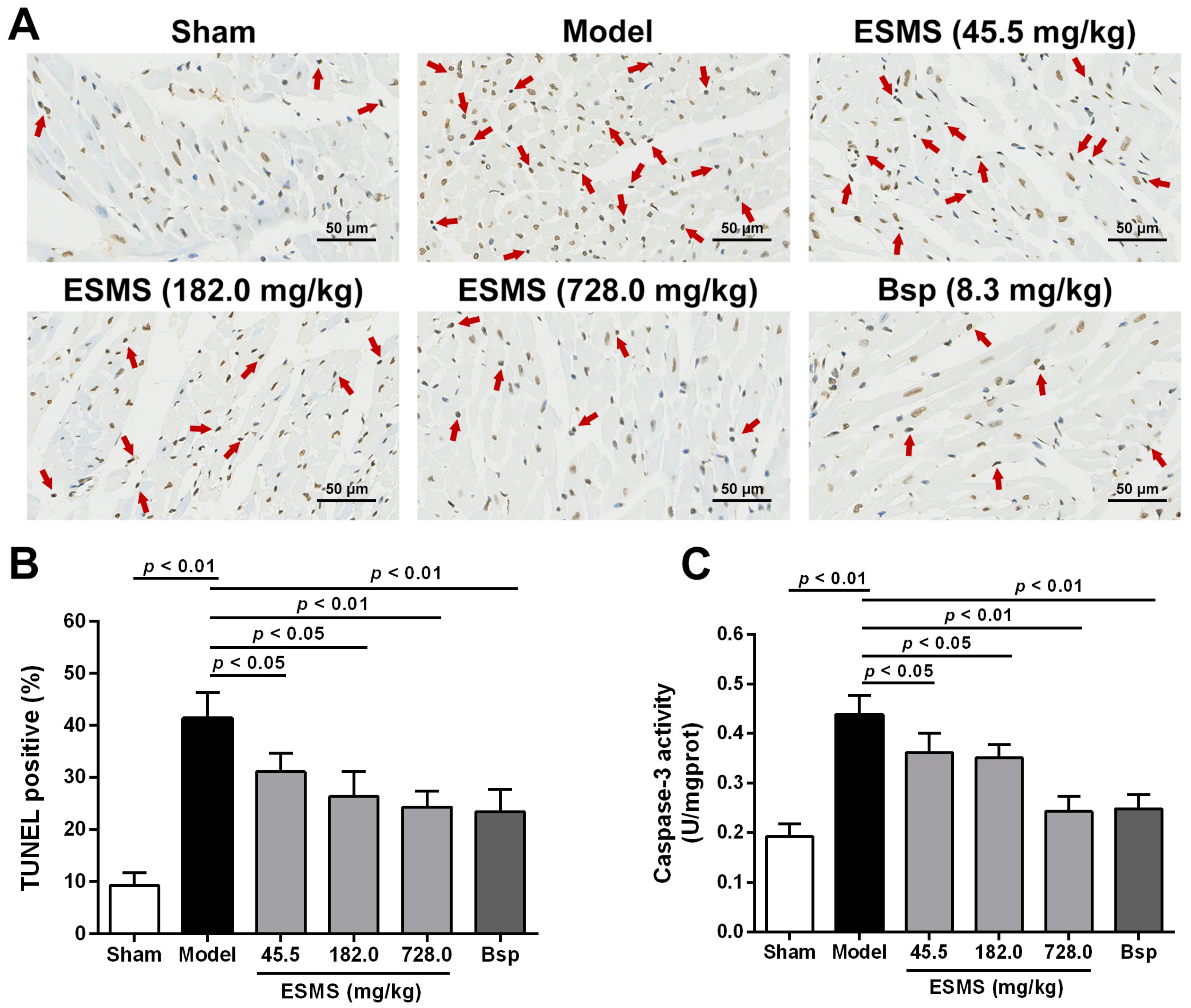
2.4. ESMS Inhibited Myocardial Apoptosis in MI-Induced HF Mice
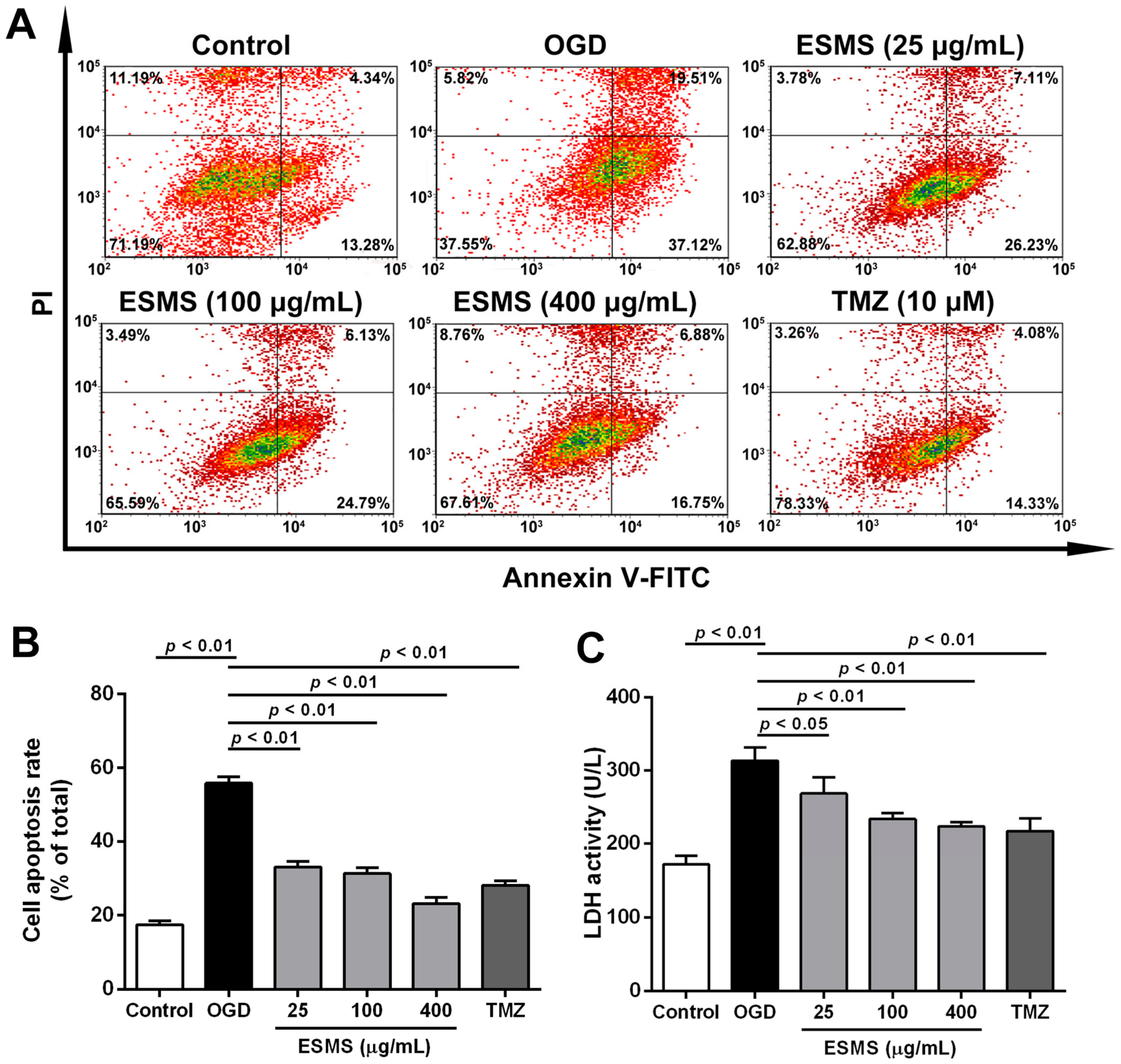
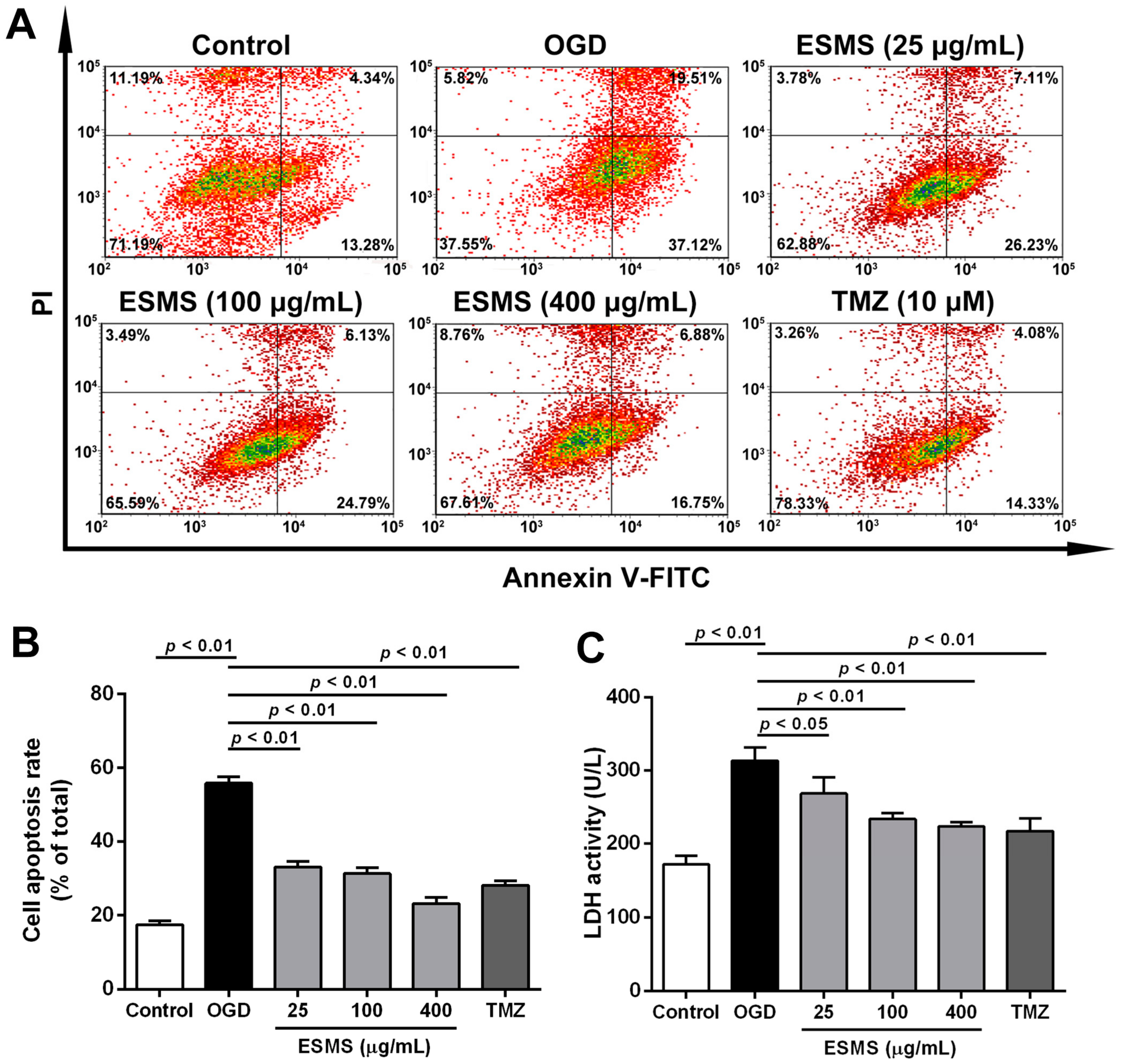
2.5. ESMS Decreased Cardiomyocyte Injury and Apoptosis in H9c2 Cells Subjected to Oxygen Glucose Deprivation (OGD)
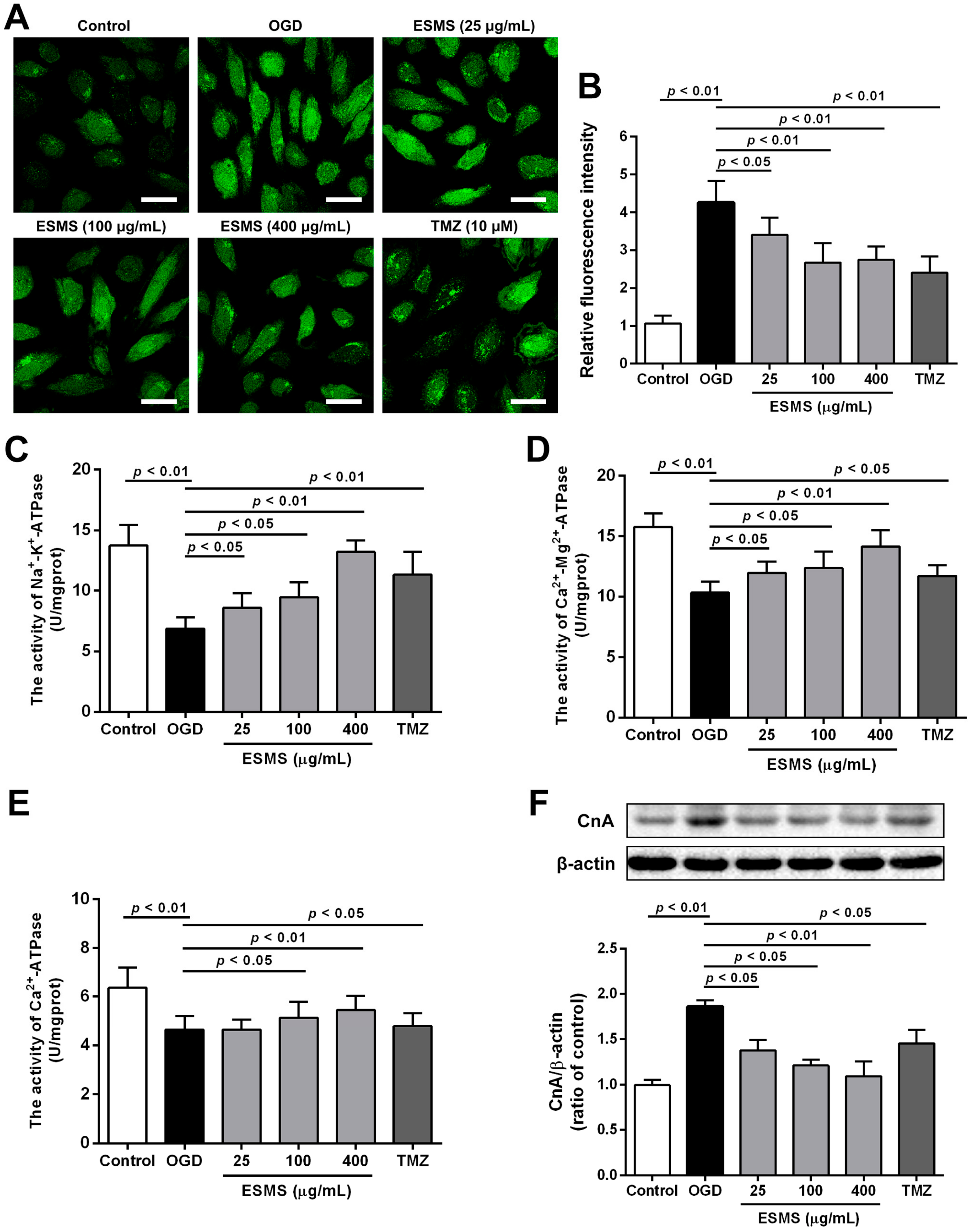
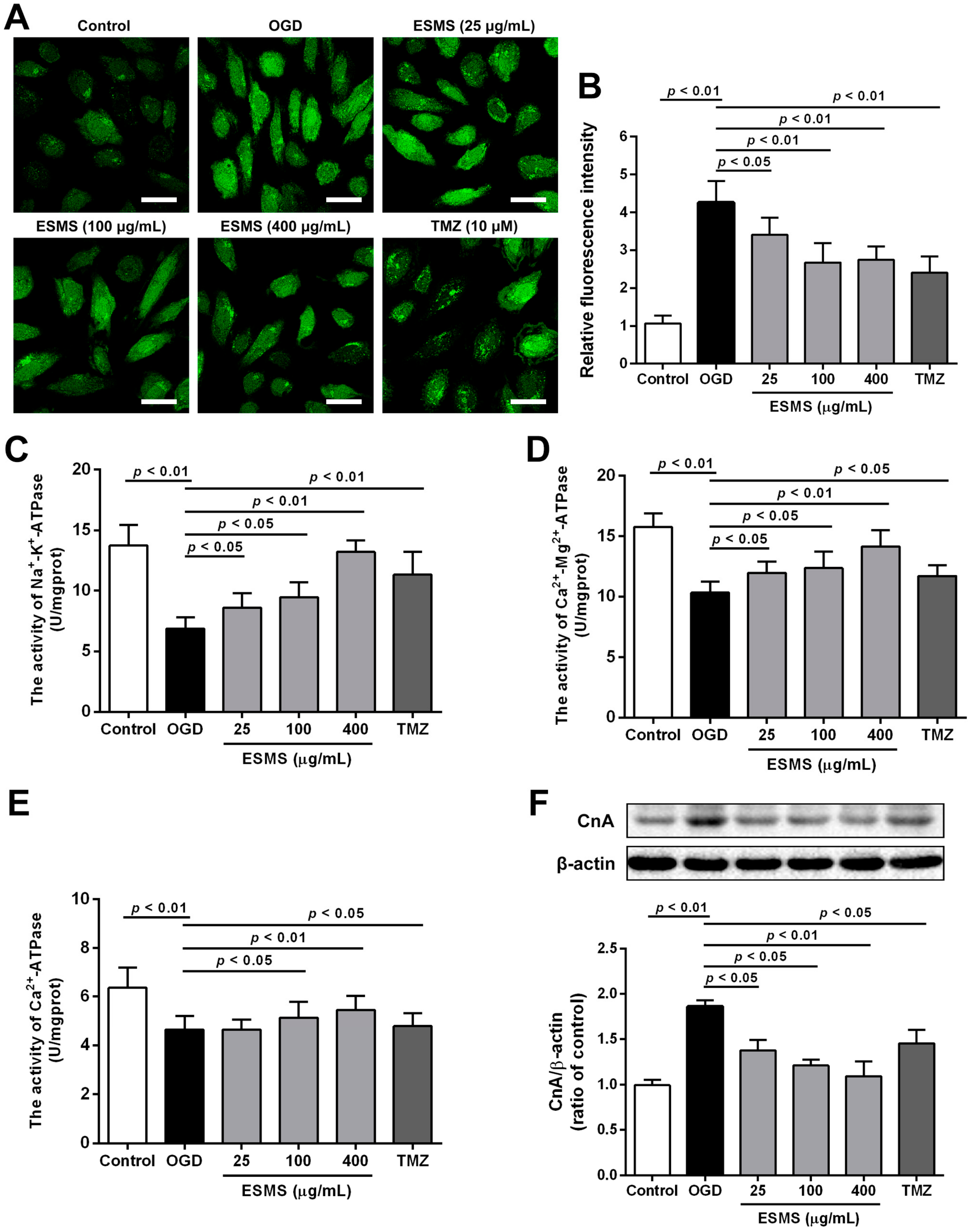
2.6. ESMS Reduced Intracellular Ca2+ Levels, Increased ATPase Activity, and Decreased the Expression of Calcineurin A (CnA) in H9c2 Cells Subjected to OGD
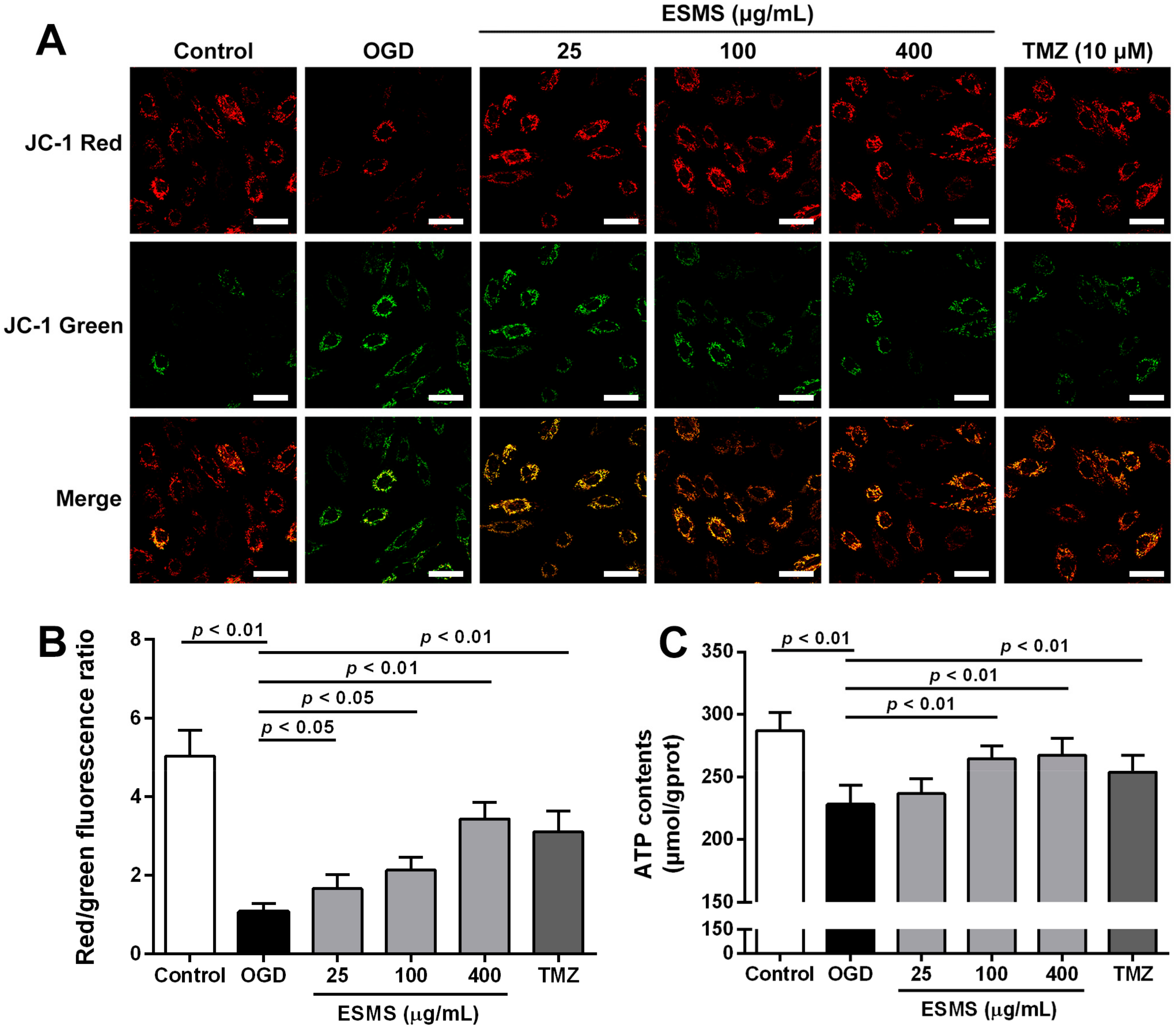
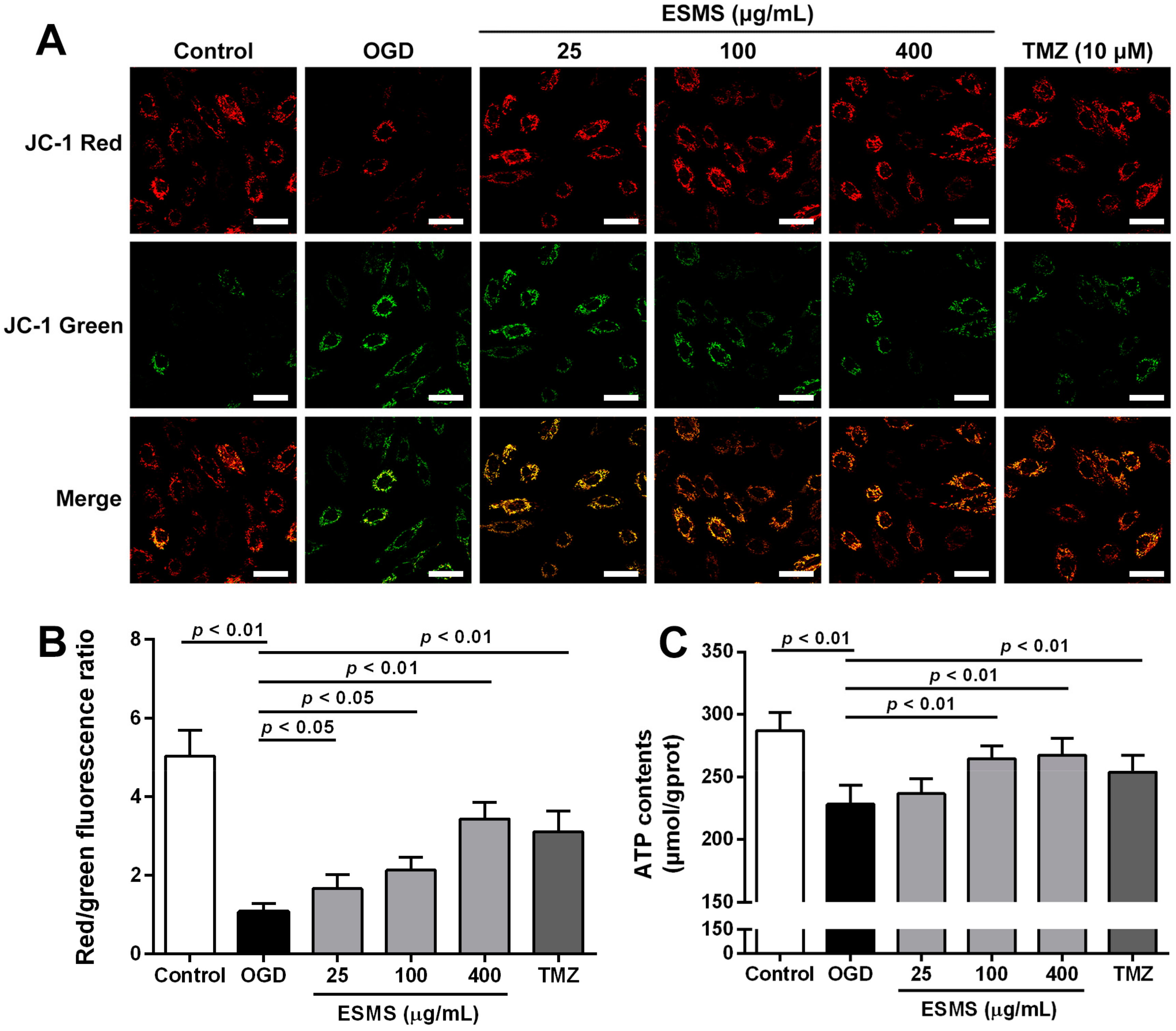
2.7. ESMS Restored Mitochondrial Membrane Potential and Cellular ATP in H9c2 Cells Subjected to OGD
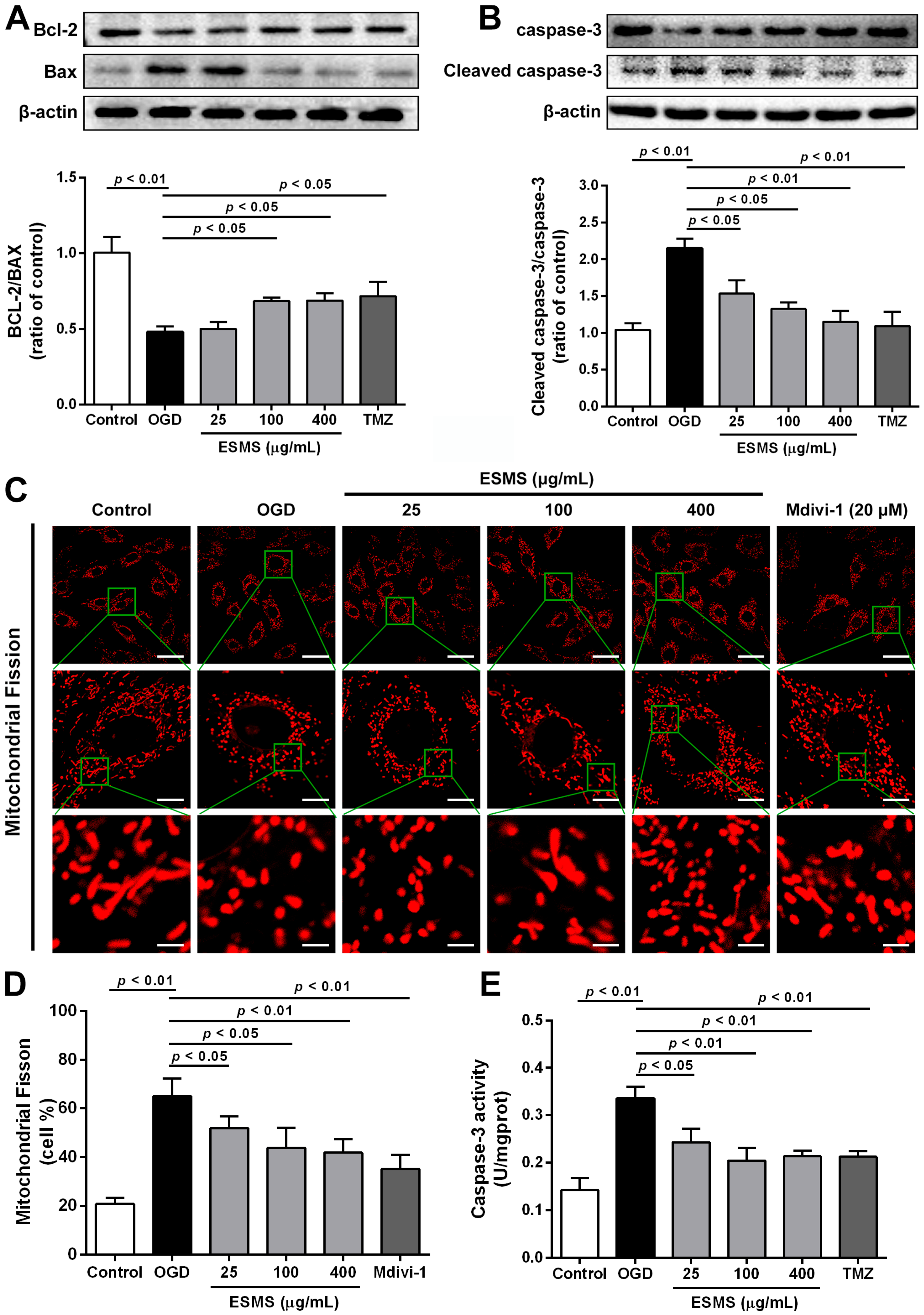
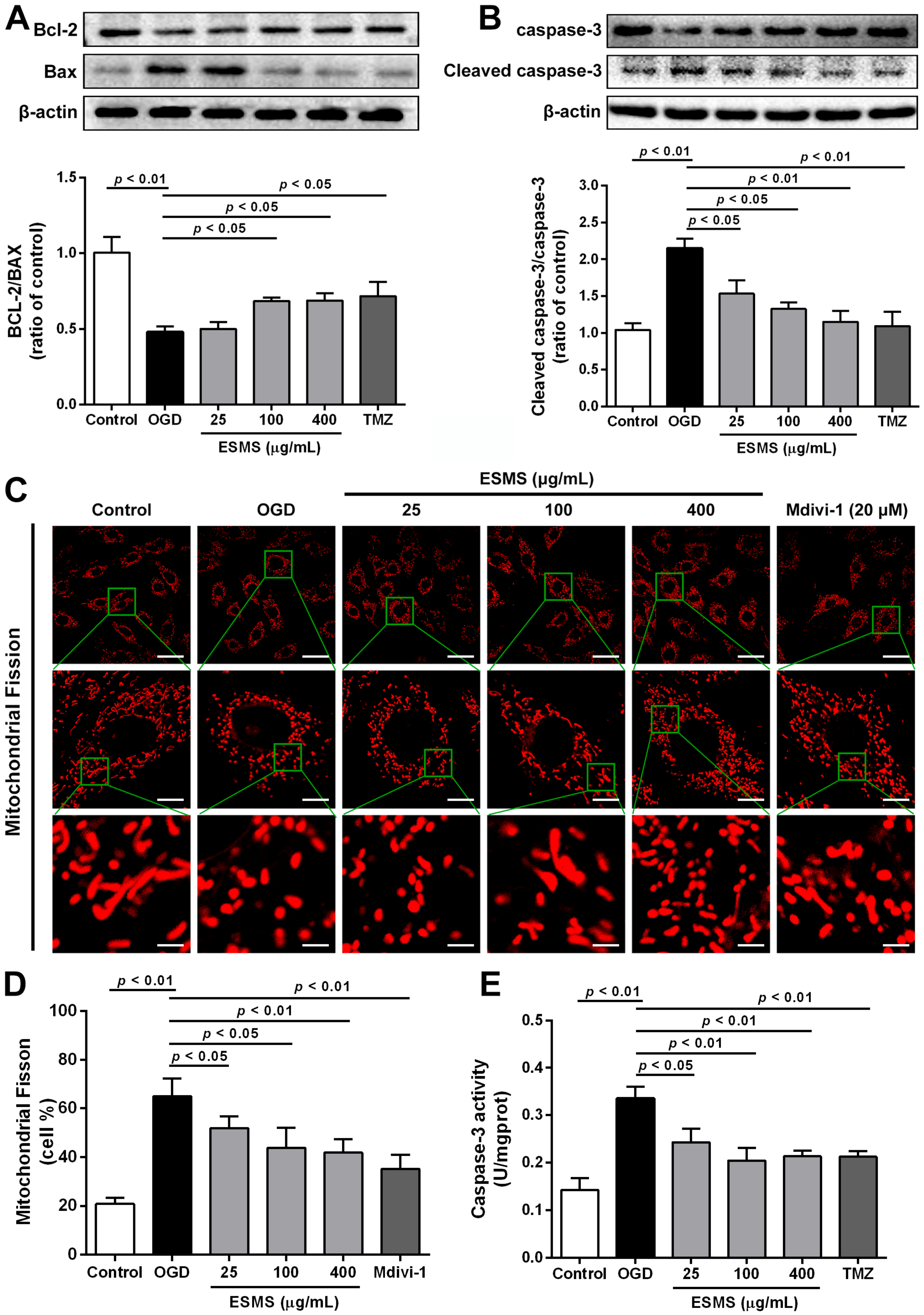
2.8. ESMS Ameliorated the Expression of Mitochondrial Mediated Apoptosis Proteins and Reduced the Caspase-3 Activity in H9c2 Cells Subjected to OGD
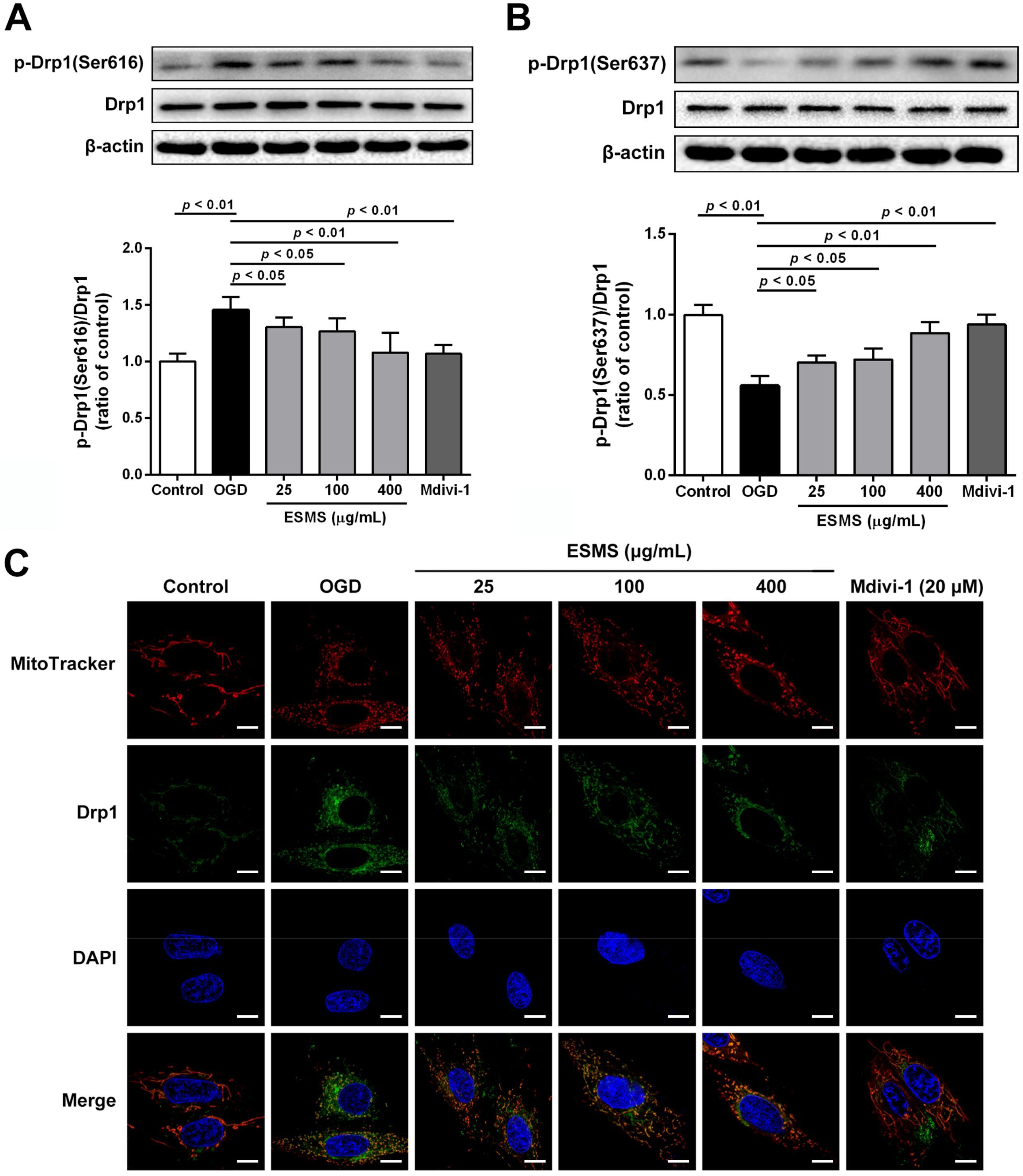
2.9. ESMS Inhibited Mitochondrial Fission and Regulated Drp1 Phosphorylation and Translocation
3. Discussion
4. Materials and Methods
4.1. Drugs and Reagents
4.2. Animals
4.3. Surgical Preparation
4.4. Echocardiographic Analysis
4.5. Histologic Examination
4.6. Evaluation of LDH, CK Activities and BNP Levels in Serum
4.7. Measurement of ATPase Activities, Ca2+ Levels, and Caspase-3 Activity in Heart Tissues
4.8. TUNEL Staining for Apoptosis In Vivo
4.9. Cell Culture
4.10. Oxygen-Glucose Deprivation (OGD) and Drug Treatment
4.11. Cell Viability and LDH Assays
4.12. Flow-Cytometric Analysis for Apoptosis
4.13. Measurement of ATPase Activities, ATP Content and Caspase-3 Activity In Vitro
4.14. Measurement of Intracellular Ca2+ Concentration
4.15. Measurement of Mitochondria Membrane Potential
4.16. Mitochondrial Fission Analysis
4.17. Western Blotting Analysis
4.18. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HF | Heart failure |
| MI | Myocardial ischemia |
| ESMS | Extract of Sheng-Mai-San |
| OGD | Oxygen-glucose deprivation |
| LVEF | Left ventricular ejection fraction |
