A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy

Abstract
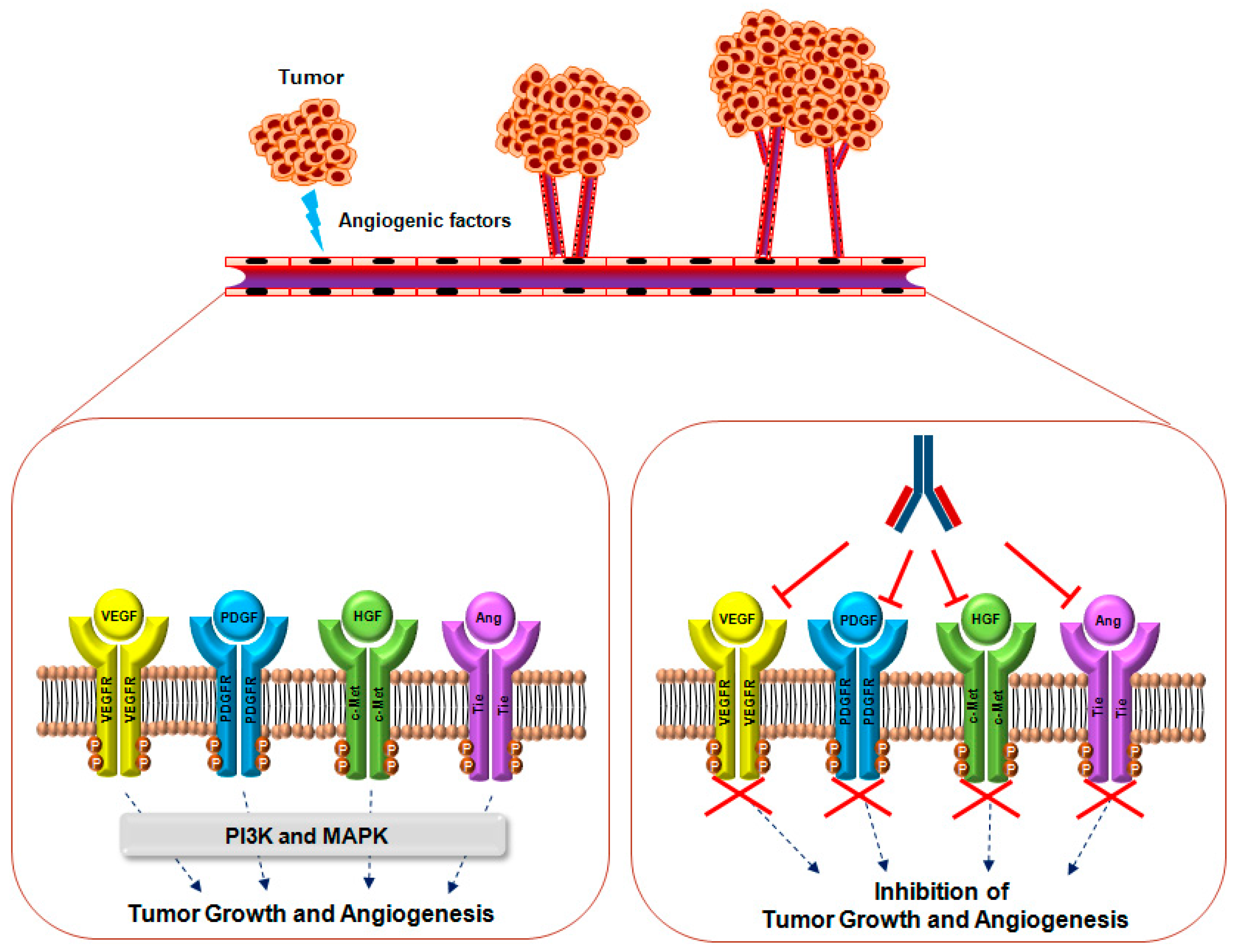
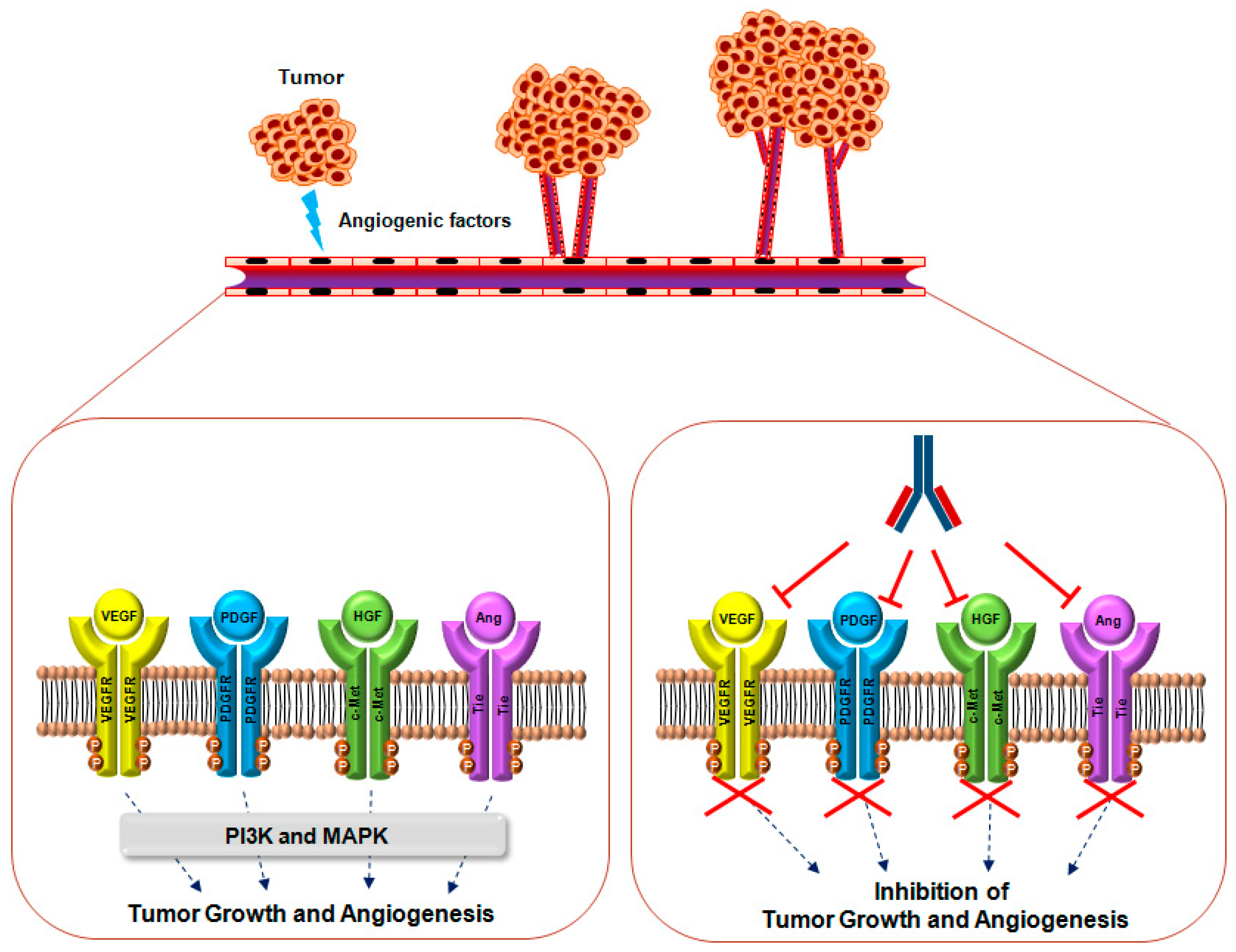
:1. Introduction
2. VEGF Signaling
2.1. Overview
2.2. Relevance of VEGF and VEGFR in Cancer
2.3. Monoclonal Antibodies Targeting VEGF and VEGFR
3. PDGF Signaling
3.1. Overview
3.2. The Role and Relevance of PDGF and PDGFR in Cancer
3.3. Monoclonal Antibodies Targeting PDGF and PDGFR
4. Ang Signaling
4.1. Overview
4.2. The Role and Relevance of Ang and the Tie Receptor in Cancer
4.3. Monoclonal Antibodies Targeting Ang and Tie Receptors
5. HGF Signaling
5.1. Overview
5.2. The Role and Relevance of HGF and c-MET Signaling in Cancer
5.3. Monoclonal Antibodies Targeting HGF and c-MET
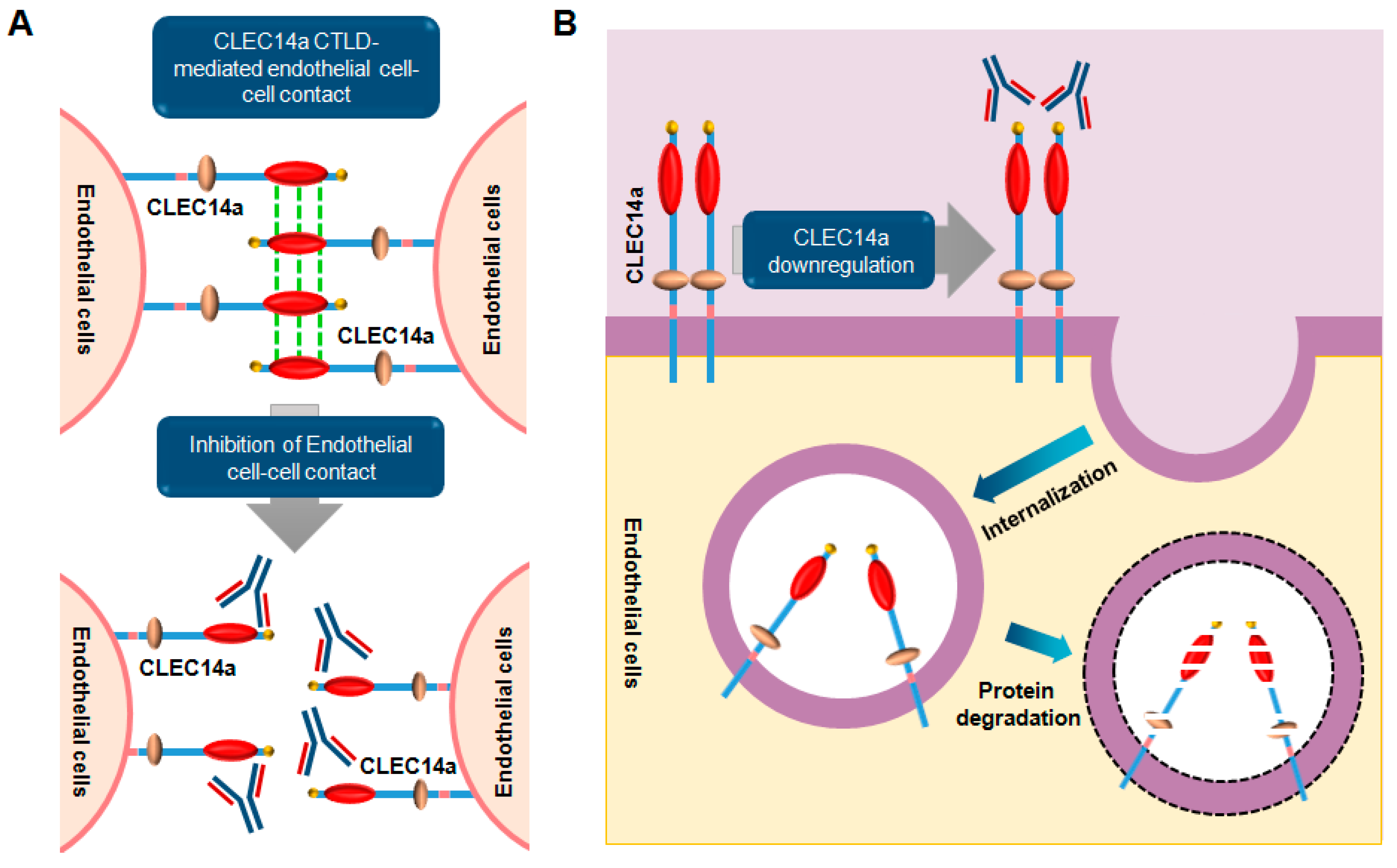
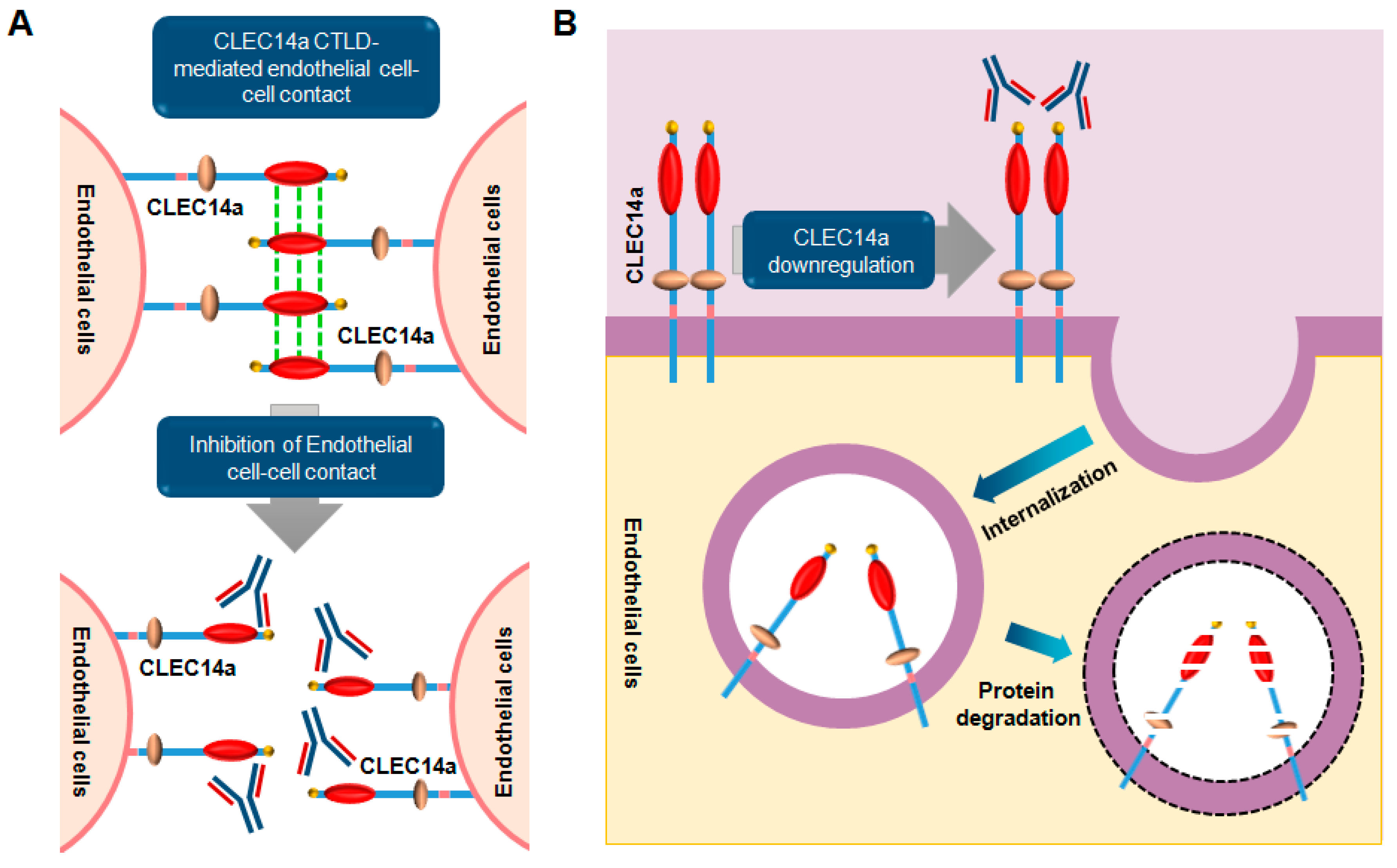
6. CLEC14a Signaling
6.1. Overview
6.2. Role and Relevance of CLEC14a in Cancer
6.3. Monoclonal Antibodies Targeting CLEC14a
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Ang | Angiopoietin |
| CLEC14a | C-type domain family 14 member |
| CTLD | C-type lectin-like domain |
| HGF | Hepatocyte growth factor |
| Ig | Immunoglobulin |
| PDGF | Platelet-derived growth factor |
| Tie | Tyrosine kinase with Ig-like and EGF-like |
| VEGF | Vascular endothelial growth factor |
References
- Li, B.; Xiu, R. Angiogenesis: From molecular mechanisms to translational implications. Clin. Hemorheol. Microcirc. 2013, 54, 345–355. [Google Scholar] [PubMed]
- Papetti, M.; Herman, I.M. Mechanisms of normal and tumor-derived angiogenesis. Am. J. Physiol. Cell Physiol. 2002, 282, C947–C970. [Google Scholar] [CrossRef] [PubMed]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.W.; Cachianes, G.; Kuang, W.J.; Goeddel, D.V.; Ferrara, N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 1989, 246, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Millauer, B.; Wizigmann-Voos, S.; Schnurch, H.; Martinez, R.; Moller, N.P.; Risau, W.; Ullrich, A. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993, 72, 835–846. [Google Scholar] [CrossRef]
- Battegay, E.J.; Rupp, J.; Iruela-Arispe, L.; Sage, E.H.; Pech, M. PDGF-BB modulates endothelial proliferation and angiogenesis in vitro via PDGF β-receptors. J. Cell Biol. 1994, 125, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Suri, C.; Jones, P.F.; Patan, S.; Bartunkova, S.; Maisonpierre, P.C.; Davis, S.; Sato, T.N.; Yancopoulos, G.D. Requisite role of angiopoietin-1, a ligand for the Tie2 receptor, during embryonic angiogenesis. Cell 1996, 87, 1171–1180. [Google Scholar] [CrossRef]
- Rosen, E.M.; Grant, D.S.; Kleinman, H.K.; Goldberg, I.D.; Bhargava, M.M.; Nickoloff, B.J.; Kinsella, J.L.; Polverini, P. Scatter factor (hepatocyte growth factor) is a potent angiogenesis factor in vivo. Symp. Soc. Exp. Biol. 1993, 47, 227–234. [Google Scholar] [PubMed]
- Taniguchi, E.; Nagae, Y.; Watanabe, H.; Ohashi, Y.; Kinoshita, S.; Manabe, R. The effect of recombinant epidermal growth factor in corneal angiogenesis. Nippon Ganka Gakkai Zasshi 1991, 95, 52–58. [Google Scholar] [PubMed]
- Folkman, J.; Browder, T.; Palmblad, J. Angiogenesis research: Guidelines for translation to clinical application. Thromb. Haemost. 2001, 86, 23–33. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Gabrilove, J.L. Angiogenic growth factors: Autocrine and paracrine regulation of survival in hematologic malignancies. Oncologist 2001, 6 (Suppl. S5), 4–7. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Tumor angiogenesis and accessibility: Role of vascular endothelial growth factor. Semin. Oncol. 2002, 29, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Culy, C. Bevacizumab: Antiangiogenic cancer therapy. Drugs Today 2005, 41, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Arriaga, Y.; Becerra, C.R. Adverse effects of bevacizumab and their management in solid tumors. Support. Cancer Ther. 2006, 3, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Cunningham, D. Managing patients treated with bevacizumab combination therapy. Oncology 2005, 69 (Suppl 3), 25–33. [Google Scholar] [CrossRef] [PubMed]
- Lucio-Eterovic, A.K.; Piao, Y.; de Groot, J.F. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin. Cancer Res. 2009, 15, 4589–4599. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro-Oncology 2010, 12, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Nakada, M.; Misaki, K.; Sato, Y.; Hayashi, Y.; Nakanuma, Y.; Hamada, J. Molecular analysis of a recurrent glioblastoma treated with bevacizumab. Brain Tumor Pathol. 2014, 31, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bahr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Sathornsumetee, S.; Rich, J.N. Antiangiogenic therapy in malignant glioma: Promise and challenge. Curr. Pharm. Des. 2007, 13, 3545–3558. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, S.; Pages, G. Mechanisms of resistance to anti-angiogenesis therapies. Biochimie 2013, 95, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Kliche, S.; Waltenberger, J. VEGF receptor signaling and endothelial function. IUBMB Life 2001, 52, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Woolard, J.; Bevan, H.S.; Harper, S.J.; Bates, D.O. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation 2009, 16, 572–592. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Muller, Y.A.; Li, B.; Christinger, H.W.; Wells, J.A.; Cunningham, B.C.; De Vos, A.M. Vascular endothelial growth factor: Crystal structure and functional mapping of the kinase domain receptor binding site. Proc. Natl. Acad. Sci. USA 1997, 94, 7192–7197. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Li, X. VEGF-B: A thing of beauty. Cell Res. 2010, 20, 741–744. [Google Scholar] [CrossRef] [PubMed]
