
Matrix Metalloproteinase Gene Activation Resulting from Disordred Epigenetic Mechanisms in Rheumatoid Arthritis


Abstract
:1. Introduction
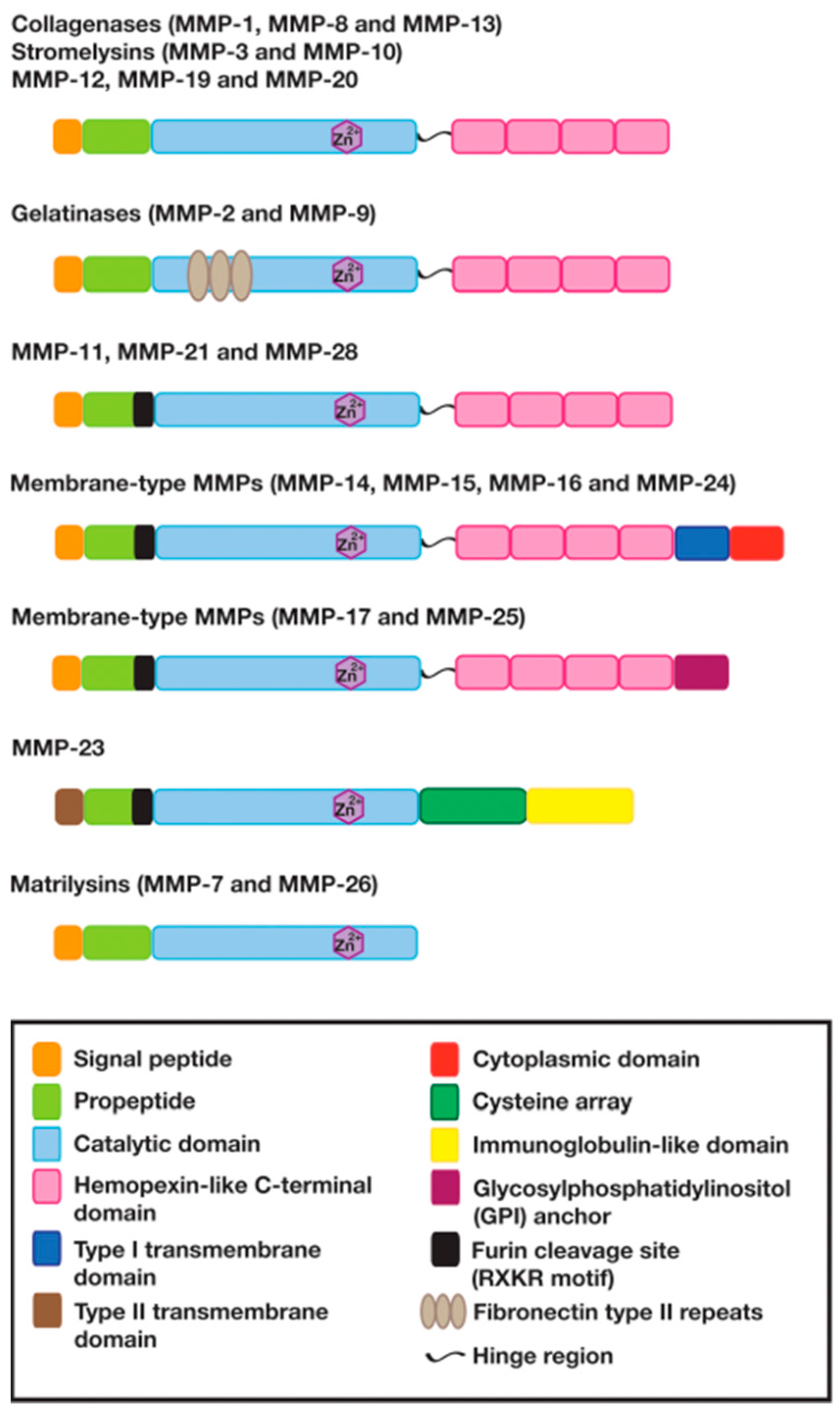
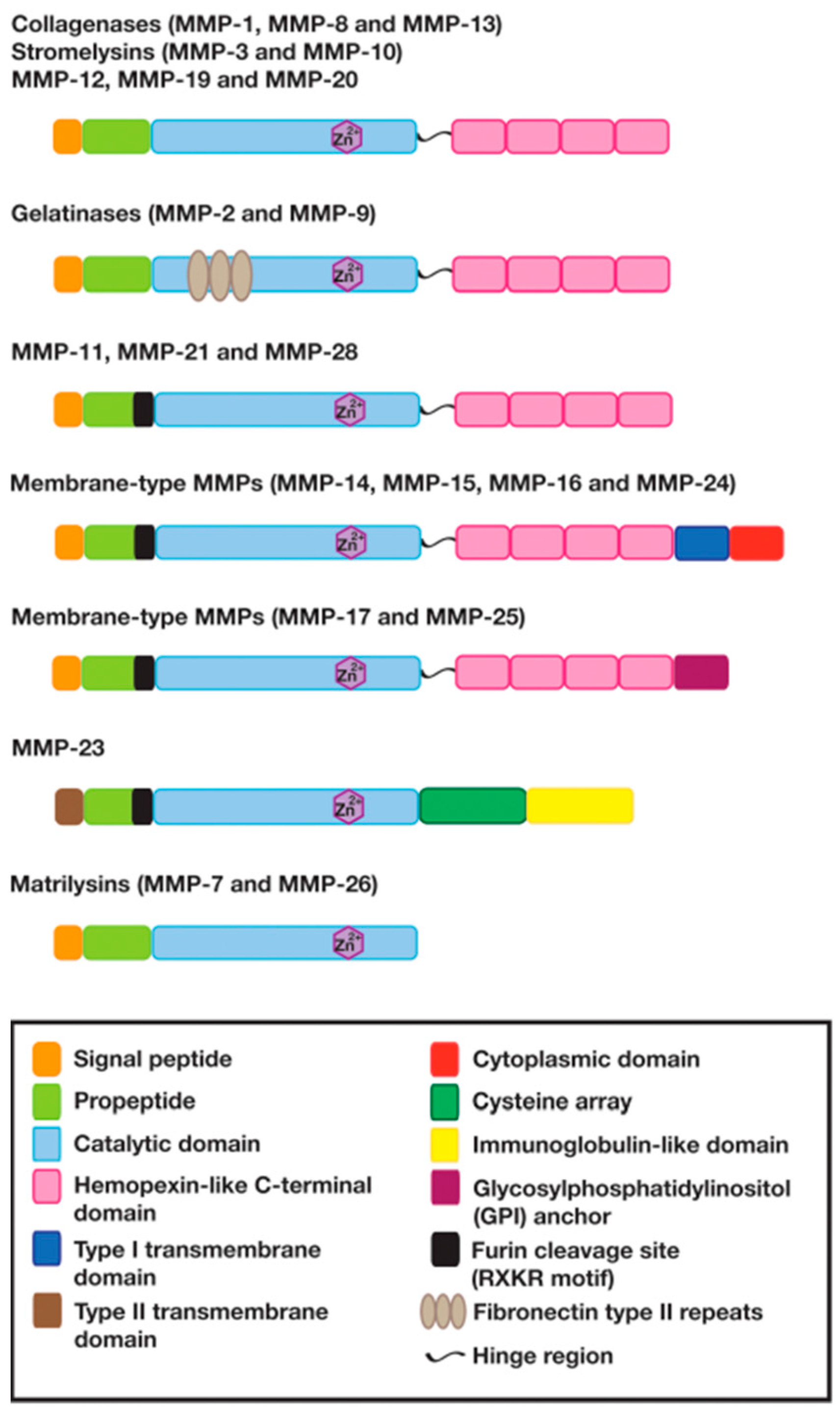
2. The Structure and Function of Matrix Metalloproteinases
3. The Pathogenesis of Rheumatoid Arthritis
3.1. The Epidemiology and Etiology of RA
3.2. The Pathological Roles of MMPs in RA
4. Epigenetic Mechanisms
4.1. Epigenetics and Chromatin Structure
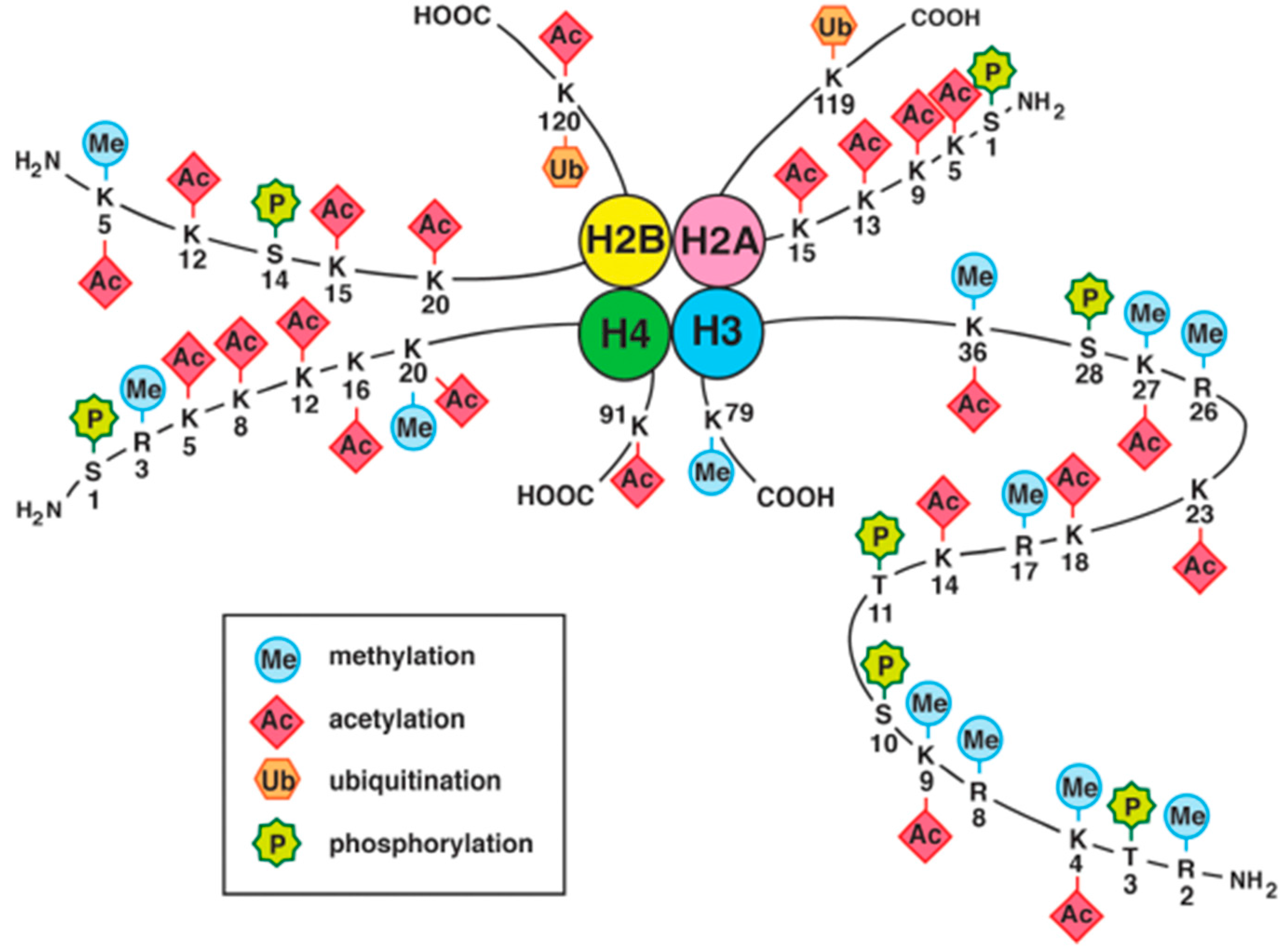
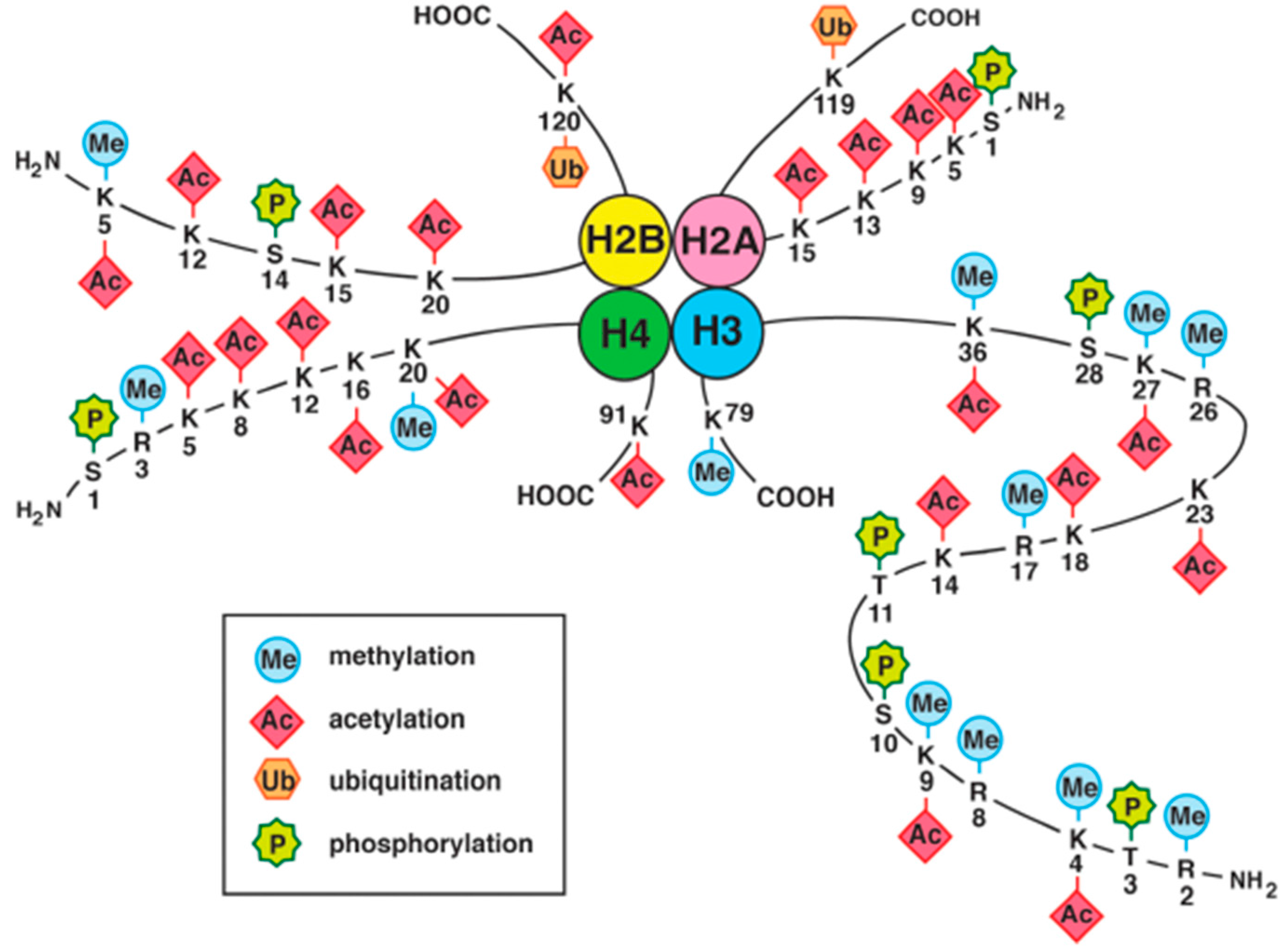
4.2. Histone Modifications
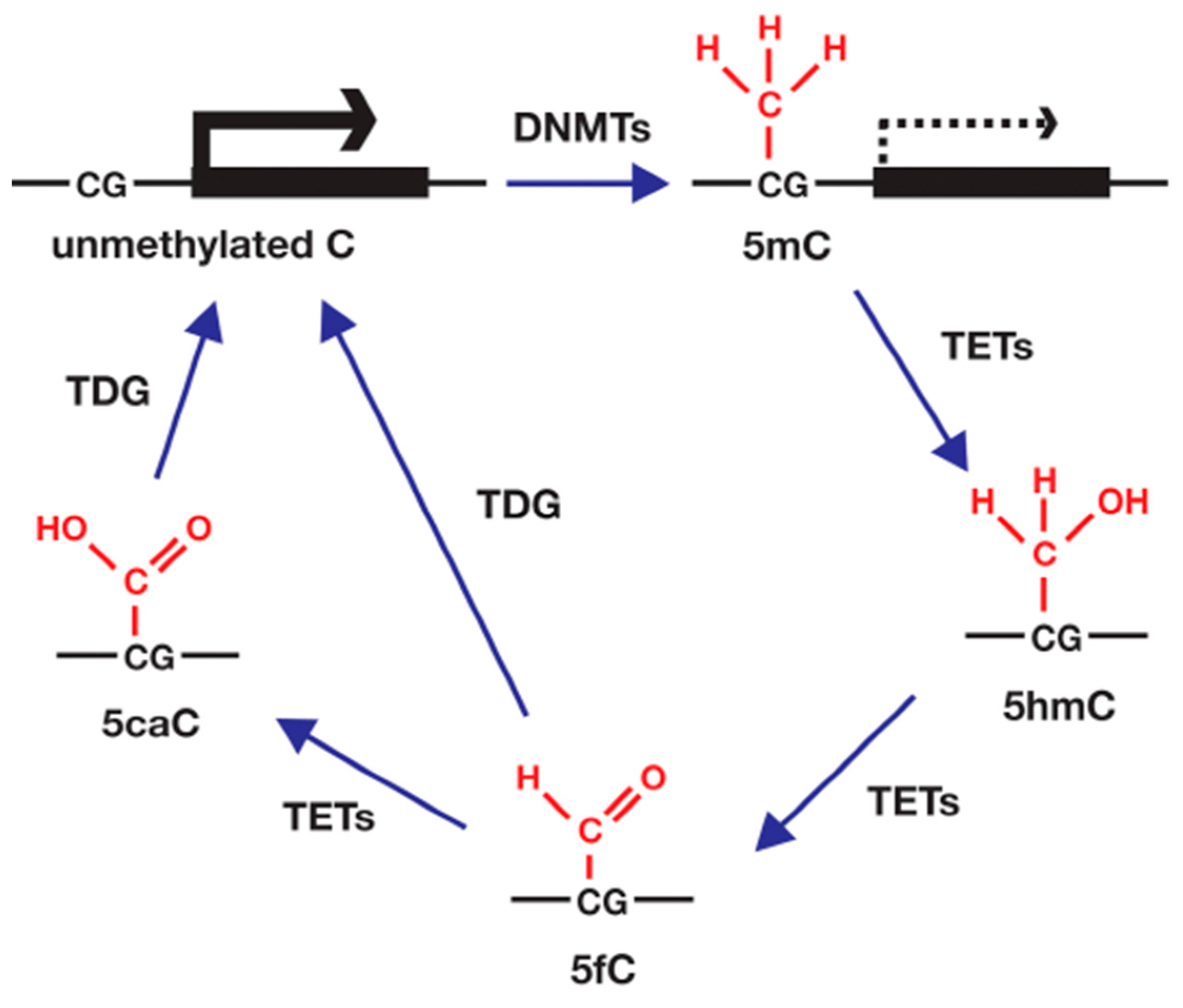
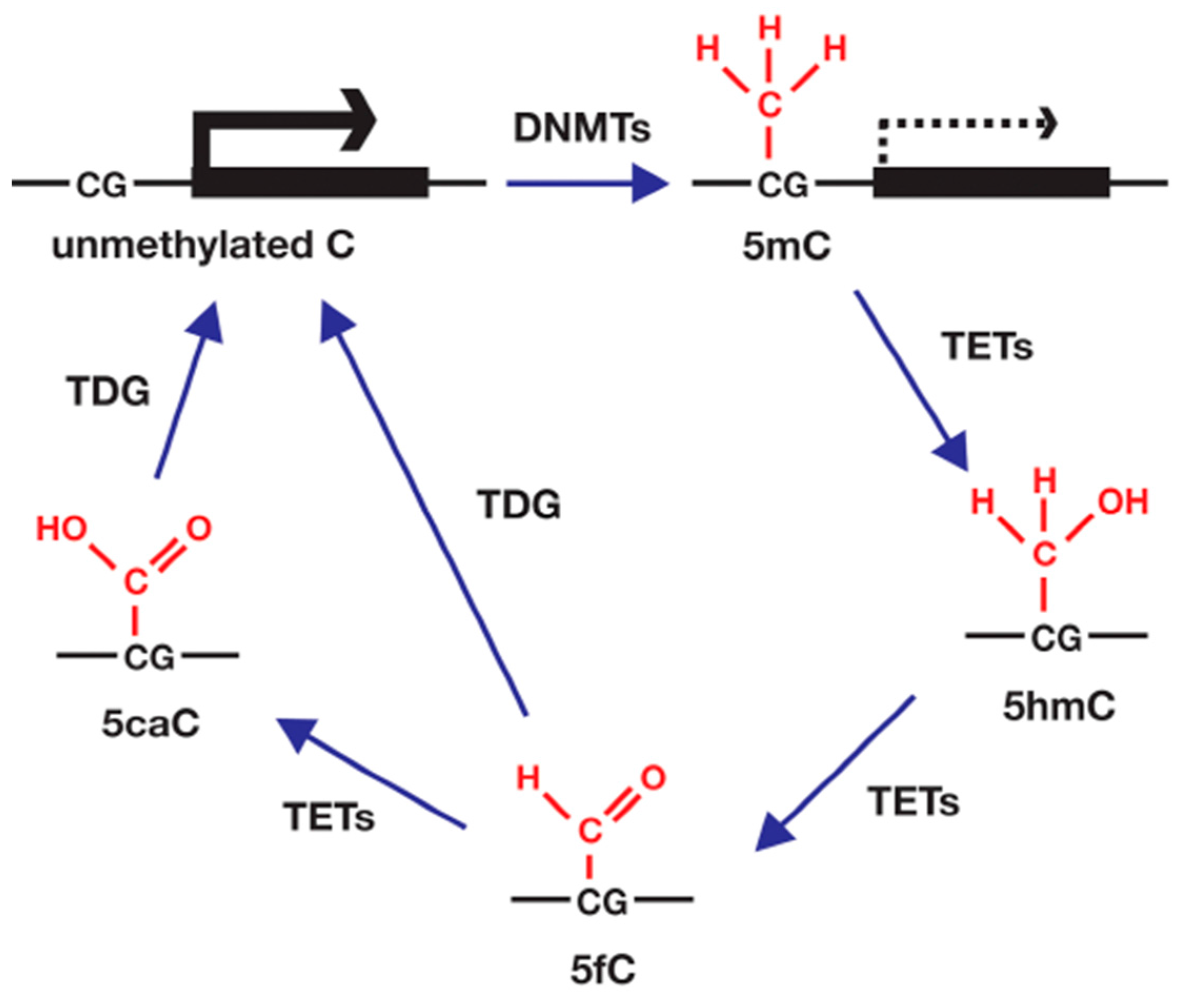
4.3. DNA Methylation
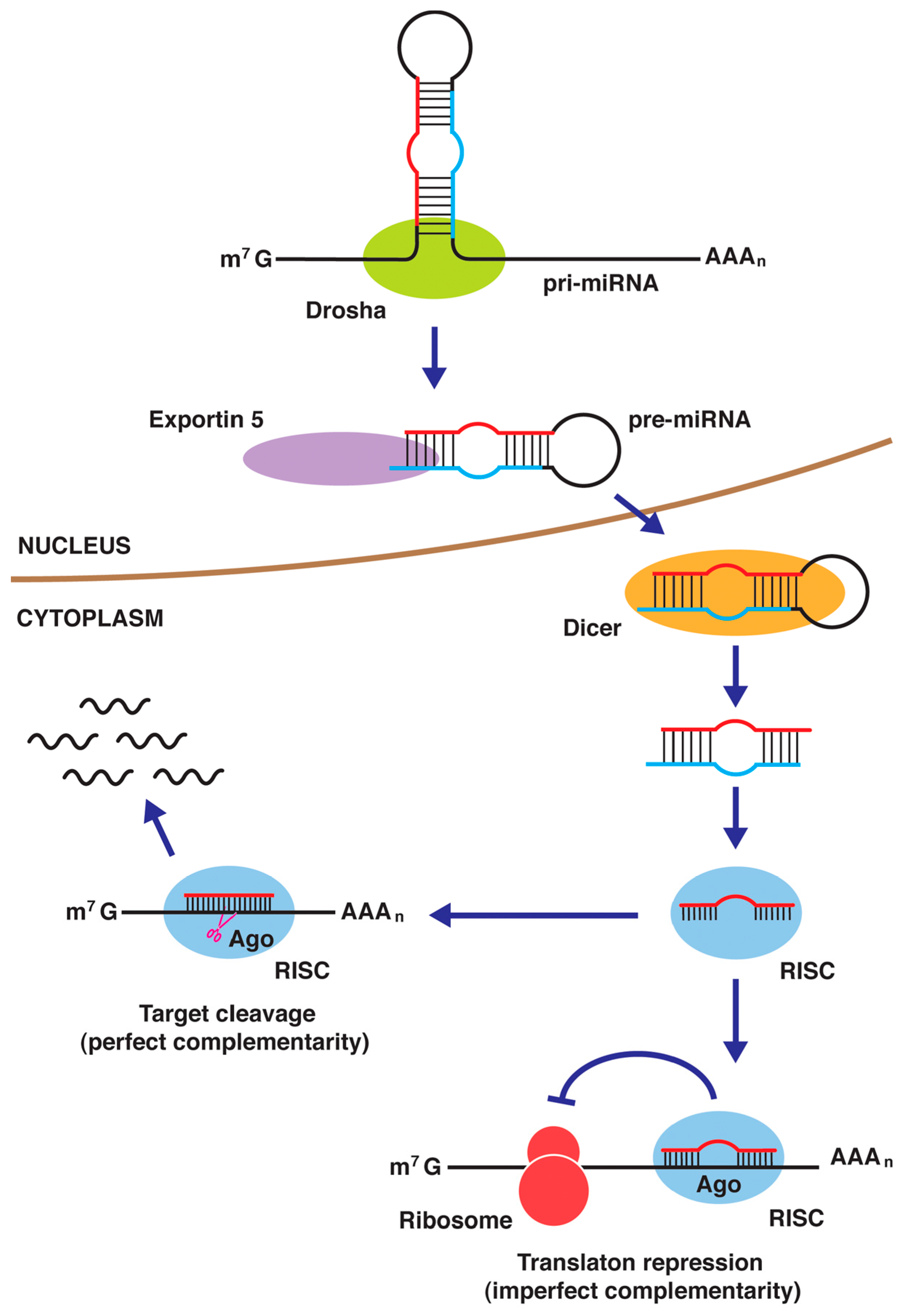
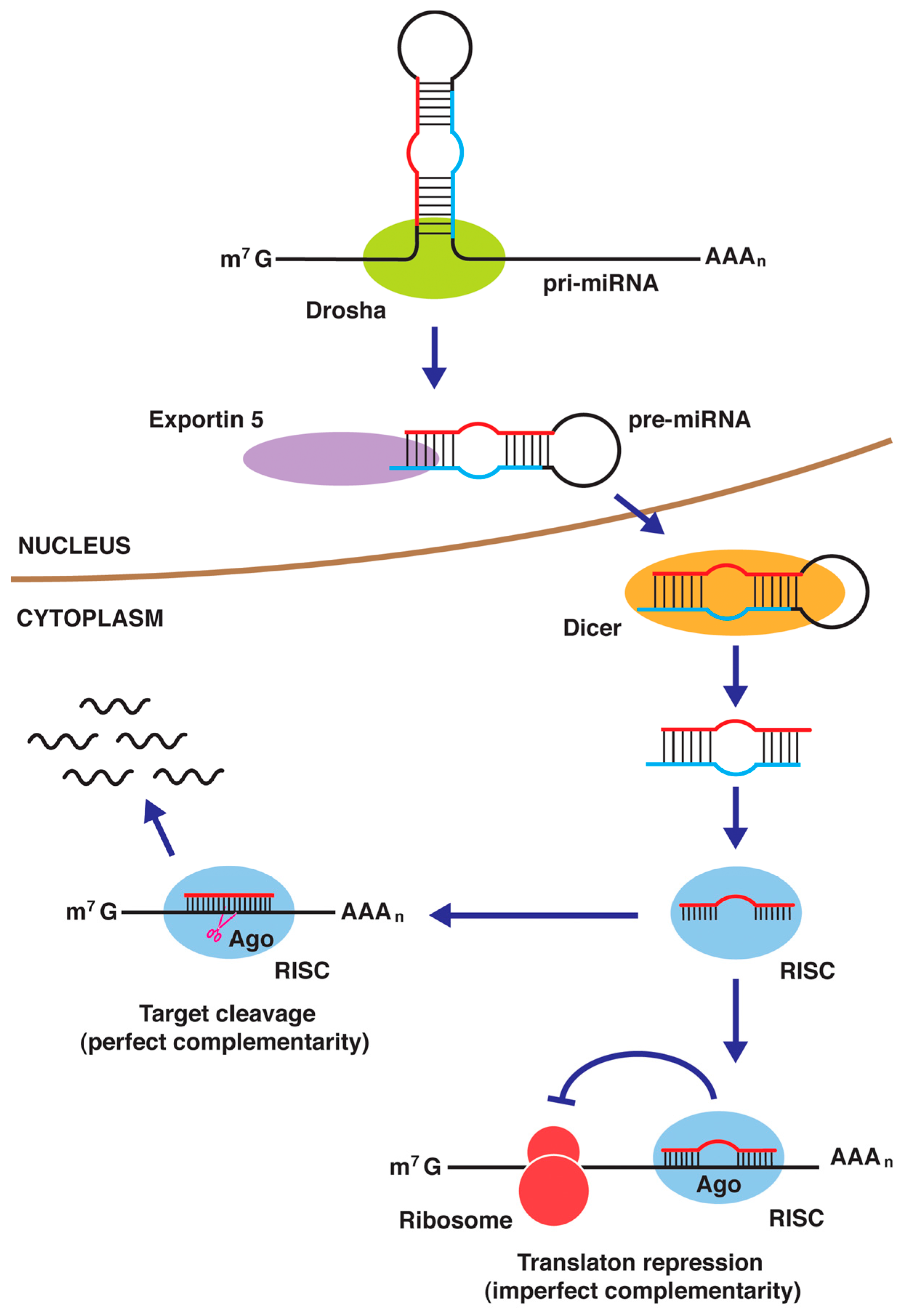
4.4. miRNAs
5. Epigenetic Mechanisms Regulating MMP Transcription in RA
5.1. Disordered Histone Modifications in MMP Genes in RA
5.2. Disordered DNA Methylation in MMP Genes in RA
5.3. Disordered miRNA Expression in MMP Genes in RA
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Marino, G.; Uria, J.A.; Puente, X.S.; Quesada, V.; Bordallo, J.; Lopez-Otin, C. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J. Biol. Chem. 2003, 278, 3671–3678. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.M. Caspases at the crossroads of immune-cell life and death. Nat. Rev. Immunol. 2006, 6, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Oikonomopoulou, K.; Hansen, K.K.; Saifeddine, M.; Vergnolle, N.; Tea, I.; Diamandis, E.P.; Hollenberg, M.D. Proteinase-mediated cell signalling: Targeting proteinase-activated receptors (PARs) by kallikreins and more. Biol. Chem. 2006, 387, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Matrisian, L.M. Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 2007, 7, 800–888. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Turk, B. Targeting proteases: Successes, failures and future prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Turk, D.; Turk, V. Protease signalling: The cutting edge. EMBO J. 2012, 31, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Overall, C.M. Protease degradomics: A new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 2002, 3, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; Blobel, C.P. In search of partners: Linking extracellular proteases to substrates. Nat. Rev. Mol. Cell Biol. 2007, 8, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Welsch, D.J.; Pelletier, J.P. Metalloproteases and inhibitors in arthritic diseases. Best Pract. Res. Clin. Rheumatol. 2001, 15, 805–829. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; Panayi, G.S. Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 2001, 344, 907–916. [Google Scholar] [PubMed]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell. Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.T.; Araki, Y.; Sato, K.; Aizaki, Y.; Yokota, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Aberrant histone acetylation contributes to elevated interleukin-6 production in rheumatoid arthritis synovial fibroblasts. Biochem. Biophys. Res. Commun. 2014, 444, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Tsuzuki Wada, T.; Aizaki, Y.; Sato, K.; Yokota, K.; Fujimoto, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Histone methylation and STAT-3 differentially regulate interleukin-6-induced matrix metalloproteinase gene activation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 2016, 68, 1111–1123. [Google Scholar] [PubMed]
- Rengel, Y.; Ospelt, C.; Gay, S. Proteinases in the joint: Clinical relevance of proteinases in joint destruction. Arthritis Res. Ther. 2007, 9, 221. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. Metalloproteinases: Potential therapeutic targets for rheumatoid arthritis. Endocr. Metab. Immune Disord. Drug Targets 2015, 15, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; Lopez-Otin, C. Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nat. Rev. Cancer 2002, 2, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.J.; Overall, C.M. TIMP independence of matrix metalloproteinase (MMP)-2 activation by membrane type 2 (MT2)-MMP is determined by contributions of both the MT2-MMP catalytic and hemopexin C domains. J. Biol. Chem. 2006, 281, 26528–26539. [Google Scholar] [CrossRef] [PubMed]
- Shofuda, K.; Yasumitsu, H.; Nishihashi, A.; Miki, K.; Miyazaki, K. Expression of three membrane-type matrix metalloproteinases (MT-MMPs) in rat vascular smooth muscle cells and characterization of MT3-MMPs with and without transmembrane domain. J. Biol. Chem. 1997, 272, 9749–9754. [Google Scholar] [PubMed]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [PubMed]
- Sakkas, L.I.; Bogdanos, D.P.; Katsiari, C.; Platsoucas, C.D. Anti-citrullinated peptides as autoantigens in rheumatoid arthritis-relevance to treatment. Autoimmun. Rev. 2014, 13, 1114–1120. [Google Scholar] [PubMed]
- Hardy, R.R.; Hayakawa, K.; Shimizu, M.; Yamasaki, K.; Kishimoto, T. Rheumatoid factor secretion from human Leu-1 + B cells. Science 1987, 236, 81–83. [Google Scholar] [PubMed]
- Tan, E.M.; Smolen, J.S. Historical observations contributing insights on etiopathogenesis of rheumatoid arthritis and role of rheumatoid factor. J. Exp. Med. 2016, 213, 1937–1950. [Google Scholar] [PubMed]
- Gibofsky, A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: A Synopsis. Am. J. Manag. Care 2014, 20, S128–S135. [Google Scholar] [PubMed]
- Nam, J.L.; Ramiro, S.; Gaujoux-Viala, C.; Takase, K.; Leon-Garcia, M.; Emery, P.; Gossec, L.; Landewe, R.; Smolen, J.S.; Buch, M.H. Efficacy of biological disease-modifying antirheumatic drugs: A systematic literature review informing the 2013 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 516–528. [Google Scholar] [PubMed]