| LVFS | Left ventricular fractional shortening |
| LDH | Lactate dehydrogenase |
| CK | Creatine kinase |
| BNP | Brain natriuretic peptide |
| CnA | Calcineurin A |
| Drp1 | Dynamin-related protein 1 |
| Bcl-2 | B cell lymphoma/lewkmia-2 |
| BAX | Bcl-2 Assaciated X protein |
| Caspase-3 | Cysteinyl aspartate specific proteinase-3 |
References
- Roger, V.L. Epidemiology of heart failure. Circ. Res. 2013, 113, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E. The war against heart failure: The lancet lecture. Lancet 2015, 385, 812–824. [Google Scholar] [CrossRef]
- O’Gara, P.T.; Kushner, F.G.; Ascheim, D.D.; Casey, D.E.; Chung, M.K.; de Lemos, J.A.; Ettinger, S.M.; Fang, J.C.; Fesmire, F.M.; Franklin, B.A.; et al. 2013 ACCF/AHA guideline for the management of stelevation myocardial infarction: Executive summary a report of the american college of cardiology foundation/american heart association task force on practice guidelines. J. Am. Coll. Cardiol. 2013, 61, 485–510. [Google Scholar] [CrossRef] [PubMed]
- Moran, A.E.; Forouzanfar, M.H.; Roth, G.A.; Mensah, G.A.; Ezzati, M.; Flaxman, A.; Murray, C.J.L.; Naghavi, M. The global burden of ischemic heart disease in 1990 and 2010. Circulation 2014, 129, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Ferrari, R.; Sharpe, N.; International Forum on Cardiac Remodeling. Cardiac remodeling-Concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart disease and stroke statistics—2017 update a report from the american heart association. Circulation 2017, 135, E146–E603. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Zhang, J.; Huang, J.; Ma, A.Q.; Yang, J.F.; Li, W.M.; Wu, Z.G.; Yao, C.; Zhang, Y.H.; Yao, W.M.; et al. A multicenter, randomized, double-blind, parallel-group, placebo-controlled study of the effects of Qili Qiangxin capsules in patients with chronic heart failure. J. Am. Coll. Cardiol. 2013, 62, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.N.; Hu, S.Y.; Li, G.X.; Xue, J.; Li, Z.M.; Liu, X.L.; Yang, X.Y.; Dong, B.; Wang, D.H.; Wang, X.F.; et al. Comparative effectiveness of di'ao xin xue kang capsule and compound danshen tablet in patients with symptomatic chronic stable angina. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Lu, B.J.; Zhao, M.H.; Rong, Y.Z.; Chen, R.M. Effect of shengmai injection on vascular endothelial and heart functions in patients with coronary heart disease complicated with diabetes mellitus. Chin. J. Integr. Med. 2008, 14, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Jiang, M.; Dong, L.Y.; Gao, J.; Hou, Y.Y.; Bai, G.; Luo, G.A. Cardioprotective effects of the yiqifumai injection and isolated compounds on attenuating chronic heart failure via NF-κB inactivation and cytokine suppression. J. Ethnopharmacol. 2013, 148, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zheng, X.J.; Fan, X.X.; Zhai, K.F.; Tan, Y.S.; Kou, J.P.; Yu, B.Y. Yiqifumai powder injection attenuates ischemia/reperfusion-induced myocardial apoptosis through ampk activation. Rejuvenation Res. 2016, 19, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.S.; Chen, H.L.; Zhang, Y.Y.; Li, F.; Liu, C.H.; Xiang, X.; Qi, J.; Chai, C.Z.; Kou, J.P.; Yu, B.Y. Yiqifumai powder injection ameliorates the oxygen-glucose deprivation-induced brain microvascular endothelial barrier dysfunction associated with the NF-κB and ROCK1/MLC signaling pathways. J. Ethnopharmacol. 2016, 183, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.Y.; Minatoguchi, S.; Arai, M.; Uno, Y.; Nishida, Y.; Hashimoto, K.; Hai, C.X.; Fukuda, K.; Akao, S.; Takemura, G.; et al. Sheng-mai-san is protective against post-ischemic myocardial dysfunction in rats through its opening of the mitochondrial katp channels. Circ. J. 2002, 66, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Wang, G.J.; Xie, H.T.; Huang, Q.; Wang, W.; Jia, Y.W. Pharmacokinetic comparisons of schizandrin after oral administration of schizandrin monomer, fructus schisandrae aqueous extract and sheng-mai-san to rats. J. Ethnopharmacol. 