Identification and Validation of New Alleles of FALSIFLORA and COMPOUND INFLORESCENCE Genes Controlling the Number of Branches in Tomato Inflorescence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
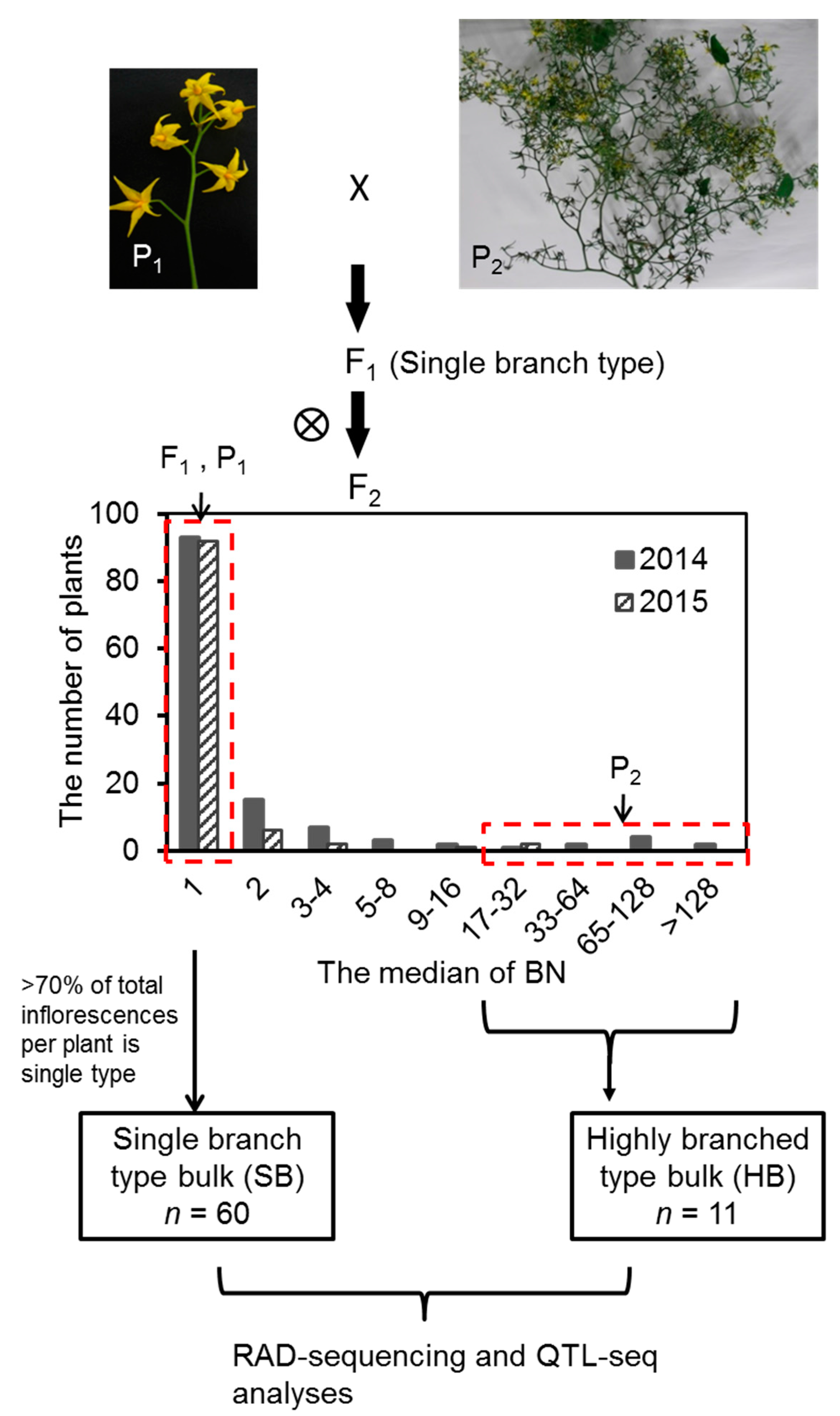
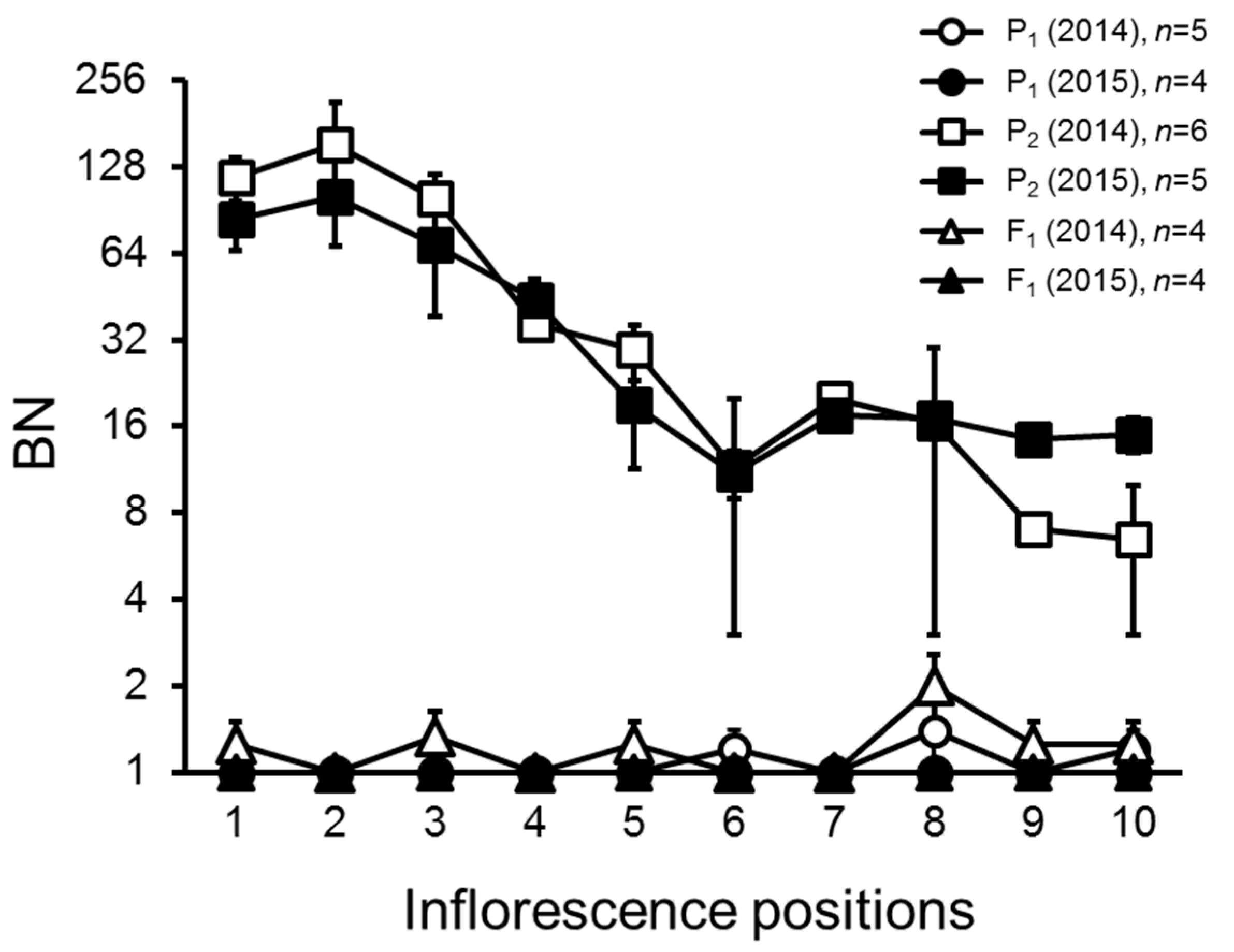
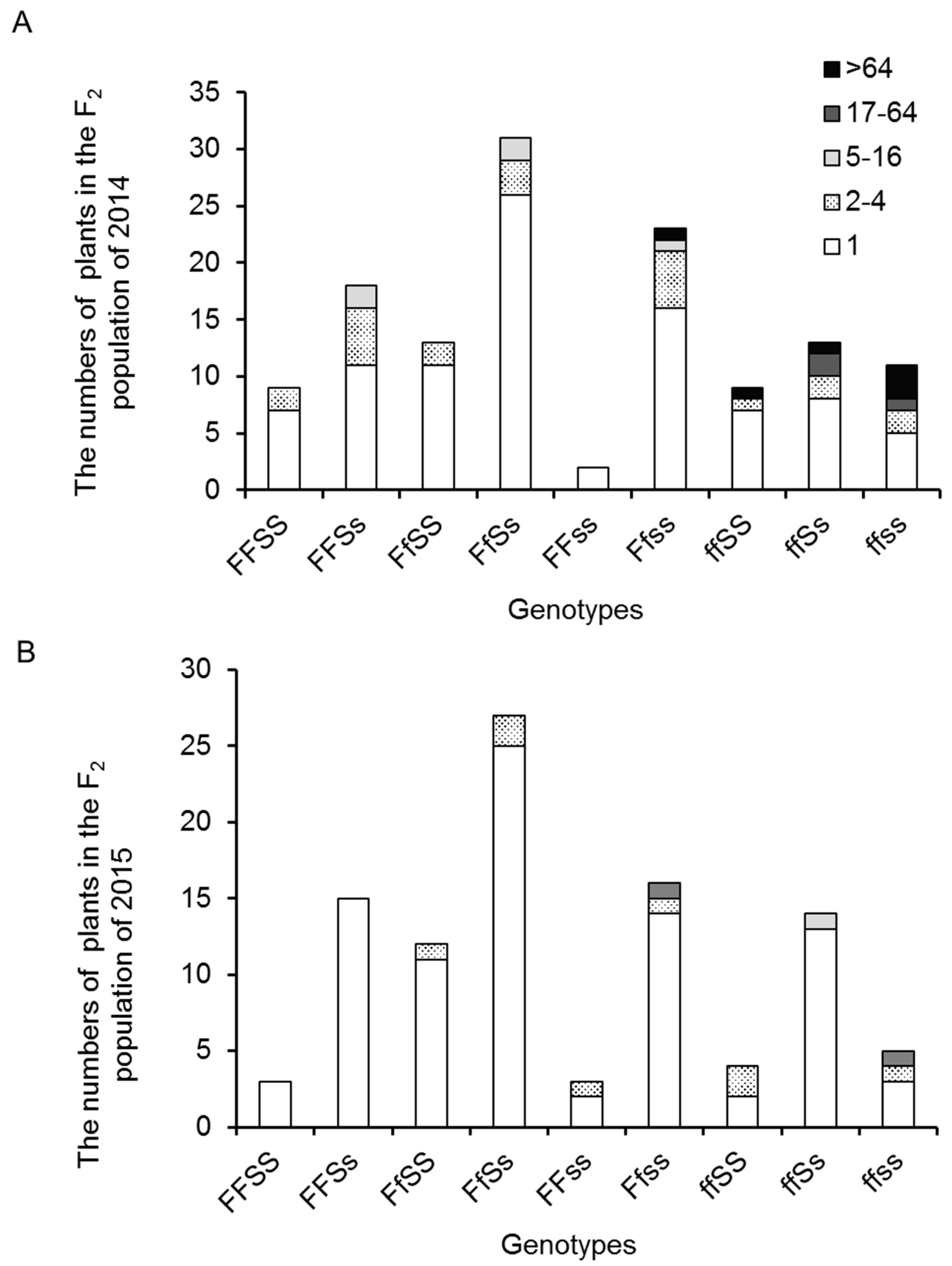
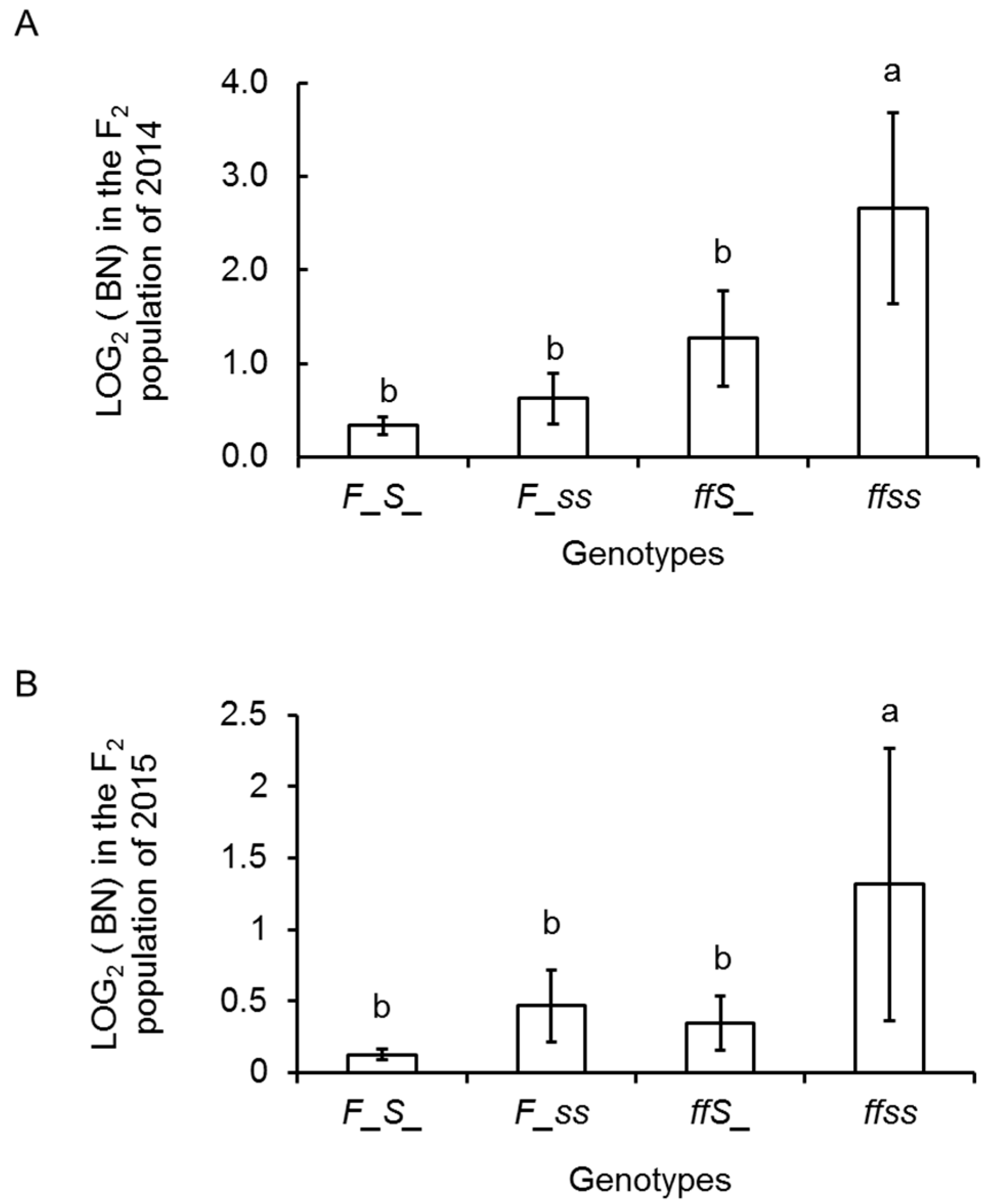
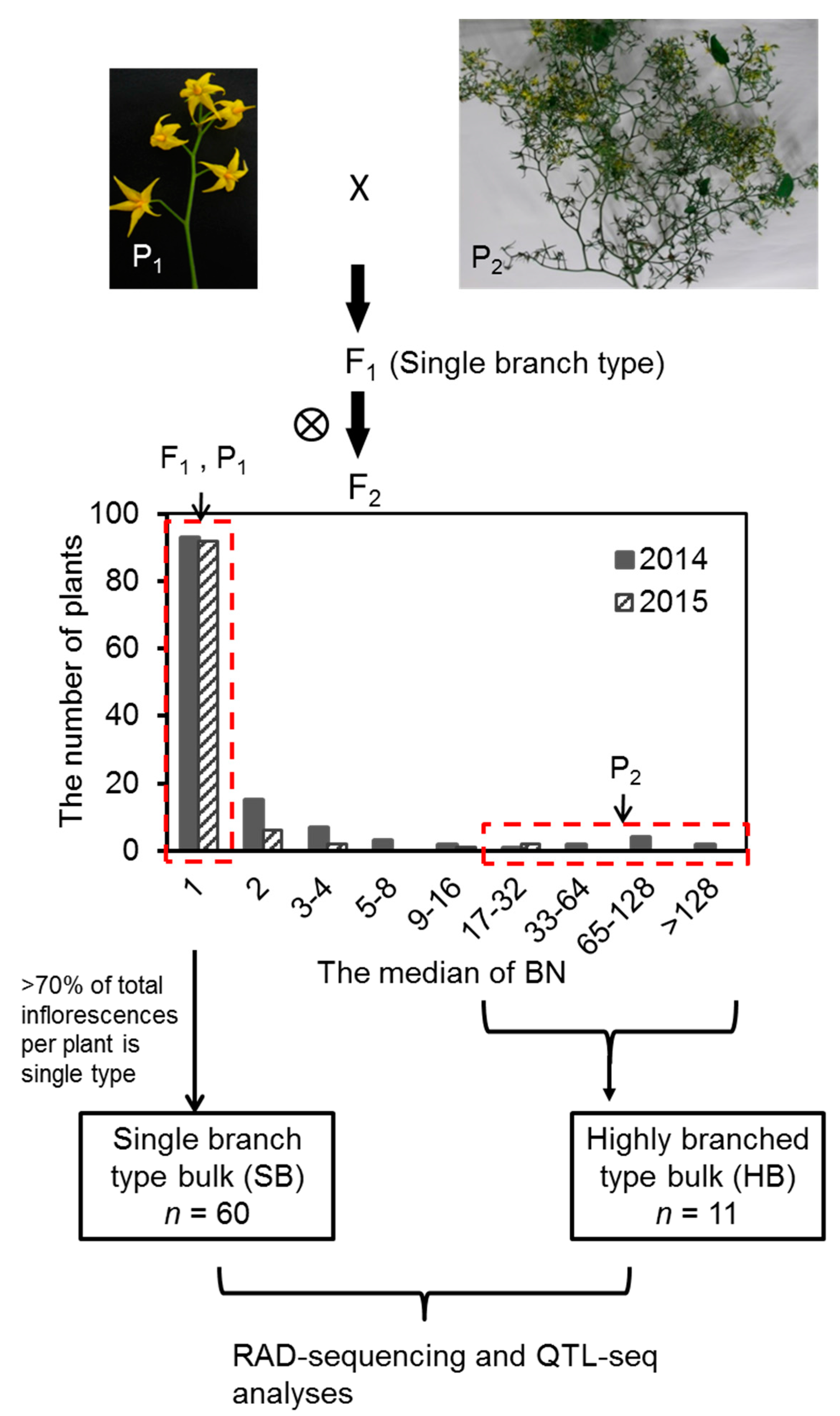
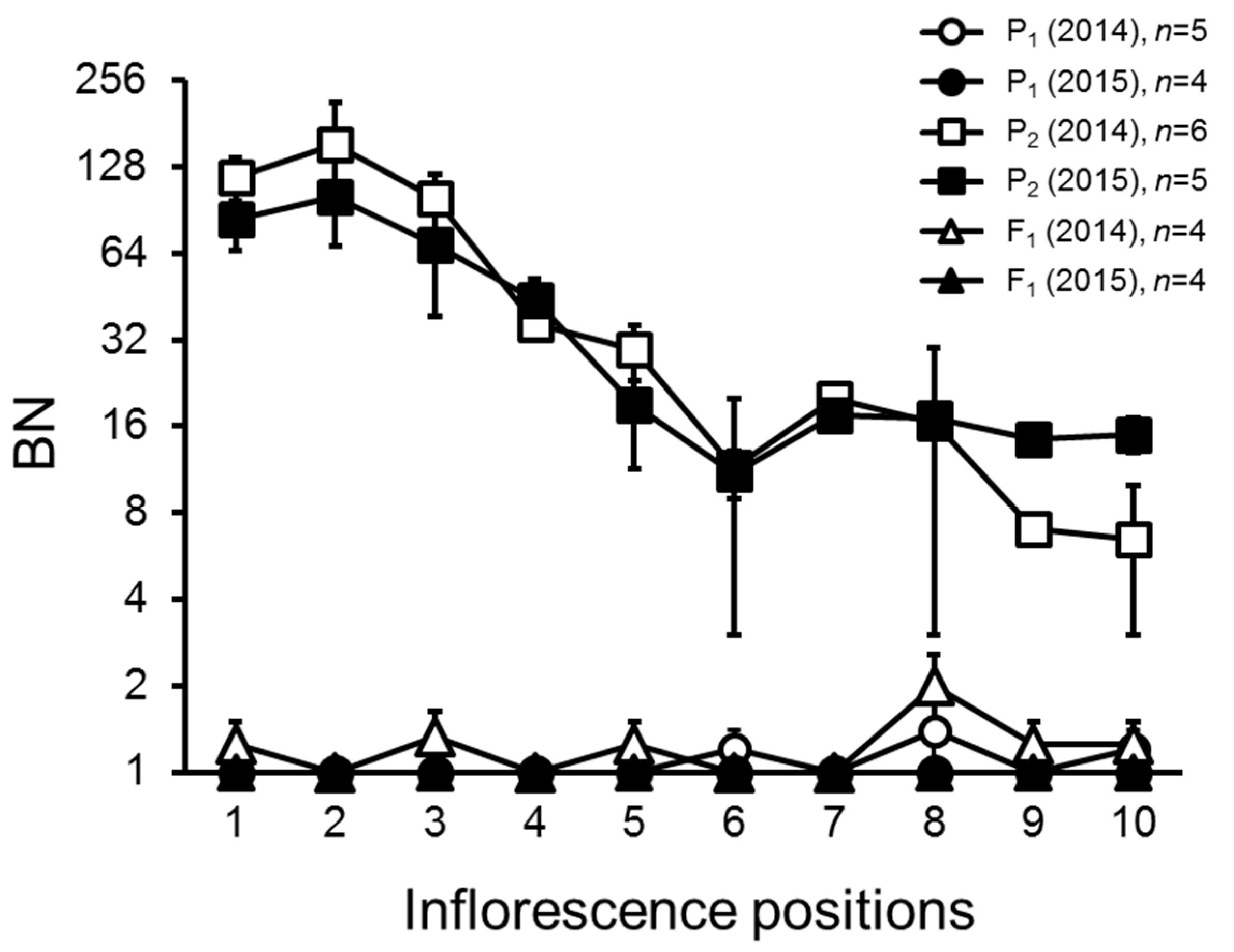
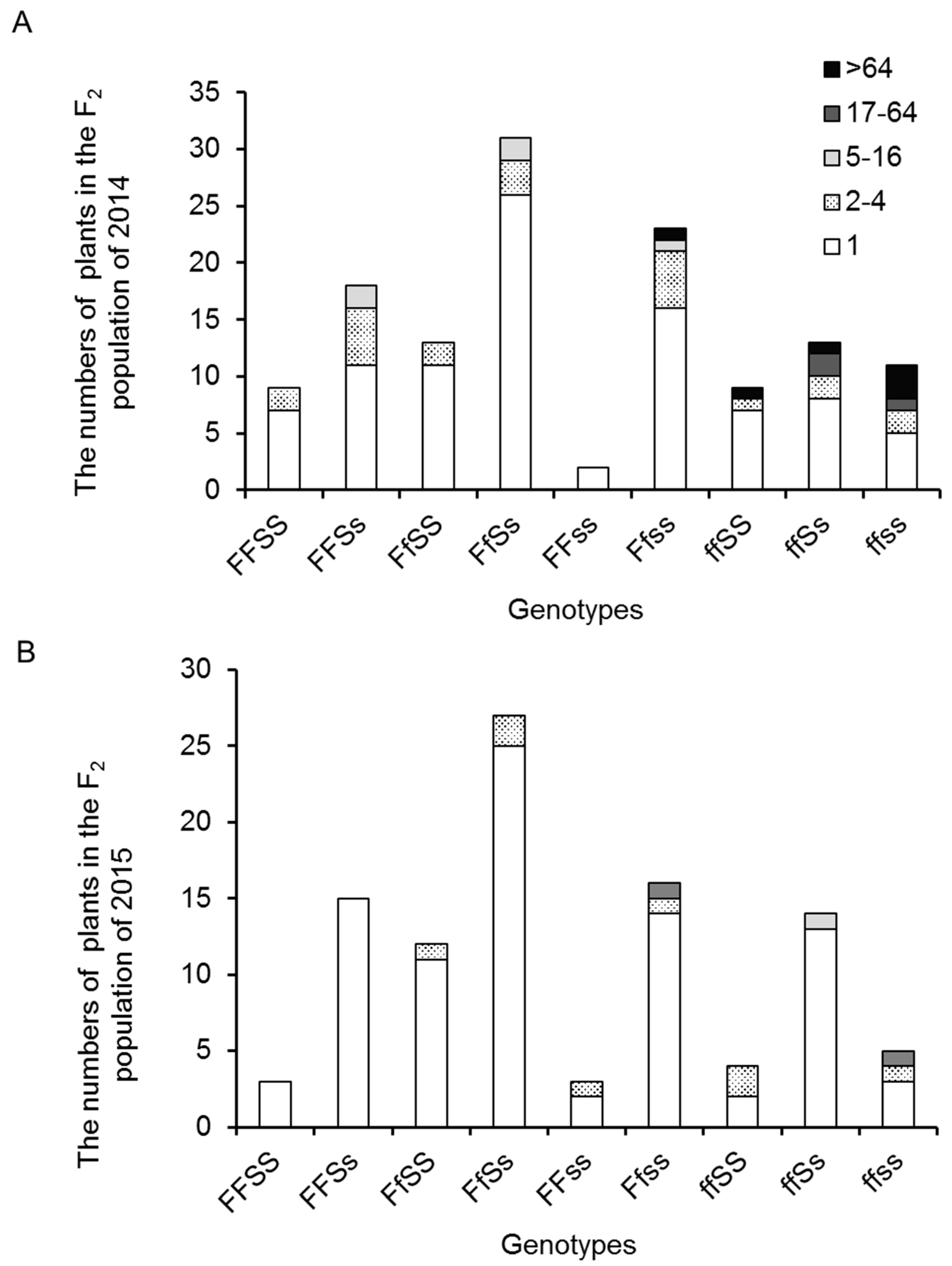
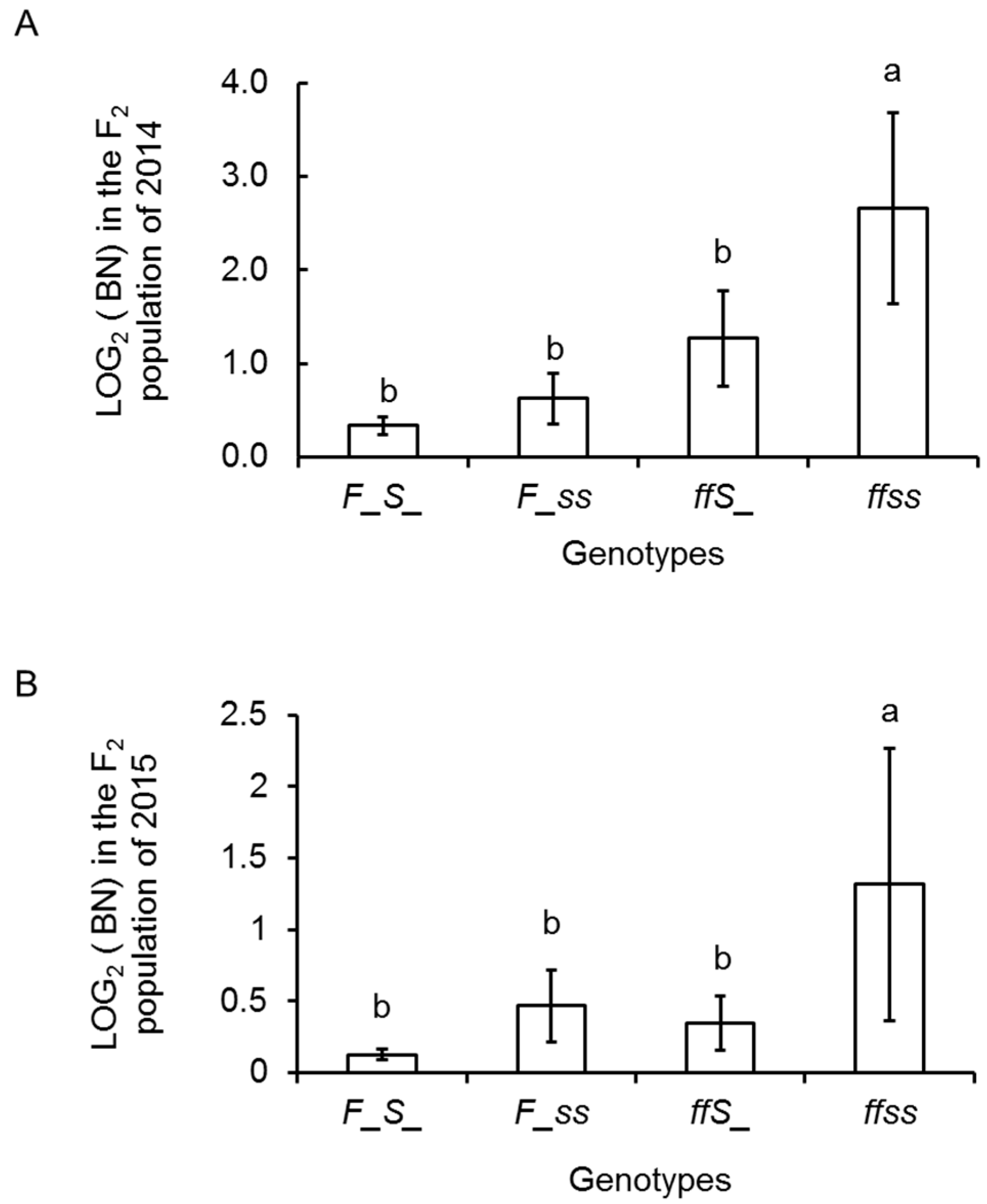
2.1. Variation and Inheritance of BN in the Two Parental Lines, F1, and F2 Populations
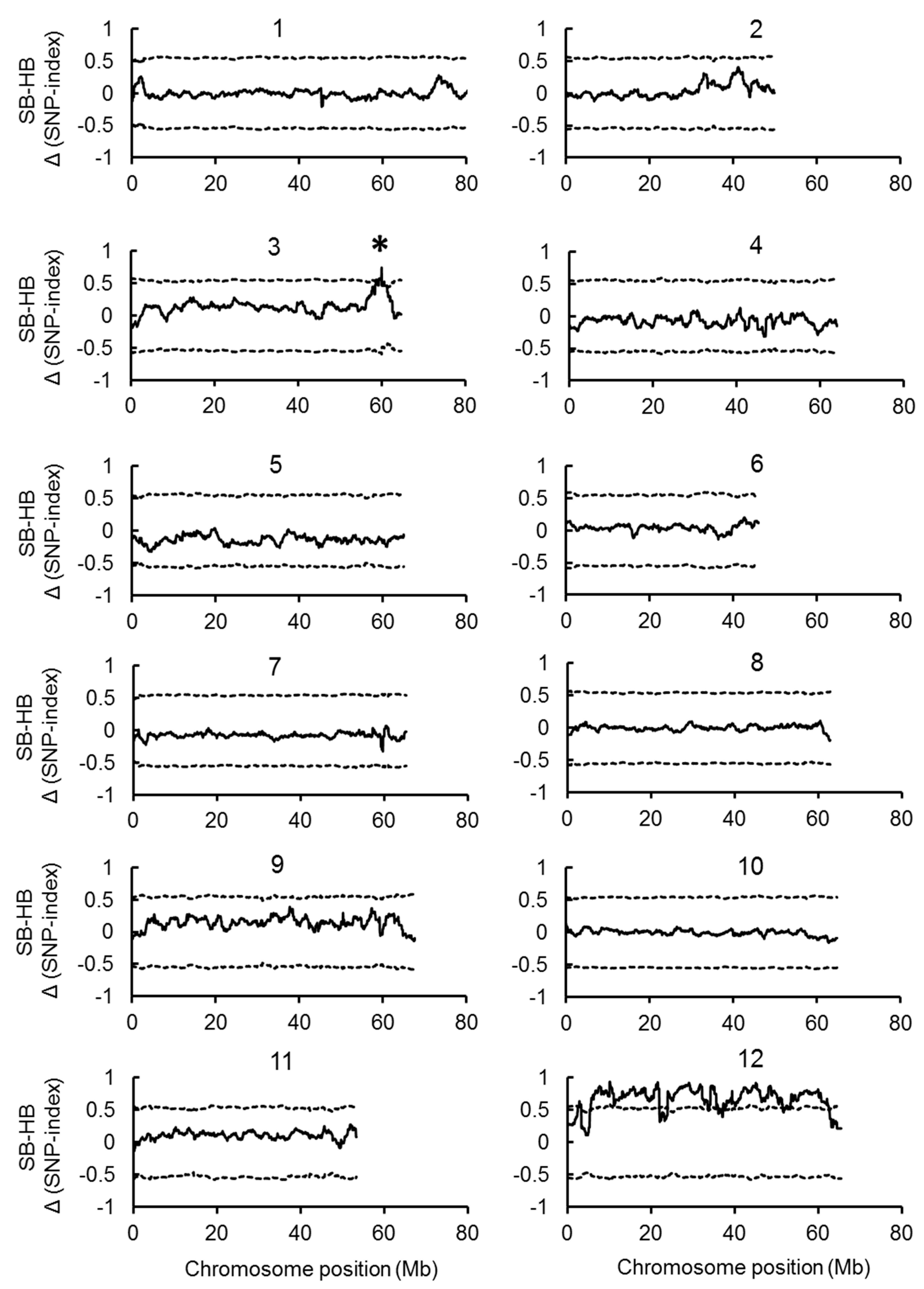
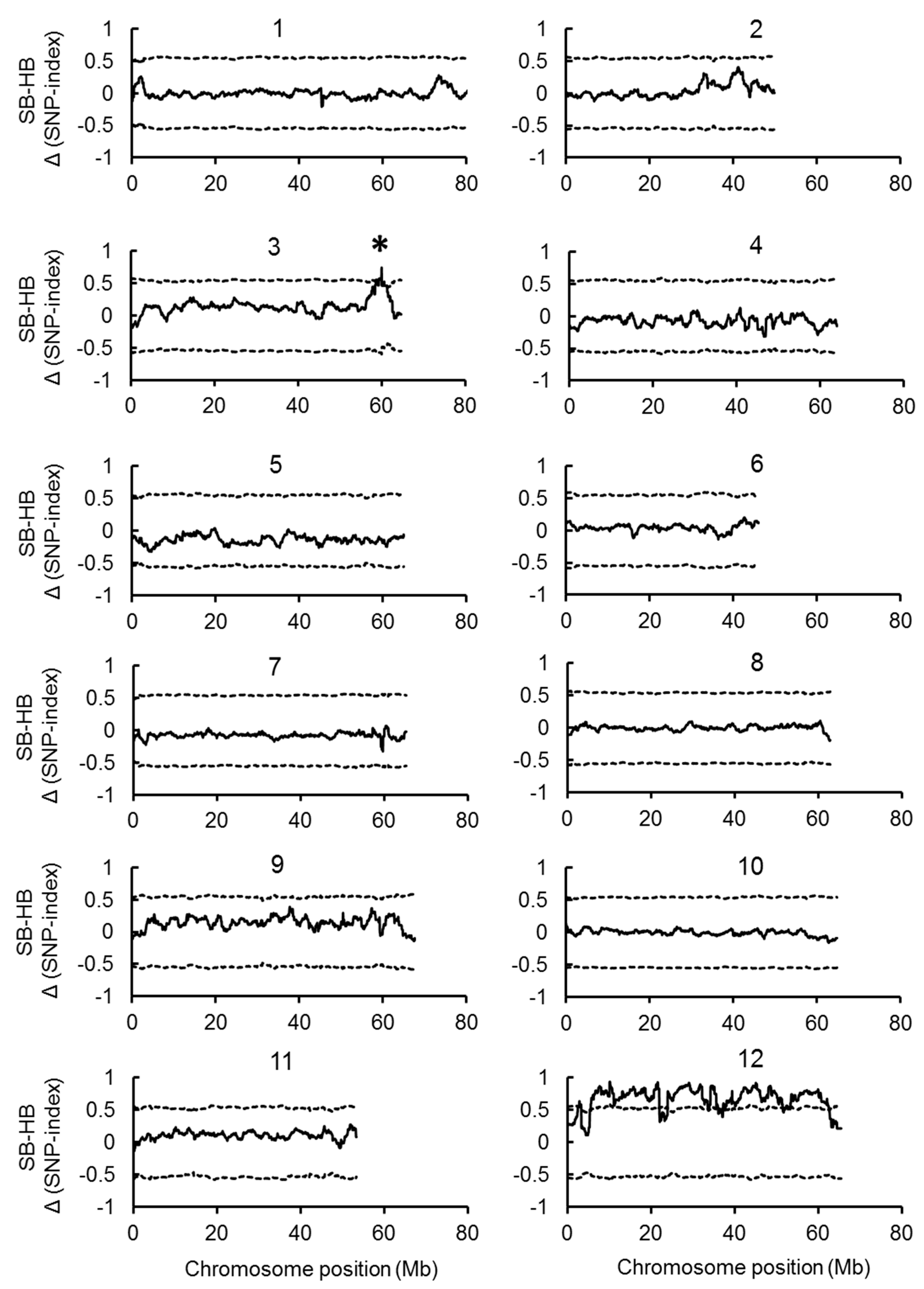
2.2. Joint QTL-seq and Bulk Segregant Analysis Used to Identify QTL Loci Controlling BN
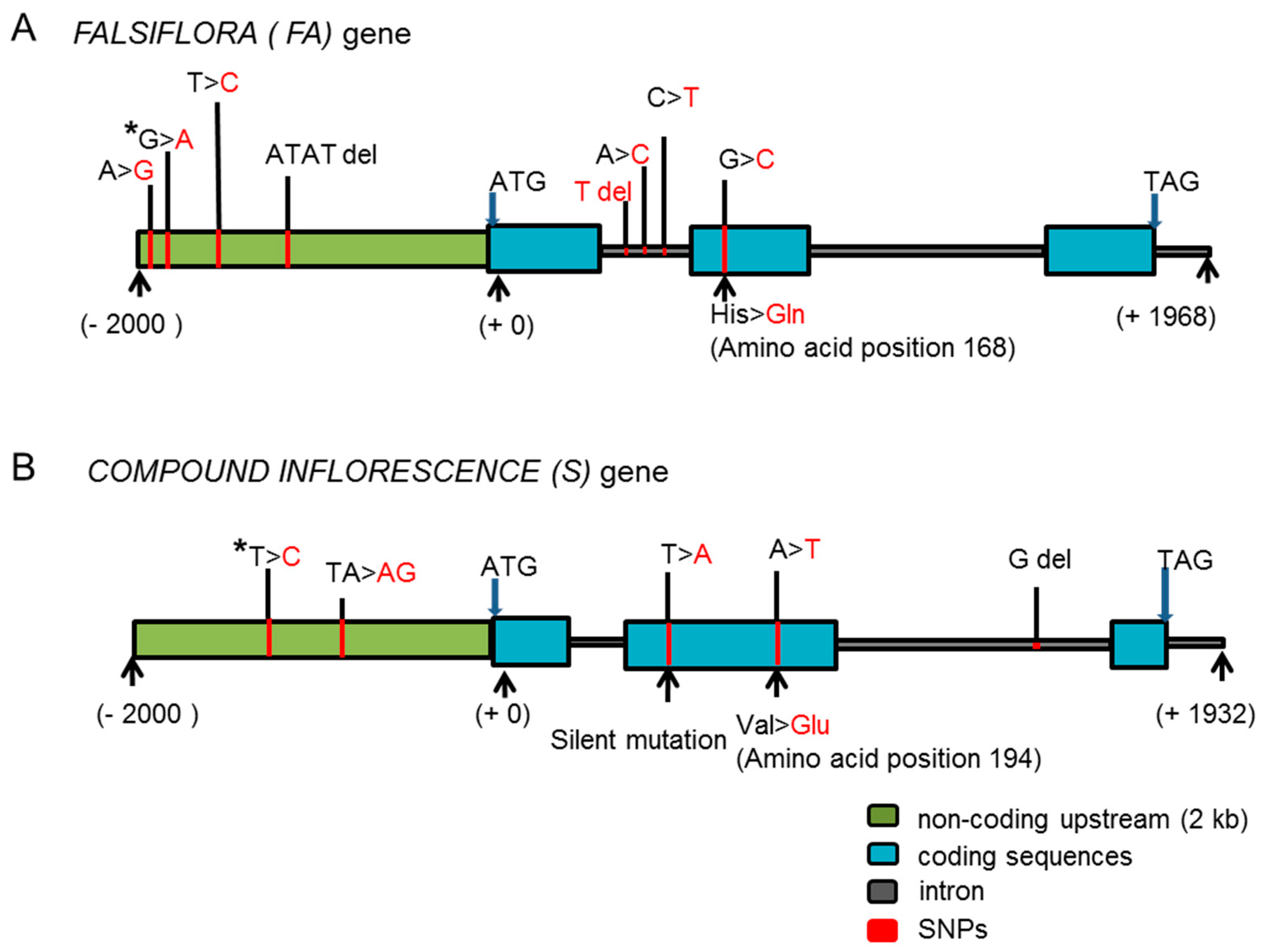
2.3. Identification of Candidate Genes for the BN Loci