- Wang, D.; Li, Y.; Liu, Y.; Shi, G. The use of biologic therapies in the treatment of rheumatoid arthritis. Curr. Pharm. Biotechnol. 2014, 15, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Yamaoka, K. JAK inhibitor tofacitinib for treating rheumatoid arthritis: From basic to clinical. Mod. Rheumatol. 2013, 23, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, P.E.; van der Heijde, D.M.; St. Clair, E.W.; Furst, D.E.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Weisman, M.; Emery, P.; Feldmann, M.; et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-tumor necrosis factor trial in rheumatoid arthritis with concomitant therapy study group. N. Engl. J. Med. 2000, 343, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Kalden, J.R.; Antoni, C.; Smolen, J.S.; Leeb, B.; Breedveld, F.C.; Macfarlane, J.D.; Bijl, H.; et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor α (cA2) versus placebo in rheumatoid arthritis. Lancet 1994, 344, 1105–1110. [Google Scholar] [CrossRef]
- Maini, R.; St Clair, E.W.; Breedveld, F.; Furst, D.; Kalden, J.; Weisman, M.; Smolen, J.; Emery, P.; Harriman, G.; Feldmann, M.; et al. Infliximab (chimeric anti-tumour necrosis factor α monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: A randomised phase III trial. ATTRACT Study Group. Lancet 1999, 354, 1932–1939. [Google Scholar] [CrossRef]
- St Clair, E.W.; van der Heijde, D.M.; Smolen, J.S.; Maini, R.N.; Bathon, J.M.; Emery, P.; Keystone, E.; Schiff, M.; Kalden, J.R.; Wang, B.; et al. Combination of infliximab and methotrexate therapy for early rheumatoid arthritis: A randomized, controlled trial. Arthritis Rheum. 2004, 50, 3432–3443. [Google Scholar] [CrossRef] [PubMed]
- Larry, W.; Moreland, M.D.; Scott, W.; Baumgartner, M.D.; Michael, H.; Schiff, M.D.; Elizabeth, A.; Tindall, M.D.; Roy, M.; Fleischmann, M.D.; et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N. Engl. J. Med. 1997, 337, 141–147. [Google Scholar]
- Moreland, L.W.; Schiff, M.H.; Baumgartner, S.W.; Tindall, E.A.; Fleischmann, R.M.; Bulpitt, K.J.; Weaver, A.L.; Keystone, E.C.; Furst, D.E.; Mease, P.J.; et al. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann. Intern. Med. 1999, 130, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Michael, E.; Weinblatt, M.D.; Joel, M.; Kremer, M.D.; Arthur, D.; Bankhurst, M.D.; Ken, J.; Bulpitt, M.D.; Roy, M.; Fleischmann, M.D.; et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N. Engl. J. Med. 1999, 340, 253–259. [Google Scholar]