- De Falco, S. The discovery of placenta growth factor and its biological activity. Exp. Mol. Med. 2012, 44, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Fearnley, G.W.; Tomlinson, D.C.; Harrison, M.A.; Ponnambalam, S. The cellular response to vascular endothelial growth factors requires co-ordinated signal transduction, trafficking and proteolysis. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yamauchi, M.; Muramatsu, M.; Osawa, T.; Tsuchida, R.; Shibuya, M. Rack1 regulates VEGF/FLT1-mediated cell migration via activation of a PI3K/Akt pathway. J. Biol. Chem. 2011, 286, 9097–9106. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Ahmad, S.; Jiang, W.G.; Huang, J.; Kontos, C.D.; Boulton, M.; Ahmed, A. Activation of vascular endothelial growth factor receptor-1 sustains angiogenesis and BCL-2 expression via the phosphatidylinositol 3-kinase pathway in endothelial cells. Diabetes 2003, 52, 2959–2968. [Google Scholar] [CrossRef] [PubMed]
- Tchaikovski, V.; Fellbrich, G.; Waltenberger, J. The molecular basis of VEGFR-1 signal transduction pathways in primary human monocytes. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Bohman, S.; Dixelius, J.; Berge, T.; Dimberg, A.; Magnusson, P.; Wang, L.; Wikner, C.; Qi, J.H.; Wernstedt, C.; et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 2005, 24, 2342–2353. [Google Scholar] [CrossRef] [PubMed]
- Salameh, A.; Galvagni, F.; Bardelli, M.; Bussolino, F.; Oliviero, S. Direct recruitment of CRK and GRB2 to VEGFR-3 induces proliferation, migration, and survival of endothelial cells through the activation of Erk, Akt, and Jnk pathways. Blood 2005, 106, 3423–3431. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhang, X.; Simons, M. Molecular controls of lymphatic VEGFR3 signaling. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Liu, L.Z. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv. Cancer Res. 2009, 102, 19–65. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jeong, D.; Han, Y.S.; Baek, M.J. Pivotal role of vascular endothelial growth factor pathway in tumor angiogenesis. Ann. Surg. Treat. Res. 2015, 89, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.P.; Levy, N.S.; Goldberg, M.A. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J. Biol. Chem. 1996, 271, 2746–2753. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Li, Y.; Jiang, G. Role of VEGF/VEGFR in the pathogenesis of leukemias and as treatment targets (review). Oncol. Rep. 2012, 28, 1935–1944. [Google Scholar] [PubMed]
- Yang, Y.; Bai, Y.; Xie, G.; Zhang, N.; Ma, Y.P.; Chen, L.J.; Jiang, Y.; Zhao, X.; Wei, Y.Q.; Deng, H.X. Efficient inhibition of non-small-cell lung cancer xenograft by systemic delivery of plasmid-encoding short-hairpin rna targeting vegf. Cancer Biother. Radiopharm. 2010, 25, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Brekken, R.A.; Hyder, S.M. Vascular endothelial growth factor induces proliferation of breast cancer cells and inhibits the anti-proliferative activity of anti-hormones. Endocr. Relat. Cancer 2006, 13, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.; Fang, J.; Yang, C. Overexpression of both VEGF-A and VEGF-C in gastric cancer correlates with prognosis, and silencing of both is effective to inhibit cancer growth. Int. J. Clin. Exp. Pathol. 2013, 6, 586–597. [Google Scholar] [PubMed]
- Ohta, Y.; Endo, Y.; Tanaka, M.; Shimizu, J.; Oda, M.; Hayashi, Y.; Watanabe, Y.; Sasaki, T. Significance of vascular endothelial growth factor messenger RNA expression in primary lung cancer. Clin. Cancer Res. 1996, 2, 1411–1416. [Google Scholar] [PubMed]
- Lu, L.; Luo, S.T.; Shi, H.S.; Li, M.; Zhang, H.L.; He, S.S.; Liu, Y.; Pan, Y.; Yang, L. Aav2-mediated gene transfer of VEGF-trap with potent suppression of primary breast tumor growth and spontaneous pulmonary metastases by long-term expression. Oncol. Rep. 2012, 28, 1332–1338. [Google Scholar] [PubMed]
- Zins, K.; Kovatchki, D.; Lucas, T.; Abraham, D. PLGF and VEGF-A regulate growth of high-risk MYCN-single copy neuroblastoma xenografts via different mechanisms. Int. J. Mol. Sci. 2016, 17, 1613. [Google Scholar] [CrossRef] [PubMed]
- De Brot, S.; Ntekim, A.; Cardenas, R.; James, V.; Allegrucci, C.; Heery, D.M.; Bates, D.O.; Odum, N.; Persson, J.L.; Mongan, N.P. Regulation of vascular endothelial growth factor in prostate cancer. Endocr. Relat. Cancer 2015, 22, R107–R123. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Li, X.; Qin, C.; Li, J. Prognostic value of VEGF in hepatocellular carcinoma patients treated with sorafenib: A meta-analysis. Med. Sci. Monit. 2015, 21, 3144–3151. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jia, M. Antibody therapies in cancer. Adv. Exp. Med. Biol. 2016, 909, 1–67. [Google Scholar] [PubMed]
- Andreakos, E.; Taylor, P.C.; Feldmann, M. Monoclonal antibodies in immune and inflammatory diseases. Curr. Opin. Biotechnol. 2002, 13, 615–620. [Google Scholar] [CrossRef]
- Martin, D.F.; Maguire, M.G.; Ying, G.S.; Grunwald, J.E.; Fine, S.L.; Jaffe, G.J. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011, 364, 1897–1908. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Braghiroli, M.I.; Sabbaga, J.; Hoff, P.M. Bevacizumab: Overview of the literature. Expert Rev. Anticancer Ther. 2012, 12, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M. Bevacizumab. Nat. Rev. Drug Discov. 2005, 4, S8–S9. [Google Scholar] [CrossRef]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. Fda drug approval summary: Bevacizumab (avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D. Bevacizumab in non-small-cell lung cancer: A review. Expert Rev. Anticancer Ther. 2011, 11, 1163–1179. [Google Scholar] [CrossRef] [PubMed]
- Rinne, M.L.; Lee, E.Q.; Nayak, L.; Norden, A.D.; Beroukhim, R.; Wen, P.Y.; Reardon, D.A. Update on bevacizumab and other angiogenesis inhibitors for brain cancer. Expert Opin. Emerg. Drugs 2013, 18, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Shih, T.; Lindley, C. Bevacizumab: An angiogenesis inhibitor for the treatment of solid malignancies. Clin. Ther. 2006, 28, 1779–1802. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Singh, H. Bevacizumab and ovarian cancer. Ther. Adv. Med. Oncol. 2013, 5, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Al-Halafi, A.M. Vascular endothelial growth factor trap-eye and trap technology: Aflibercept from bench to bedside. Oman J. Ophthalmol. 2014, 7, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Andre, T.; Chibaudel, B. Aflibercept (zaltrap (®)) approved in metastatic colorectal cancer. Bull Cancer 2013, 100, 1023–1025. [Google Scholar] [PubMed]