2008, 115, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, H.; Wang, X.J.; Konishi, T. Role of component herbs in antioxidant activity of sheng-mai-san—A traditional Chinese medicine formula preventing cerebral oxidative damage in rat. Am. J. Chin. Med. 2003, 31, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Zhang, J.Q.; Liu, C.H.; Zhu, D.N.; Yu, B.Y. Screening and identifying the myocardial-injury protective ingredients from sheng-mai-san. Pharm. Biol. 2013, 51, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.M.; Kang, L.Y.; Wang, Y.; Zhao, X.P.; Liu, X.; Xu, L.; Li, Z. A metabonomic study of cardioprotection of ginsenosides, schizandrin, and ophiopogonin d against acute myocardial infarction in rats. BMC Complement. Altern. Med. 2014, 14, 401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Liu, C.H.; Zhang, J.Q.; Zhu, D.N.; Yu, B.Y. Protective effects and active ingredients of yi-qi-fu-mai sterile powder against myocardial oxidative damage in mice. J. Pharmacol. Sci. 2013, 122, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.Q.; Ju, A.C.; Liu, C.H.; Wang, T.; Yu, B.Y.; Qi, J. Protective effect of the extract of yi-qi-fu-mai preparation on hypoxia-induced heart injury in mice. Chin. J. Nat. Med. 2016, 14, 401–406. [Google Scholar] [CrossRef]
- Ong, S.B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L. Mitochondrial dynamics—Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.H.G.M.; Rossignol, R.; Dieteren, C.E.J.; Murphy, M.P.; Koopman, W.J.H. Redox homeostasis and mitochondrial dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Zaja, I.; Bai, X.W.; Liu, Y.N.; Kikuchi, C.; Dosenovic, S.; Yan, Y.S.; Canfield, S.G.; Bosnjak, Z.J. Cdk1, PKCδ and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem. Biophys. Res. Commun. 2014, 453, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Marks, A.R. Calcium cycling proteins and heart failure: Mechanisms and therapeutics. J. Clin. Investig. 2013, 123, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Shannon, T.R. Calcium movements inside the sarcoplasmic reticulum of cardiac myocytes. J. Mol. Cell. Cardiol. 2013, 58, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Verdejo, H.E.; del Campo, A.; Troncoso, R.; Gutierrez, T.; Toro, B.; Quiroga, C.; Pedrozo, Z.; Munoz, J.P.; Garcia, L.; Castro, P.F.; et al. Mitochondria, myocardial remodeling, and cardiovascular disease. Curr. Hypertens. Rep. 2012, 14, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; de Brito, O.M.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef]