- Bathon, J.M.; Martin, R.W.; Fleischmann, R.M.; Tesser, J.R.; Schiff, M.H.; Keystone, E.C.; Genovese, M.C.; Wasko, M.C.; Moreland, L.W.; Weaver, A.L.; et al. A comparison of etanercept and methotrexate in patients with early rheumatoid arthritis. N. Engl. J. Med. 2000, 343, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Bathon, J.M.; Martin, R.W.; Fleischmann, R.M.; Tesser, J.R.; Schiff, M.H.; Keystone, E.C.; Wasko, M.C.; Moreland, L.W.; Weaver, A.L.; et al. Etanercept versus methotrexate in patients with early rheumatoid arthritis: Two-year radiographic and clinical outcomes. Arthritis Rheum. 2002, 46, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Breedveld, F.C.; Hall, S.; Durez, P.; Chang, D.J.; Robertson, D.; Singh, A.; Pedersen, R.D.; Koenig, A.S.; Freundlich, B. Comparison of methotrexate monotherapy with a combination of methotrexate and etanercept in active, early, moderate to severe rheumatoid arthritis (COMET): A randomised, double-blind, parallel treatment trial. Lancet 2008, 372, 375–382. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Keystone, E.C.; Furst, D.E.; Moreland, L.W.; Weisman, M.H.; Birbara, C.A.; Fischkoff, S.A.; Chartash, E.K. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: The ARMADA trial. Arthritis Rheum. 2003, 48, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Keystone, E.C.; Kavanaugh, A.F.; Sharp, J.T.; Tannenbaum, H.; Hua, Y.; Teoh, L.S.; Fischkoff, S.A.; Chartash, E.K. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: A randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004, 50, 1400–1411. [Google Scholar] [PubMed]
- Breedveld, F.C.; Weisman, M.H.; Kavanaugh, A.F.; Cohen, S.B.; Pavelka, K.; van Vollenhoven, R.; Sharp, J.; Perez, J.L.; Spencer-Green, G.T. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006, 54, 26–37. [Google Scholar] [PubMed]
- Kay, J.; Matteson, E.L.; Dasgupta, B.; Nash, P.; Durez, P.; Hall, S.; Hsia, E.C.; Han, J.; Wagner, C.; Xu, Z.; et al. Golimumab in patients with active rheumatoid arthritis despite treatment with methotrexate: A randomized, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2008, 58, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Keystone, E.C.; Genovese, M.C.; Klareskog, L.; Hsia, E.C.; Hall, S.T.; Miranda, P.C.; Pazdur, J.; Bae, S.C.; Palmer, W.; Zrubek, J.; et al. Golimumab, a human antibody to tumour necrosis factor α given by monthly subcutaneous injections, in active rheumatoid arthritis despite methotrexate therapy: The GO-FORWARD Study. Ann. Rheum. Dis. 2009, 68, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Keystone, E.; Genovese, M.C.; Klareskog, L.; Hsia, E.C.; Hall, S.; Miranda, P.C.; Pazdur, J.; Bae, S.C.; Palmer, W.; Xu, S.; et al. Golimumab in patients with active rheumatoid arthritis despite methotrexate therapy: 52-week results of the GO-FORWARD study. Ann. Rheum. Dis. 2010, 69, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Fleischmann, R.M.; Moreland, L.W.; Hsia, E.C.; Strusberg, I.; Durez, P.; Nash, P.; Amante, E.J.; Churchill, M.; Park, W.; et