- Sarwar, S.; Clearfield, E.; Soliman, M.K.; Sadiq, M.A.; Baldwin, A.J.; Hanout, M.; Agarwal, A.; Sepah, Y.J.; Do, D.V.; Nguyen, Q.D. Aflibercept for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 2016, 2, CD011346. [Google Scholar] [PubMed]
- Ashraf, M.; Souka, A.A.R. Aflibercept in age-related macular degeneration: Evaluating its role as a primary therapeutic option. Eye 2017. [Google Scholar] [CrossRef] [PubMed]
- Moreno, T.A.; Kim, S.J. Ranibizumab (lucentis) versus bevacizumab (avastin) for the treatment of age-related macular degeneration: An economic disparity of eye health. Semin. Ophthalmol. 2016, 31, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Bevacizumab (avastin) and age-related macular degeneration. Lower cost does not justify taking risks. Prescrire Int. 2015, 24, 201–204.
- Spratlin, J.L.; Mulder, K.E.; Mackey, J.R. Ramucirumab (IMC-1121B): A novel attack on angiogenesis. Future Oncol. 2010, 6, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Spratlin, J. Ramucirumab (imc-1121b): Monoclonal antibody inhibition of vascular endothelial growth factor receptor-2. Curr. Oncol. Rep. 2011, 13, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Daga, H. Ramucirumab for the treatment of advanced or metastatic non-small cell lung cancer. Expert Opin. Biol. Ther. 2016, 16, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.M.; Vaidya, A. Ramucirumab: First global approval. Drugs 2014, 74, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Zatarain-Barron, Z.L.; Cardona, A.F.; Carmona, A.; Lopez-Mejia, M. Ramucirumab in the treatment of non-small cell lung cancer. Expert Opin. Drug Saf. 2017, 16, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Park, J.O.; Ryoo, B.Y.; Yen, C.J.; Poon, R.; Pastorelli, D.; Blanc, J.F.; Chung, H.C.; Baron, A.D.; Pfiffer, T.E.; et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (reach): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 859–870. [Google Scholar] [CrossRef]
- Mackey, J.R.; Ramos-Vazquez, M.; Lipatov, O.; McCarthy, N.; Krasnozhon, D.; Semiglazov, V.; Manikhas, A.; Gelmon, K.A.; Konecny, G.E.; Webster, M.; et al. Primary results of ROSE/TRIO-12, a randomized placebo-controlled phase III trial evaluating the addition of ramucirumab to first-line docetaxel chemotherapy in metastatic breast cancer. J. Clin. Oncol. 2015, 33, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tagawa, S.T.; Kohli, M.; Eisen, A.; Canil, C.; Sridhar, S.S.; Spira, A.; Yu, E.Y.; Burke, J.M.; Shaffer, D.; et al. Docetaxel as monotherapy or combined with ramucirumab or icrucumab in second-line treatment for locally advanced or metastatic urothelial carcinoma: An open-label, three-arm, randomized controlled phase II trial. J. Clin. Oncol. 2016, 34, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H. Tanibirumab (ttac-0001): A fully human monoclonal antibody targets vascular endothelial growth factor receptor 2 (VEGFR-2). Arch. Pharm. Res. 2011, 34, 1223–1226. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Lee, S.Y.; Lee, W.S.; Yoo, J.S.; Sun, J.M.; Lee, J.; Park, S.H.; Park, J.O.; Ahn, M.J.; Lim, H.Y.; et al. Phase I trial and pharmacokinetic study of tanibirumab, a fully human monoclonal antibody to vascular endothelial growth factor receptor 2, in patients with refractory solid tumors. Investig. New Drugs 2017. [Google Scholar] [CrossRef] [PubMed]
- Raica, M.; Cimpean, A.M. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals 2010, 3, 572–599. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Lipton, A.; Klinger, I. Serum factor requirements of normal and simian virus 40-transformed 3T3 mouse fibroplasts. Proc. Natl. Acad. Sci. USA 1971, 68, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Ek, B.; Heldin, C.H. Characterization of a tyrosine-specific kinase activity in human fibroblast membranes stimulated by platelet-derived growth factor. J. Biol. Chem. 1982, 257, 10486–10492. [Google Scholar] [PubMed]
- Heldin, C.H. Targeting the pdgf signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef] [PubMed]
- Shim, A.H.; Liu, H.; Focia, P.J.; Chen, X.; Lin, P.C.; He, X. Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc. Natl. Acad. Sci. USA 2010, 107, 11307–11312. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Chen, X.; He, X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim. Biophys. Acta 2013, 1834, 2176–2186. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, L.; Li, H.; Eriksson, U. The pdgf family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004, 15, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Heldin, C.H.; Ostman, A. Immunoglobulin-like domain 4-mediated receptor-receptor interactions contribute to platelet-derived growth factor-induced receptor dimerization. J. Biol. Chem. 1997, 272, 12676–12682. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yuzawa, S.; Schlessinger, J. Contacts between membrane proximal regions of the PDGF receptor ectodomain are required for receptor activation but not for receptor dimerization. Proc. Natl. Acad. Sci. USA 2008, 105, 7681–7686. [Google Scholar] [CrossRef] [PubMed]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Yang, S.I.; Chan, T.O.; Datta, K.; Kazlauskas, A.; Morrison, D.K.; Kaplan, D.R.; Tsichlis, P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 1995, 81, 727–736. [Google Scholar] [CrossRef]
- Akimoto, K.; Takahashi, R.; Moriya, S.; Nishioka, N.; Takayanagi, J.; Kimura, K.; Fukui, Y.; Osada, S.; Mizuno, K.; Hirai, S.; et al. EGF or PDGF receptors activate atypical pkclambda through phosphatidylinositol 3-kinase. EMBO J. 1996, 15, 788–798. [Google Scholar] [PubMed]
- Chung, J.; Grammer, T.C.; Lemon, K.P.; Kazlauskas, A.; Blenis, J. PDGF- and insulin-dependent pp70s6k activation mediated by phosphatidylinositol-3-OH kinase. Nature 1994, 370, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ilasaca, M.; Li, W.; Uren, A.; Yu, J.C.; Kazlauskas, A.; Gutkind, J.S.; Heidaran, M.A. Requirement of phosphatidylinositol-3 kinase for activation of JNK/SAPKs by PDGF. Biochem. Biophys. Res. Commun. 1997, 232, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.T.; Eguinoa, A.; Qiu, R.G.; Stokoe, D.; Cooke, F.T.; Walters, R.; Wennstrom, S.; Claesson-Welsh, L.; Evans, T.; Symons, M.; et al. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr. Biol. 1995, 5, 393–403. [Google Scholar] [CrossRef]