- Pennanen, C.; Parra, V.; Lopez-Crisosto, C.; Morales, P.E.; del Campo, A.; Gutierrez, T.; Rivera-Mejias, P.; Kuzmicic, J.; Chiong, M.; Zorzano, A.; et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell Sci. 2014, 127, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Korge, P.; Honda, H.M.; Ping, P.P. Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 2003, 93, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Costa, V.; Scorrano, L. Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010, 17, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, A.A.; Butler, J.; Schelbert, E.B.; Greene, S.J.; Quyyumi, A.A.; Bonow, R.O.; Cohen, I.; Gheorghiade, M.; Lipinski, M.J.; Sun, W.; et al. Mechanisms contributing to the progression of ischemic and nonischemic dilated cardiomyopathy possible modulating effects of paracrine activities of stem cells. J. Am. Coll. Cardiol. 2015, 66, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the american society of echocardiography and the european association of cardiovascular imaging. J. Am. Soc. Echocardiogr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gao, E.; Lei, Y.H.; Shang, X.Y.; Huang, Z.M.; Zuo, L.; Boucher, M.; Fan, Q.A.; Chuprun, J.K.; Ma, X.L.; Koch, W.J. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 2010, 107, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.; Lygate, C.A.; Saunders, J.; Schneider, J.E.; Ye, X.J.; Hulbert, K.; Noble, J.A.; Neubauer, S. Quantitative 3-dimensional echocardiography for accurate and rapid cardiac phenotype characterization in mice. Circulation 2004, 110, 1632–1637. [Google Scholar] [CrossRef] [PubMed]
- Mo, W.L.; Chai, C.Z.; Kou, J.P.; Yan, Y.O.; Yu, B.Y. Sheng-mai-san attenuates contractile dysfunction and structural damage induced by chronic intermittent hypoxia in mice. Chin. J. Nat. Med. 2015, 13, 743–750. [Google Scholar] [CrossRef]