al. Golimumab, a human anti-tumor necrosis factor alpha monoclonal antibody, injected subcutaneously every four weeks in methotrexate-naive patients with active rheumatoid arthritis: Twenty-four-week results of a phase III, multicenter, randomized, double-blind, placebo-controlled study of golimumab before methotrexate as first-line therapy for early-onset rheumatoid arthritis. Arthritis Rheum. 2009, 60, 2272–2283. [Google Scholar] [PubMed]
- Kremer, J.; Ritchlin, C.; Mendelsohn, A.; Baker, D.; Kim, L.; Xu, Z.; Han, J.; Taylor, P. Golimumab, a new human anti-tumor necrosis factor alpha antibody, administered intravenously in patients with active rheumatoid arthritis: Forty-eight-week efficacy and safety results of a phase III randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2010, 62, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Keystone, E.; van der Heijde, D.; Mason, D.; Landewe, R., Jr.; Vollenhoven, R.R.; Combe, B.; Emery, P.; Strand, V.; Mease, P.; Desai, C.; et al. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: Findings of a fifty-two-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 2008, 58, 3319–3329. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.; Landewe, R.R.; Mease, P.; Brzezicki, J.; Mason, D.; Luijtens, K.; van Vollenhoven, R.F.; Kavanaugh, A.; Schiff, M.; Burmester, G.R.; et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: The RAPID 2 study. A randomised controlled trial. Ann. Rheum. Dis. 2009, 68, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.; Vencovsky, J.; van Vollenhoven, R.R.; Borenstein, D.; Box, J.; Coteur, G.; Goel, N.; Brezinschek, H.P.; Innes, A.; Strand, V. Efficacy and safety of certolizumab pegol monotherapy every 4 weeks in patients with rheumatoid arthritis failing previous disease-modifying antirheumatic therapy: The FAST4WARD study. Ann. Rheum. Dis. 2009, 68, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.E.; Hazleman, B.; Smith, M.; Moss, K.; Lisi, L.; Scott, D.G.; Patel, J.; Sopwith, M.; Isenberg, D.A. Efficacy of a novel PEGylated humanized anti-TNF fragment (CDP870) in patients with rheumatoid arthritis: A phase II double-blinded, randomized, dose-escalating trial. Rheumatology 2002, 41, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.J.; Szczepanski, L.; Szechinski, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close, D. R.; Stevens, R.M.; Shaw, T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 2004, 350, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.S.; Emery, P.; Greenwald, M.M.; Dougados, M.; Furie, R.R.; Genovese, M.C.; Keystone, E.C.; Loveless, J.E.; Burmester, G.R.; Cravets, M.W.; et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006, 54, 2793–2806. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.J.; Westhovens, R.; Leon, M.; Di Giorgio, E.; Alten, R.; Steinfeld, S.; Russell, A.; Dougados, M.; Emery, P.; Nuamah, I.F.; et al. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N. Engl. J. Med. 2003, 349, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.J.; Genant, H.H.; Moreland, L.L.; Russell, A.A.; Emery, P.; Abud-Mendoza, C.; Szechinski, J.; Li, T.; Ge, Z.; Becker, J.C.; et al. Effects of abatacept in patients with methotrexate-resistant active rheumatoid arthritis: A randomized trial. Ann. Intern. Med. 2006, 144, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.M.; Becker, J.J.; Schiff, M.; Luggen, M.; Sherrer, Y.; Kremer, J.; Birbara, C.; Box, J.; Natarajan, K.; et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. N. Engl. J. Med. 2005, 353, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.J.; Genant, H.H.; Moreland, L.L.; Russell, A.A.; Emery, P.; Abud-Mendoza, C.; Szechiński, J.; Li, T.; Teng, J.; Becker, J.C.; et al. Results of a two-year followup study of patients with rheumatoid arthritis who received a combination of abatacept and methotrexate. Arthritis Rheum. 2008, 58, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.J.; Dougados, M.; Emery, P.; Durez, P.; Sibilia, J.; Shergy, W.; Steinfeld, S.; Tindall, E.; Becker, J.C.; Li, T.; et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: Twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005, 52, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Westhovens, R.; Robles, M.; Ximenes, A.A.; Nayiager, S.; Wollenhaupt, J.; Durez, P.; Gomez-Reino, J.; Grassi, W.; Haraoui, B.; Shergy, W.; et al. Clinical efficacy and safety of abatacept in methotrexate-naive patients with early rheumatoid arthritis and poor prognostic factors. Ann. Rheum. Dis. 2009, 68, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Bresnihan, B.; Alvaro-Gracia, J.J.; Cobby, M.; Doherty, M.; Domljan, Z.; Emery, P.; Nuki, G.; Pavelka, K.; Rau, R.; Rozman, B.; et al. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 1998, 41, 2196–2204. [Google Scholar] [CrossRef]
- Maini, R.R.; Taylor, P.P.; Szechinski, J.; Pavelka, K.; Broll, J.; Balint, G.; Emery, P.; Raemen, F.; Petersen, J.; Smolen, J.; et al. Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 2006, 54, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.J.; Beaulieu, A.; Rubbert-Roth, A.; Ramos-Remus, C.; Rovensky, J.; Alecock, E.; Woodworth, T.; Alten, R.; OPTION Investigators. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): A double-blind, placebo-controlled, randomised trial. Lancet 2008, 371, 987–997. [Google Scholar] [CrossRef]