- Li, W.L.; Yamada, Y.; Ueno, M.; Nishikawa, S.; Takakura, N. Platelet derived growth factor receptor α is essential for establishing a microenvironment that supports definitive erythropoiesis. J. Biochem. 2006, 140, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.F.; Mustoe, T.A.; Altrock, B.W.; Deuel, T.F.; Thomason, A. Role of platelet-derived growth factor in wound healing. J. Cell Biochem. 1991, 45, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Ataliotis, P.; Mercola, M. Distribution and functions of platelet-derived growth factors and their receptors during embryogenesis. Int. Rev. Cytol. 1997, 172, 95–127. [Google Scholar] [PubMed]
- Caplan, A.I.; Correa, D. PDGF in bone formation and regeneration: New insights into a novel mechanism involving MSCS. J. Orthop. Res. 2011, 29, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Board, R.; Jayson, G.C. Platelet-derived growth factor receptor (PDGFR): A target for anticancer therapeutics. Drug Resist. Updates 2005, 8, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Brakenhielm, E.; Li, X.; Pietras, K.; Widenfalk, J.; Ostman, A.; Eriksson, U.; Cao, Y. Angiogenesis stimulated by PDGF-CC, a novel member in the PDGF family, involves activation of PDGFR-αα and -αβ receptors. FASEB J. 2002, 16, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ponten, A.; Aase, K.; Karlsson, L.; Abramsson, A.; Uutela, M.; Backstrom, G.; Hellstrom, M.; Bostrom, H.; Li, H.; et al. PDGF-c is a new protease-activated ligand for the PDGF α-receptor. Nat. Cell Biol. 2000, 2, 302–309. [Google Scholar] [PubMed]
- Li, X.; Kumar, A.; Zhang, F.; Lee, C.; Li, Y.; Tang, Z.; Arjuna, P. VEGF-independent angiogenic pathways induced by PDGF-c. Oncotarget 2010, 1, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Uutela, M.; Wirzenius, M.; Paavonen, K.; Rajantie, I.; He, Y.; Karpanen, T.; Lohela, M.; Wiig, H.; Salven, P.; Pajusola, K.; et al. PDGF-d induces macrophage recruitment, increased interstitial pressure, and blood vessel maturation during angiogenesis. Blood 2004, 104, 3198–3204. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hou, X.; Lee, C.; Li, Y.; Maminishkis, A.; Tang, Z.; Zhang, F.; Langer, H.F.; Arjunan, P.; Dong, L.; et al. Platelet-derived growth factor-DD targeting arrests pathological angiogenesis by modulating glycogen synthase kinase-3β phosphorylation. J. Biol. Chem. 2010, 285, 15500–15510. [Google Scholar] [CrossRef] [PubMed]
- Manzat Saplacan, R.M.; Balacescu, L.; Gherman, C.; Chira, R.I.; Craiu, A.; Mircea, P.A.; Lisencu, C.; Balacescu, O. The role of PDGFs and PDGFRs in colorectal cancer. Mediat. Inflamm. 2017, 2017, 4708076. [Google Scholar] [CrossRef] [PubMed]
- Di Tomaso, E.; London, N.; Fuja, D.; Logie, J.; Tyrrell, J.A.; Kamoun, W.; Munn, L.L.; Jain, R.K. PDGF-c induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PLoS ONE 2009, 4, e5123. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Seo, S.Y.; Ha, M.; Ku, J.H.; Kim, H.H.; Kwak, C. Inhibition of prostate cancer using RNA interference-directed knockdown of platelet-derived growth factor receptor. Urology 2011, 77, e1509–e1515. [Google Scholar] [CrossRef] [PubMed]
- Gotzmann, J.; Fischer, A.N.; Zojer, M.; Mikula, M.; Proell, V.; Huber, H.; Jechlinger, M.; Waerner, T.; Weith, A.; Beug, H.; et al. A crucial function of PDGF in TGF-β-mediated cancer progression of hepatocytes. Oncogene 2006, 25, 3170–3185. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, E.; Song, Y.H.; Krishnappa, S.; Alt, E. Epithelial-mesenchymal transition in breast cancer lines is mediated through PDGF-d released by tissue-resident stem cells. Int. J. Cancer 2012, 131, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- De Falco, S. Antiangiogenesis therapy: An update after the first decade. Korean J. Intern. Med. 2014, 29, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Loizos, N.; Xu, Y.; Huber, J.; Liu, M.; Lu, D.; Finnerty, B.; Rolser, R.; Malikzay, A.; Persaud, A.; Corcoran, E.; et al. Targeting the platelet-derived growth factor receptor alpha with a neutralizing human monoclonal antibody inhibits the growth of tumor xenografts: Implications as a potential therapeutic target. Mol. Cancer Ther. 2005, 4, 369–379. [Google Scholar] [PubMed]
- Wagner, A.J.; Kindler, H.; Gelderblom, H.; Schoffski, P.; Bauer, S.; Hohenberger, P.; Kopp, H.G.; Lopez-Martin, J.A.; Peeters, M.; Reichardt, P.; et al. A phase II study of a human anti-PDGFRα monoclonal antibody (olaratumab, IMC-3G3) in previously treated patients with metastatic gastrointestinal stromal tumors. Ann. Oncol. 2017, 28, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Liu, Q.; Fatatis, A. Targeting the α receptor for platelet-derived growth factor as a primary or combination therapy in a preclinical model of prostate cancer skeletal metastasis. Clin. Cancer Res. 2010, 16, 5002–5010. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; Sweeney, C.; Youssoufian, H.; Qin, A.; Dontabhaktuni, A.; Loizos, N.; Nippgen, J.; Amato, R. A phase I study of olaratumab, an anti-platelet-derived growth factor receptor α (PDGFRα) monoclonal antibody, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Jones, R.L.; Van Tine, B.A.; Chmielowski, B.; Elias, A.D.; Adkins, D.; Agulnik, M.; Cooney, M.M.; Livingston, M.B.; Pennock, G.; et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: An open-label phase 1b and randomised phase 2 trial. Lancet 2016, 388, 488–497. [Google Scholar] [CrossRef]
- Brindle, N.P.; Saharinen, P.; Alitalo, K. Signaling and functions of angiopoietin-1 in vascular protection. Circ. Res. 2006, 98, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Fagiani, E.; Christofori, G. Angiopoietins in angiogenesis. Cancer Lett. 2013, 328, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Aldrich, T.H.; Jones, P.F.; Acheson, A.; Compton, D.L.; Jain, V.; Ryan, T.E.; Bruno, J.; Radziejewski, C.; Maisonpierre, P.C.; et al. Isolation of angiopoietin-1, a ligand for the Tie2 receptor, by secretion-trap expression cloning. Cell 1996, 87, 1161–1169. [Google Scholar] [CrossRef]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, D.M.; Griffiths, J.A.; Rojas, J.; Aldrich, T.H.; Jones, P.F.; Zhou, H.; McClain, J.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; et al. Angiopoietins 3 and 4: Diverging gene counterparts in mice and humans. Proc. Natl. Acad. Sci. USA 1999, 96, 1904–1909. [Google Scholar] [CrossRef] [PubMed]