- Kawabe, M.; Sato, A.; Hoshi, T.; Endo, M.; Yoshida, I.; Aonuma, K. Incremental value of b-type natriuretic peptide for detection and risk reclassification of obstructive coronary artery disease on computed tomography angiography. J. Cardiol. 2017, 69, 671–677. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Bohm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. J. Heart Fail. 2012, 14, 803–869. [Google Scholar] [PubMed]
- Wassilew, K.; Terziev, D.; Wassilew, G.; Fitzl, G.; Frolich, K.; Kandolf, R.; Fried, A. Ultrastructural morphometric findings of cardiomyocytes in patients with impaired ventricular function—A comparative clinicopathological study. Cardiovasc. Pathol. 2016, 25, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kirshenbaum, L.A.; Li, T.; Danelisen, I.; Singal, P.K. Apoptosis in adriamycin cardiomyopathy and its modulation by probucol. Antioxid. Redox Signal. 2001, 3, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Mork, H.K.; Sjaastad, I.; Sejersted, O.M.; Louch, W.E. Slowing of cardiomyocyte Ca2+ release and contraction during heart failure progression in postinfarction mice. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1069–H1079. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Chen, X.; Baines, C.P.; Klevitsky, R.; Zhang, X.; Zhang, H.; Jaleel, N.; Chua, B.H.; Hewett, T.E.; Robbins, J.; et al. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Investig. 2007, 117, 2431–2444. [Google Scholar] [CrossRef] [PubMed]
- Roe, A.T.; Frisk, M.; Louch, W.E. Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr. Pharm. Des. 2015, 21, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Mork, H.K.; Sjaastad, L.; Sande, J.B.; Periasamy, M.; Sejersted, O.M.; Louch, W.E. Increased cardiomyocyte function and Ca2+ transients in mice during early congestive heart failure. J. Mol. Cell. Cardiol. 2007, 43, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Saini, H.K.; Dhalla, N.S. Sarcolemmal cation channels and exchangers modify the increase in intracellular calcium in cardiomyocytes on inhibiting Na+-k+-ATPase. Am. J. Physiol.Heart C 2007, 293, H169–H181. [Google Scholar] [CrossRef] [PubMed]