- Emery, P.; Keystone, E.; Tony, H.H.; Cantagrel, A.; van Vollenhoven, R.; Sanchez, A.; Alecock, E.; Lee, J.; Kremer, J. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: Results from a 24-week multicentre randomised placebo-controlled trial. Ann. Rheum. Dis. 2008, 67, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.M.; McKay, J.J.; Nasonov, E.E.; Mysler, E.E.; da Silva, N.N.; Alecock, E.; Woodworth, T.; Gomez-Reino, J.J. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: The tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum. 2008, 58, 2968–2980. [Google Scholar] [PubMed]
- Jones, G.; Sebba, A.; Gu, J.; Lowenstein, M.M.; Calvo, A.; Gomez-Reino, J.J.; Siri, D.A.; Tomsic, M.; Alecock, E.; Woodworth, T.; et al. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: The AMBITION study. Ann. Rheum. Dis. 2010, 69, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.J.; Blanco, R.; Brzosko, M.; Burgos-Vargas, R.; Halland, A.A.; Vernon, E.; Ambs, P.; Fleischmann, R. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate responses to methotrexate: Results from the double-blind treatment phase of a randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum. 2011, 63, 609–621. [Google Scholar] [PubMed]
- Fleischmann, R.; Kremer, J.; Cush, J.; Schulze-Koops, H.; Connell, C.C.; Bradley, J.D.; Gruben, D.; Wallenstein, G.V.; Zwillich, S.H.; et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Van Vollenhoven, R.R.; Fleischmann, R.; Cohen, S.; Lee, E.E.; Garcia Meijide, J.J.; Wagner, S.; Forejtova, S; Zwillich, S.H.; Gruben, D.; Koncz, T.; et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N. Engl. J. Med. 2012, 367, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijde, D.; Tanaka, Y.; Fleischmann, R.; Keystone, E.; Kremer, J.; Zerbini, C.; Cardiel, M.H.; Cohen, S.; Nash, P.; Song, Y.W.; et al. Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: Twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum. 2013, 65, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.G.; Blanco, R.; Charles-Schoeman, C.; Wollenhaupt, J.; Zerbini, C.; Benda, B.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; Zwillich, S.H.; et al. Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: A randomised phase 3 trial. Lancet 2013, 381, 451–460. [Google Scholar] [CrossRef]
- Kremer, J.; Li, Z.Z.; Hall, S.; Fleischmann, R.; Genovese, M.; Martin-Mola, E.; Isaacs, J.D.; Gruben, D.; Wallenstein, G.; Krishnaswami, S.; et al. Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: A randomized trial. Ann. Intern. Med. 2013, 159, 253–261. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Aho, K.; Koskenvuo, M.; Tuominen, J.; Kaprio, J. Occurrence of rheumatoid arthritis in a nationwide series of twins. J. Rheumatol. 1986, 13, 899–902. [Google Scholar] [PubMed]