- Saharinen, P.; Kerkela, K.; Ekman, N.; Marron, M.; Brindle, N.; Lee, G.M.; Augustin, H.; Koh, G.Y.; Alitalo, K. Multiple angiopoietin recombinant proteins activate the Tie1 receptor tyrosine kinase and promote its interaction with Tie2. J. Cell Biol. 2005, 169, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Marron, M.B.; Hughes, D.P.; Edge, M.D.; Forder, C.L.; Brindle, N.P. Evidence for heterotypic interaction between the receptor tyrosine kinases Tie-1 and Tie-2. J. Biol. Chem. 2000, 275, 39741–39746. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Augustin, H.G. The role of the angiopoietins in vascular morphogenesis. Angiogenesis 2009, 12, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G. Role of angiopoietins and tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis. Cell Tissue Res. 2003, 314, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, U.; Krissl, T.; Koidl, S.; Weiss, C.; Koblizek, T.; Deutsch, U.; Martiny-Baron, G.; Marme, D.; Augustin, H.G. Angiopoietin-1 and angiopoietin-2 share the same binding domains in the Tie-2 receptor involving the first Ig-like loop and the epidermal growth factor-like repeats. J. Biol. Chem. 2003, 278, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Sandhya, V.K.; Singh, P.; Parthasarathy, D.; Kumar, A.; Advani, J.; Gattu, R.; Ranjit, D.V.; Vaidyanathan, R.; Mathur, P.P.; et al. Signaling network map of endothelial Tek tyrosine kinase. J. Signal Transduct. 2014, 2014, 173026. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.D.; Stauffer, T.P.; Yang, W.P.; York, J.D.; Huang, L.; Blanar, M.A.; Meyer, T.; Peters, K.G. Tyrosine 1101 of Tie2 is the major site of association of p85 and is required for activation of phosphatidylinositol 3-kinase and Akt. Mol. Cell Biol. 1998, 18, 4131–4140. [Google Scholar] [CrossRef] [PubMed]
- Makinde, T.; Agrawal, D.K. Intra and extravascular transmembrane signalling of angiopoietin-1-Tie2 receptor in health and disease. J. Cell Mol. Med. 2008, 12, 810–828. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.; Wong, V.; Burova, E.; Wei, Y.; Zabski, S.; Griffiths, J.; Lai, K.M.; Lin, H.C.; Ioffe, E.; Yancopoulos, G.D.; et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev. 2004, 18, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Chen, S.H.; Sturk, C.; Master, Z.; Tran, J.; Kerbel, R.S.; Dumont, D.J. A unique autophosphorylation site on Tie2/Tek mediates Dok-R phosphotyrosine binding domain binding and function. Mol. Cell Biol. 2003, 23, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.P.; Marron, M.B.; Brindle, N.P. The antiinflammatory endothelial tyrosine kinase Tie2 interacts with a novel nuclear factor-κb inhibitor ABIN-2. Circ. Res. 2003, 92, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Tadros, A.; Hughes, D.P.; Dunmore, B.J.; Brindle, N.P. Abin-2 protects endothelial cells from death and has a role in the antiapoptotic effect of angiopoietin-1. Blood 2003, 102, 4407–4409. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Kim, W.J.; Koh, Y.J.; Piao, S.; Jin, H.R.; Lee, S.W.; Choi, M.J.; Shin, H.Y.; Kwon, M.H.; Jung, K.; et al. Designed angiopoietin-1 variant, comp-angiopoietin-1, rescues erectile function through healthy cavernous angiogenesis in a hypercholesterolemic mouse. Sci. Rep. 2015, 5, 9222. [Google Scholar] [CrossRef] [PubMed]
- Lekas, M.; Lekas, P.; Mei, S.H.; Deng, Y.; Dumont, D.J.; Stewart, D.J. Tie2-dependent neovascularization of the ischemic hindlimb is mediated by angiopoietin-2. PLoS ONE 2012, 7, e43568. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.D.; Liu, X.E.; Wu, J.M.; Cai, X.J.; Mou, Y.P.; Li, J.D. Expression and significance of angiopoietin-2 in gastric cancer. World J. Gastroenterol. 2004, 10, 1382–1385. [Google Scholar] [PubMed]
- Mitsuhashi, N.; Shimizu, H.; Ohtsuka, M.; Wakabayashi, Y.; Ito, H.; Kimura, F.; Yoshidome, H.; Kato, A.; Nukui, Y.; Miyazaki, M. Angiopoietins and Tie-2 expression in angiogenesis and proliferation of human hepatocellular carcinoma. Hepatology 2003, 37, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.A.; Liu, W.; Jung, Y.D.; Fan, F.; Wilson, M.; Reinmuth, N.; Shaheen, R.M.; Bucana, C.D.; Ellis, L.M. The effects of angiopoietin-1 and -2 on tumor growth and angiogenesis in human colon cancer. Cancer Res. 2001, 61, 1255–1259. [Google Scholar] [PubMed]
- Helfrich, I.; Edler, L.; Sucker, A.; Thomas, M.; Christian, S.; Schadendorf, D.; Augustin, H.G. Angiopoietin-2 levels are associated with disease progression in metastatic malignant melanoma. Clin. Cancer Res. 2009, 15, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Takanami, I. Overexpression of ang-2 mrna in non-small cell lung cancer: Association with angiogenesis and poor prognosis. Oncol. Rep. 2004, 12, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Mazzieri, R.; Pucci, F.; Moi, D.; Zonari, E.; Ranghetti, A.; Berti, A.; Politi, L.S.; Gentner, B.; Brown, J.L.; Naldini, L.; et al. Targeting the Ang2/Tie2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 2011, 19, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Holopainen, T.; Saharinen, P.; D’Amico, G.; Lampinen, A.; Eklund, L.; Sormunen, R.; Anisimov, A.; Zarkada, G.; Lohela, M.; Helotera, H.; et al. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J. Natl. Cancer Inst. 2012, 104, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Nasarre, P.; Thomas, M.; Kruse, K.; Helfrich, I.; Wolter, V.; Deppermann, C.; Schadendorf, D.; Thurston, G.; Fiedler, U.; Augustin, H.G. Host-derived angiopoietin-2 affects early stages of tumor development and vessel maturation but is dispensable for later stages of tumor growth. Cancer Res. 2009, 69, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Machein, M.R.; Knedla, A.; Knoth, R.; Wagner, S.; Neuschl, E.; Plate, K.H. Angiopoietin-1 promotes tumor angiogenesis in a rat glioma model. Am. J. Pathol. 2004, 165, 1557–1570. [Google Scholar] [CrossRef]