2.4. Inflorescence Trait Variations Associated with FA and S Loci
3. Discussion
3.1. The New Allele Identified for the FALSIFLORA Gene Is the Candidate Gene Involved in the Control of BN in Tomato
3.2. Effects of FA and S Depend on the Genetic and Environmental Background
4. Materials and Methods
4.1. Phenotyping of Mapping Population for Inflorescence Traits
4.2. Mapping and QTL Analyses
4.3. Sequence Analysis of Candidate Genes and Single-Nucleotide Polymorphism (SNP) Genotyping of F2 Individuals
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siriwardana, N.S.; Lamb, R.S. The poetry of reproduction: The role of LEAFY in Arabidopsis thaliana flower formation. Int. J. Dev. Biol. 2012, 56, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Teo, Z.W.N.; Song, S.; Wang, Y.-Q.; Liu, J.; Yu, H. New insights into the regulation of inflorescence architecture. Trends Plant Sci. 2014, 19, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Jiang, K.; Schatz, M.C.; Lippman, Z.B. Rate of meristem maturation determines inflorescence architecture in tomato. Proc. Natl. Acad. Sci. USA 2012, 109, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Crane, M. Heredity of types of inflorescence and fruits in tomato. J. Genet. 1915, 5, 1–11. [Google Scholar] [CrossRef]
- Lippman, Z.B.; Cohen, O.; Alvarez, J.P.; Abu-Abied, M.; Pekker, I.; Paran, I.; Eshed, Y.; Zamir, D. The making of a compound inflorescence in tomato and related nightshades. PLoS Biol. 2008, 6, e288. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Molinero-Rosales, N.; Jamilena, M.; Zurita, S.; Gómez, P.; Capel, J.; Lozano, R. FALSIFLORA, the tomato orthologue of FLORICAULA and LEAFY, controls flowering time and floral meristem identity. Plant J. 1999, 20, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.G. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 2012, 485, 635–641. [Google Scholar] [Green Version]
- Winter, C.M.; Austin, R.S.; Blanvillain-Baufume, S.; Reback, M.A.; Monniaux, M.; Wu, M.-F.; Sang, Y.; Yamaguchi, A.; Yamaguchi, N.; Parker, J.E. LEAFY target genes reveal floral regulatory logic, cis motifs, and a link to biotic stimulus response. Dev. Cell 2011, 20, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Schultz, E.A.; Haughn, G.W. LEAFY, a homeotic gene that regulates inflorescence development in Arabidopsis. Plant Cell 1991, 3, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Moyroud, E.; Kusters, E.; Monniaux, M.; Koes, R.; Parcy, F. LEAFY blossoms. Trends Plant Sci. 2010, 15, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Yamaguchi, A.; Abe, M.; Wagner, D.; Komeda, Y. LEAFY controls Arabidopsis pedicel length and orientation by affecting adaxial–abaxial cell fate. Plant J. 2012, 69, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Weigel, D.; Alvarez, J.; Smyth, D.R.; Yanofsky, M.F.; Meyerowitz, E.M. LEAFY controls floral meristem identity in Arabidopsis. Cell 1992, 69, 843–859. [Google Scholar] [CrossRef]
- Coen, E.S.; Romero, J.; Doyle, S.; Elliott, R.; Murphy, G.; Carpenter, R. Floricaula: A homeotic gene required for flower development in Antirrhinum majus. Cell 1990, 63, 1311–1322. [Google Scholar] [CrossRef]
- Weigel, D.; Nilsson, O. A developmental switch sufficient for flower initiation in diverse plants. Nature 1995, 377, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, M.A.; Weigel, D. Independent regulation of flowering by phytochrome B and gibberellins in Arabidopsis. Plant Physiol. 