- Marin-Garcia, J.; Akhmedov, A.T. Mitochondrial dynamics and cell death in heart failure. Heart Fail. Rev. 2016, 21, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Camperchioli, A.; Mariani, M.; Bartollino, S.; Petrella, L.; Persico, M.; Orteca, N.; Scambia, G.; Shahabi, S.; Ferlini, C.; Fattorusso, C. Investigation of the Bcl-2 multimerisation process: Structural and functional implications. BBA-Mol. Cell Res. 2011, 1813, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Mayorga, M.; Bahi, N.; Ballester, M.; Comella, J.X.; Sanchis, D. Bcl-2 is a key factor for cardiac fibroblast resistance to programmed cell death. J. Biol. Chem. 2004, 279, 34882–34889. [Google Scholar] [CrossRef] [PubMed]
- Landes, T.; Martinou, J.C. Mitochondrial outer membrane permeabilization during apoptosis: The role of mitochondrial fission. BBA-Mol. Cell Res. 2011, 1813, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.A.; Gan, X.T.; Haist, J.V.; Rajapurohitam, V.; Zeidan, A.; Faruq, S.; Karmazyn, M. Ginseng inhibits cardiomyocyte hypertrophy and heart failure via NHE-1 inhibition and attenuation of calcineurin activation. Circ. Heart Fail 2011, 4, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Zhao, B.; Wu, Y.; Hou, J.B.; Zhang, L.; Xu, J.J.; Xia, Z.Y. Ginsenoside Rb1 preconditioning enhances enos expression and attenuates myocardial ischemia/reperfusion injury in diabetic rats. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.S.; Huang, X.N.; Dai, Z.K.; Yang, G.Z.; Zhou, Q.X.; Shi, J.S.; Wu, Q. Inhibitory effect of ginsenoside Rb1 on cardiac hypertrophy induced by monocrotaline in rat. J. Ethnopharmacol. 2007, 111, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lv, X.T.; Wu, Q.; Huang, X.N. Ginsenoside Rg1 inhibits rat left ventricular hypertrophy induced by abdominal aorta coarctation: Involvement of calcineurin and mitogen-activated protein kinase signalings. Eur. J. Pharmacol. 2009, 608, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.X.; Sunami, A.; Namiki, T.; Sawanobori, T.; Furukawa, T. Electrophysiological effects of ginseng and ginsenoside Re in guinea pig ventricular myocytes. Eur. J. Pharmacol. 2003, 476, 35–44. [Google Scholar] [CrossRef]