- Nakayama, T.; Yao, L.; Tosato, G. Mast cell-derived angiopoietin-1 plays a critical role in the growth of plasma cell tumors. J. Clin. Investig. 2004, 114, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Hawighorst, T.; Skobe, M.; Streit, M.; Hong, Y.K.; Velasco, P.; Brown, L.F.; Riccardi, L.; Lange-Asschenfeldt, B.; Detmar, M. Activation of the Tie2 receptor by angiopoietin-1 enhances tumor vessel maturation and impairs squamous cell carcinoma growth. Am. J. Pathol. 2002, 160, 1381–1392. [Google Scholar] [CrossRef]
- Hayes, A.J.; Huang, W.Q.; Yu, J.; Maisonpierre, P.C.; Liu, A.; Kern, F.G.; Lippman, M.E.; McLeskey, S.W.; Li, L.Y. Expression and function of angiopoietin-1 in breast cancer. Br. J. Cancer 2000, 83, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Stoeltzing, O.; Ahmad, S.A.; Liu, W.; McCarty, M.F.; Wey, J.S.; Parikh, A.A.; Fan, F.; Reinmuth, N.; Kawaguchi, M.; Bucana, C.D.; et al. Angiopoietin-1 inhibits vascular permeability, angiogenesis, and growth of hepatic colon cancer tumors. Cancer Res. 2003, 63, 3370–3377. [Google Scholar] [PubMed]
- Tait, C.R.; Jones, P.F. Angiopoietins in tumours: The angiogenic switch. J. Pathol. 2004, 204, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Kelley, R.K.; Tolcher, A.W.; Razak, A.R.; Van Loon, K.; Patnaik, A.; Bedard, P.L.; Alfaro, A.A.; Beeram, M.; Adriaens, L.; et al. A phase I first-in-human study of nesvacumab (REGN910), a fully human Anti-Angiopoietin-2 (Ang2) monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2016, 22, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.; Eichten, A.; Castanaro, C.; Pasnikowski, E.; Adler, A.; Lalani, A.S.; Papadopoulos, N.; Kyle, A.H.; Minchinton, A.I.; Yancopoulos, G.D.; et al. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 2013, 73, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Bupathi, M.; Kaseb, A.; Janku, F. Angiopoietin 2 as a therapeutic target in hepatocellular carcinoma treatment: Current perspectives. Onco Targets Ther. 2014, 7, 1927–1932. [Google Scholar] [PubMed]
- Dowlati, A.; Vlahovic, G.; Natale, R.B.; Rasmussen, E.; Singh, I.; Hwang, Y.C.; Rossi, J.; Bass, M.B.; Friberg, G.; Pickett, C.A. A phase I, first-in-human study of amg 780, an angiopoietin-1 and -2 inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2016, 22, 4574–4584. [Google Scholar] [CrossRef] [PubMed]
- Kienast, Y.; Klein, C.; Scheuer, W.; Raemsch, R.; Lorenzon, E.; Bernicke, D.; Herting, F.; Yu, S.; The, H.H.; Martarello, L.; et al. Ang-2-VEGF-A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF-A and Ang-2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin. Cancer Res. 2013, 19, 6730–6740. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.C.; Boult, J.K.; Thomas, M.; Koehler, A.; Nayak, T.; Tessier, J.; Ooi, C.H.; Birzele, F.; Belousov, A.; Zajac, M.; et al. Acute tumour response to a bispecific ang-2-VEGF-a antibody: Insights from multiparametric MRI and gene expression profiling. Br. J. Cancer 2016, 115, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Leow, C.C.; Coffman, K.; Inigo, I.; Breen, S.; Czapiga, M.; Soukharev, S.; Gingles, N.; Peterson, N.; Fazenbaker, C.; Woods, R.; et al. Medi3617, a human anti-angiopoietin 2 monoclonal antibody, inhibits angiogenesis and tumor growth in human tumor xenograft models. Int. J. Oncol. 2012, 40, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nawa, K.; Ichihara, A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun. 1984, 122, 1450–1459. [Google Scholar] [CrossRef]
- Venepalli, N.K.; Goff, L. Targeting the HGF-cMET axis in hepatocellular carcinoma. Int. J. Hepatol. 2013, 2013, 341636. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tsao, M.S. An overview of the c-Met signaling pathway. Ther. Adv. Med. Oncol. 2011, 3, S7–S19. [Google Scholar] [CrossRef] [PubMed]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-Met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of Met inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Ponzetto, C.; Comoglio, P.M. Identification of functional domains in the hepatocyte growth factor and its receptor by molecular engineering. J. Biotechnol. 1994, 37, 109–122. [Google Scholar] [CrossRef]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. The hepatocyte growth factor receptor: Structure, function and pharmacological targeting in cancer. Curr. Signal Transduct. Ther. 2011, 6, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Fixman, E.D.; Fournier, T.M.; Kamikura, D.M.; Naujokas, M.A.; Park, M. Pathways downstream of Shc and Grb2 are required for cell transformation by the tpr-Met oncoprotein. J. Biol. Chem. 1996, 271, 13116–13122. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, G.; Giordano, S.; Zhen, Z.; Salcini, A.E.; Lanfrancone, L.; Bardelli, A.; Panayotou, G.; Waterfield, M.D.; Ponzetto, C.; Pelicci, P.G.; et al. The motogenic and mitogenic responses to hgf are amplified by the shc adaptor protein. Oncogene 1995, 10, 1631–1638. [Google Scholar] [PubMed]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; Dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef]
- Paumelle, R.; Tulasne, D.; Kherrouche, Z.; Plaza, S.; Leroy, C.; Reveneau, S.; Vandenbunder, B.; Fafeur, V. Hepatocyte growth factor/scatter factor activates the ETS1 transcription factor by a RAS-RAF-MEK-ERK signaling pathway. Oncogene 2002, 21, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.H.; Jeffers, M.; Bellacosa, A.; Mitsuuchi, Y.; Vande Woude, G.F.; Testa, J.R. Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.Y.; Meens, J.A.; Schick, C.; Organ, S.L.; Qiao, H.; Tremblay, E.A.; Schaeffer, E.; Uniyal, S.; Chan, B.M.; Elliott, B.E. Src and fak mediate cell-matrix adhesion-dependent activation of met during transformation of breast epithelial cells. J. Cell Biochem. 2009, 107, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, N.; Hung, W.; Tremblay, E.; Saulnier, R.; Elliott, B. C-src kinase activity is required for hepatocyte growth factor-induced motility and anchorage-independent growth of mammary carcinoma cells. J. Biol. Chem. 1998, 273, 33714–33721. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V.; Chen, L.; Sleeman, J.P.; Herrlich, P.; Ponta, H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 2002, 16, 3074–3086. [Google Scholar] [CrossRef] [PubMed]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. A signaling adapter function for α6β4 integrin in the control of HGF-dependent invasive growth. Cell 2001, 107, 643–654. [Google Scholar] [CrossRef]