1999, 120, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, M.A.; Soowal, L.N.; Lee, I.; Weigel, D. LEAFY expression and flower initiation in Arabidopsis. Development 1997, 124, 3835–3844. [Google Scholar] [PubMed]
- Hempel, F.D.; Weigel, D.; Mandel, M.A.; Ditta, G.; Zambryski, P.C.; Feldman, L.J.; Yanofsky, M.F. Floral determination and expression of floral regulatory genes in Arabidopsis. Development 1997, 124, 3845–3853. [Google Scholar] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Singh, M.; Srivastava, R.; Bajaj, D.; Saxena, M.S.; Rana, J.C.; Bansal, K.C.; Tyagi, A.K.; Parida, S.K. mQTL-seq delineates functionally relevant candidate gene harbouring a major QTL regulating pod number in chickpea. DNA Res. 2016, 23, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Bomblies, K.; Wang, R.-L.; Ambrose, B.A.; Schmidt, R.J.; Meeley, R.B.; Doebley, J. Duplicate FLORICAULA/LEAFY homologs zfl1 and zfl2 control inflorescence architecture and flower patterning in maize. Development 2003, 130, 2385–2395. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Kosugi, S.; Yoshida, K.; Natsume, S.; Takagi, H.; Kanzaki, H.; Matsumura, H.; Yoshida, K.; Mitsuoka, C.; Tamiru, M. Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 2012, 30, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Woffelman, C. Lynon Biosoft, Institute of Molecular Plant. Sciences; DNAMAN for Windows, Version 5.2.10; Leiden University: Leiden, The Netherlands, 2004. [Google Scholar]
- Swindell, S.R.; Plasterer, T.N. SEQMAN: Contig assembly. In Sequence Data Analysis Guidebook; Springer: Berlin, Germany, 1997; pp. 75–89. [Google Scholar]
- Reed, G.H.; Wittwer, C.T. Sensitivity and specificity of single-nucleotide polymorphism scanning by high-resolution melting analysis. Clin. Chem. 2004, 50, 1748–1754. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, H.; Kawabata, S. Identification and Validation of New Alleles of FALSIFLORA and COMPOUND INFLORESCENCE Genes Controlling the Number of Branches in Tomato Inflorescence. Int. J. Mol. Sci. 2017, 18, 1572. https://doi.org/10.3390/ijms18071572
Zheng H, Kawabata S. Identification and Validation of New Alleles of FALSIFLORA and COMPOUND INFLORESCENCE Genes Controlling the Number of Branches in Tomato Inflorescence. International Journal of Molecular Sciences. 2017; 18(7):1572. https://doi.org/10.3390/ijms18071572
Chicago/Turabian StyleZheng, Huan, and Saneyuki Kawabata. 2017. "Identification and Validation of New Alleles of FALSIFLORA and COMPOUND INFLORESCENCE Genes Controlling the Number of Branches in Tomato Inflorescence" International Journal of Molecular Sciences 18, no. 7: 1572. https://doi.org/10.3390/ijms18071572