- Choi, S.H.; Lee, B.H.; Kim, H.J.; Jung, S.W.; Kim, H.S.; Shin, H.C.; Lee, J.H.; Kim, H.C.; Rhim, H.; Hwang, S.H.; et al. Ginseng gintonin activates the human cardiac delayed rectifier k+ channel: Involvement of Ca2+/calmodulin binding sites. Mol. Cells 2014, 37, 656–663. [Google Scholar] [CrossRef] [PubMed]
- You, W.T.; Zhou, T.; Ma, Z.C.; Liang, Q.D.; Xiao, C.R.; Tang, X.L.; Tan, H.L.; Zhang, B.L.; Wang, Y.G.; Gao, Y. Ophiopogonin D maintains Ca2+ homeostasis in rat cardiomyocytes in vitro by upregulating CYP2J3/EETs and suppressing ER stress. Acta Pharmacol. Sin. 2016, 37, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Yim, T.K.; Ko, K.M. Schisandrin B protects against myocardial ischemia-reperfusion injury by enhancing myocardial glutathione antioxidant status. Mol. Cell. Biochem. 1999, 196, 151–156. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Tian, Y.; Hu, S.; Bi, S.; Li, S.; Hu, Y.; Kou, J.; Qi, J.; Yu, B. Extract of Sheng-Mai-San Ameliorates Myocardial Ischemia-Induced Heart Failure by Modulating Ca2+-Calcineurin-Mediated Drp1 Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 1825. https://doi.org/10.3390/ijms18091825
Yang Y, Tian Y, Hu S, Bi S, Li S, Hu Y, Kou J, Qi J, Yu B. Extract of Sheng-Mai-San Ameliorates Myocardial Ischemia-Induced Heart Failure by Modulating Ca2+-Calcineurin-Mediated Drp1 Signaling Pathways. International Journal of Molecular Sciences. 2017; 18(9):1825. https://doi.org/10.3390/ijms18091825
Chicago/Turabian StyleYang, Ye, Yushan Tian, Siyao Hu, Suxia Bi, Suxia Li, Yuanjia Hu, Junping Kou, Jin Qi, and Boyang Yu. 2017. "Extract of Sheng-Mai-San Ameliorates Myocardial Ischemia-Induced Heart Failure by Modulating Ca2+-Calcineurin-Mediated Drp1 Signaling Pathways" International Journal of Molecular Sciences 18, no. 9: 1825. https://doi.org/10.3390/ijms18091825