- Fischer, O.M.; Giordano, S.; Comoglio, P.M.; Ullrich, A. Reactive oxygen species mediate met receptor transactivation by G protein-coupled receptors and the epidermal growth factor receptor in human carcinoma cells. J. Biol. Chem. 2004, 279, 28970–28978. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Sung, E.S.; Ahn, J.H.; An, S.; Huh, J.; You, W.K. Development of antibody-based c-Met inhibitors for targeted cancer therapy. ImmunoTargets Ther. 2015, 4, 35–44. [Google Scholar] [PubMed]
- Zhu, H.; Naujokas, M.A.; Park, M. Receptor chimeras indicate that the met tyrosine kinase mediates the motility and morphogenic responses of hepatocyte growth/scatter factor. Cell Growth Differ. 1994, 5, 359–366. [Google Scholar] [PubMed]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Bladt, F.; Goedecke, S.; Brinkmann, V.; Zschiesche, W.; Sharpe, M.; Gherardi, E.; Birchmeier, C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995, 373, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.S.; Park, M.; Blair, D.G.; Tainsky, M.A.; Huebner, K.; Croce, C.M.; Vande Woude, G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984, 311, 29–33. [Google Scholar] [CrossRef]
- Liang, T.J.; Reid, A.E.; Xavier, R.; Cardiff, R.D.; Wang, T.C. Transgenic expression of TPR-MET oncogene leads to development of mammary hyperplasia and tumors. J. Clin. Investig. 1996, 97, 2872–2877. [Google Scholar] [CrossRef] [PubMed]
- Soman, N.R.; Correa, P.; Ruiz, B.A.; Wogan, G.N. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc. Natl. Acad. Sci. USA 1991, 88, 4892–4896. [Google Scholar] [CrossRef] [PubMed]
- Corso, S.; Comoglio, P.M.; Giordano, S. Cancer therapy: Can the challenge be met? Trends Mol. Med. 2005, 11, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ferrell, L.D.; Faouzi, S.; Maher, J.J.; Bishop, J.M. Activation of the met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Biol. 2001, 153, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; LaRochelle, W.J.; Sharp, R.; Otsuka, T.; Kriebel, P.; Anver, M.; Aaronson, S.A.; Merlino, G. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc. Natl. Acad. Sci. USA 1997, 94, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.S.; Sweeney, C.S.; Mendelson, D.S.; Eckhardt, S.G.; Anderson, A.; Beaupre, D.M.; Branstetter, D.; Burgess, T.L.; Coxon, A.; Deng, H.; et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin. Cancer Res. 2010, 16, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, T.; Ma, M.; Zhuang, Y.; Patton, A.; Hu, Z.; Mounho, B. Pharmacokinetics and safety of a fully human hepatocyte growth factor antibody, AMG 102, in cynomolgus monkeys. Pharm. Res. 2007, 24, 1910–1918. [Google Scholar] [CrossRef] [PubMed]
- Burgess, T.; Coxon, A.; Meyer, S.; Sun, J.; Rex, K.; Tsuruda, T.; Chen, Q.; Ho, S.Y.; Li, L.; Kaufman, S.; et al. Fully human monoclonal antibodies to hepatocyte growth factor with therapeutic potential against hepatocyte growth factor/c-Met-dependent human tumors. Cancer Res. 2006, 66, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Iveson, T.; Donehower, R.C.; Davidenko, I.; Tjulandin, S.; Deptala, A.; Harrison, M.; Nirni, S.; Lakshmaiah, K.; Thomas, A.; Jiang, Y.; et al. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: An open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 2014, 15, 1007–1018. [Google Scholar] [CrossRef]
- D’Arcangelo, M.; Cappuzzo, F. Focus on the potential role of ficlatuzumab in the treatment of non-small cell lung cancer. Biologics 2013, 7, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Weiss, G.J.; Papadopoulos, K.P.; Hofmeister, C.C.; Tibes, R.; Tolcher, A.; Isaacs, R.; Jac, J.; Han, M.; Payumo, F.C.; et al. Phase I ficlatuzumab monotherapy or with erlotinib for refractory advanced solid tumours and multiple myeloma. Br. J. Cancer 2014, 111, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Bann, D.V.; Deschler, D.G.; Goyal, N. Novel immunotherapeutic approaches for head and neck squamous cell carcinoma. Cancers 2016, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, W.; Okamoto, I.; Tanaka, K.; Hatashita, E.; Yamada, Y.; Kuwata, K.; Yamaguchi, H.; Arao, T.; Nishio, K.; Fukuoka, M.; et al. Tak-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation. Mol. Cancer Ther. 2010, 9, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Parikh, R.A.; Wang, P.; Beumer, J.H.; Chu, E.; Appleman, L.J. The potential roles of hepatocyte growth factor (HGF)-met pathway inhibitors in cancer treatment. OncoTargets Ther. 2014, 7, 969–983. [Google Scholar]
- Merchant, M.; Ma, X.; Maun, H.R.; Zheng, Z.; Peng, J.; Romero, M.; Huang, A.; Yang, N.Y.; Nishimura, M.; Greve, J.; et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc. Natl. Acad. Sci. USA 2013, 110, E2987–E2996. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Mocci, S.; Phan, S.; Shames, D.S.; Smith, D.; Yu, W.; Paton, V.E.; et al. Results from the phase III randomized trial of onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV non-small-cell lung cancer: Metlung. J. Clin. Oncol. 2017, 35, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Ervin, T.J.; Ramlau, R.A.; Daniel, D.B.; Goldschmidt, J.H., Jr.; Blumenschein, G.R., Jr.; Krzakowski, M.J.; Robinet, G.; Godbert, B.; Barlesi, F.; et al. Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 4105–4114. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Goldman, J.W.; Algazi, A.P.; Turner, P.K.; Moser, B.; Hu, T.; Wang, X.A.; Tuttle, J.; Wacheck, V.; Wooldridge, J.E.; et al. A first-in-human phase I study of a bivalent MET antibody, emibetuzumab (ly2875358), as monotherapy and in combination with erlotinib in advanced cancer. Clin. Cancer Res. 2017, 23, 1910–1919. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, W.; Wortinger, M.A.; Yan, S.B.; Cornwell, P.; Peek, V.L.; Stephens, J.R.; Tetreault, J.W.; Xia, J.; Manro, J.R.; et al. Ly2875358, a neutralizing and internalizing anti-met bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth. Clin. Cancer Res. 2014, 20, 6059–6070. [Google Scholar] [CrossRef] [PubMed]
- Zelensky, A.N.; Gready, J.E. The C-type lectin-like domain superfamily. FEBS J. 2005, 272, 6179–6217. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.; Swain, R.K.; Zhuang, X.; Vorschmitt, H.; Reynolds, G.; Durant, S.; Beesley, J.F.; Herbert, J.M.; Sheldon, H.; Andre, M.; et al. Identification and angiogenic role of the novel tumor endothelial marker CLEC14A. Oncogene 2012, 31, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Rho, S.S.; Choi, H.J.; Min, J.K.; Lee, H.W.; Park, H.; Kim, Y.M.; Kwon, Y.G. Clec14a is specifically expressed in endothelial cells and mediates cell to cell adhesion. Biochem. Biophys. Res. Commun. 2011, 404, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Ki, M.K.; Jeoung, M.H.; Choi, J.R.; Rho, S.S.; Kwon, Y.G.; Shim, H.; Chung, J.; Hong, H.J.; Song, B.D.; Lee, S. Human antibodies targeting the C-type lectin-like domain of the tumor endothelial cell marker CLEC14A regulate angiogenic properties in vitro. Oncogene 2013, 32, 5449–5457. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Naylor, A.J.; Khan, A.; Noy, P.J.; Mambretti, M.; Lodhia, P.; Athwal, J.; Korzystka, A.; Buckley, C.D.; Willcox, B.E.; et al. Multimerin-2 is a ligand for group 14 family C-type lectins CLEC14A, CD93 and CD248 spanning the endothelial pericyte interface. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Noy, P.J.; Lodhia, P.; Khan, K.; Zhuang, X.; Ward, D.G.; Verissimo, A.R.; Bacon, A.; Bicknell, R. Blocking CLEC14A-MMRN2 binding inhibits sprouting angiogenesis and tumour growth. Oncogene 2015, 34, 5821–5831. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Kim, M.R.; Lee, W.R.; Kim, J.H.; Heo, K.; Lee, S. CLEC14A-hsp70-1a interaction regulates hsp70-1a-induced angiogenesis. Sci. Rep. 2017. under revision. [Google Scholar]
- Zanivan, S.; Maione, F.; Hein, M.Y.; Hernandez-Fernaud, J.R.; Ostasiewicz, P.; Giraudo, E.; Mann, M. Silac-based proteomics of human primary endothelial cell morphogenesis unveils tumor angiogenic markers. Mol. Cell Proteom. 2013, 12, 3599–3611. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Quarna, J.; Antoniotti, P.; Cuppini, L.; Finocchiaro, G.; Eoli, M.; Rosti, V.; et al. A subpopulation of circulating endothelial cells express CD109 and is enriched in the blood of cancer patients. PLoS ONE 2014, 9, e114713. [Google Scholar] [CrossRef] [PubMed]
- Krishna Priya, S.; Kumar, K.; Hiran, K.R.; Bindhu, M.R.; Nagare, R.P.; Vijaykumar, D.K.; Ganesan, T.S. Expression of a novel endothelial marker, C-type lectin 14a, in epithelial ovarian cancer and its prognostic significance. Int. J. Clin. Oncol. 2017, 22, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Park, C.S.; Jang, J.H.; Kim, M.R.; Na, H.; Lee, K.; Yoo, B.C.; Kim, Y.; Lee, J.; Kim, S.J.; et al. Inhibition of VEGF-dependent angiogenesis and tumor angiogenesis by an optimized antibody targeting CLEC14A. Mol. Oncol. 2017. under revision. [Google Scholar]

{kind=link}
{kind=link}
| Target | Antibody | Company/Institute | Antibody Type | Status | Indication |
|---|---|---|---|---|---|
| VEGF-A | Bevacizumab (Avastin) | Genentech | Humanized, IgG1 | FDA approval | Colorectal cancer, NSCLC, glioblastoma, breast cancer, renal carcinoma, epithelial ovarian cancer |
| VEGF-A, -B, PGF | Aflibercept (Zaltrap) | Regeneron | Fully human, IgG1 | FDA approval | Colorectal cancer |
| VEGFR2 | Ramucirumab (Cyramza) | Imclone | Fully human, IgG1 | FDA approval, Phase II | Advanced gastric or gastro-esophageal junction adenocarcinoma, NSCLC, advanced or metastatic urothelial carcinoma |
| VEGFR2 | Tanibirumab | Pharmabcine | Fully human, IgG1 | Phase II | Recurrent glioblastoma |
| PDGFRα | Olaratumab (Lartruvo) | Eli Lilly | Fully human, IgG1 | FDA approval | Soft tissue sarcoma |
| Ang-2 | Nesvacumab (REGN910) | Regeneron | Fully human, IgG1 | Phase I | Advanced solid tumors |
| Ang-1, -2 | AMG780 | Amgen | Fully human, IgG2 | Phase I | Advanced solid tumors |
| Ang-2 | MEDI3617 | MedImmune LLC | Fully human, IgG1 | Phase I | Advanced solid tumors |
| Ang-2 and VEGF | Vanucizumab | Genentech | Bispecific, IgG1 | Phase II | Colorectal cancer |
| HGF | Rilotumumab (AMG102) | Amgen | Fully human, IgG2 | Phase I, II, III | Gastric and esophagogastric junction cancer, NSCLC, metastatic colorectal cancer, prostate cancer, renal cell carcinoma |
| HGF | Ficlatuzumab (SCH900105) | AVEO Pharmaceuticals | Humanized, IgG1 | Phase I, II | Head and neck squamous carcinoma, NSCLC |
| HGF | TAK-701 | Galaxy Biotech | Humanized, IgG1 | Phase I | Advanced solid tumors |
| Tyrosine-protein kinase Met (c-MET) | Onartuzumab (MetMab) | Genentech | Humanized, IgG1 | Phase I, II, III | NSCLC, glioblastoma, gastric cancer, hepatocellular carcinoma, breast cancer |
| c-MET | Emibetuzumab (LY-2875358) | Eli Lilly | Humanized, IgG4 | Phase II | NSCLC, gastric cancer |
| CLEC14a | - | Univ. of Birmingham | Mouse | Preclinical | Undetermined as yet |
| CLEC14a | - | Scripps Korea Antibody Institute | Fully human, IgG1 | Preclinical | Undetermined as yet |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, D.-H.; Kim, M.R.; Jang, J.H.; Na, H.-J.; Lee, S. A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1786. https://doi.org/10.3390/ijms18081786
Kong D-H, Kim MR, Jang JH, Na H-J, Lee S. A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy. International Journal of Molecular Sciences. 2017; 18(8):1786. https://doi.org/10.3390/ijms18081786
Chicago/Turabian StyleKong, Deok-Hoon, Mi Ra Kim, Ji Hye Jang, Hee-Jun Na, and Sukmook Lee. 2017. "A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy" International Journal of Molecular Sciences 18, no. 8: 1786. https://doi.org/10.3390/ijms18081786
APA StyleKong, D.-H., Kim, M. R., Jang, J. H., Na, H.-J., & Lee, S. (2017). A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy. International Journal of Molecular Sciences, 18(8), 1786. https://doi.org/10.3390/ijms18081786