- Silman, A.A.; MacGregor, A.A.; Thomson, W.; Holligan, S.; Carthy, D.; Farhan, A.; Ollier, W.E. Twin concordance rates for rheumatoid arthritis: Results from a nationwide study. Br. J. Rheumatol. 1993, 32, 903–907. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, A.A.; Snieder, H.; Rigby, A.A.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Hemminki, K.; Li, X.; Sundquist, J.; Sundquist, K. Familial associations of rheumatoid arthritis with autoimmune diseases and related conditions. Arthritis Rheum. 2009, 60, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gay, M.A.; Garcia-Porrua, C.; Hajeer, A.H. Influence of human leukocyte antigen-DRB1 on the susceptibility and severity of rheumatoid arthritis. Semin. Arthritis Rheum. 2002, 31, 355–360. [Google Scholar] [CrossRef] [PubMed]
- van Gaalen, F.F.; van Aken, J.; Huizinga, T.T.; Schreuder, G.G.; Breedveld, F.F.; Zanelli, E.; van Venrooij, W.J.; Verweij, C.L.; Toes, R.E.; de Vries, R.R. Association between HLA class II genes and autoantibodies to cyclic citrullinated peptides (CCPs) influences the severity of rheumatoid arthritis. Arthritis Rheum. 2004, 50, 2113–2121. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Okada, Y.; Suzuki, A.; Kochi, Y. Genetic studies of rheumatoid arthritis. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2015, 91, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Saag, K.G.; Cerhan, J.R.; Kolluri, S.; Ohashi, K.; Hunninghake, G.W.; Schwartz, D.A. Cigarette smoking and rheumatoid arthritis severity. Ann. Rheum. Dis. 1997, 56, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, D.; Shepstone, L.; Moots, R.; Lear, J.T.; Lynch, M.P. Heavy cigarette smoking is strongly associated with rheumatoid arthritis (RA), particularly in patients without a family history of RA. Ann. Rheum. Dis. 2001, 60, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Alspaugh, M.A.; Jensen, F.C.; Rabin, H.; Tan, E.M. Lymphocytes transformed by Epstein-–Barr virus. Induction of nuclear antigen reactive with antibody in rheumatoid arthritis. J. Exp. Med. 1978, 147, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Costenbader, K.H.; Karlson, E.W. Epstein–Barr virus and rheumatoid arthritis: Is there a link? Arthritis Res. Ther. 2006, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, K.; Wegner, N.; Yucel-Lindberg, T.; Venables, P.J. Periodontitis in RA-the citrullinated enolase connection. Nat. Rev. Rheumatol. 2010, 6, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, E.D.; Greenwald, R.A.; Kushner, L.J.; Weissmann, G. Hypothesis: The humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation 2004, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Accardo, S. Sex hormones, HLA and rheumatoid arthritis. Clin. Exp. Rheumatol. 1991, 9, 641–646. [Google Scholar] [PubMed]
- Cutolo, M.; Villaggio, B.; Craviotto, C.; Pizzorni, C.; Seriolo, B.; Sulli, A. Sex hormones and rheumatoid arthritis. Autoimmun. Rev. 2002, 1, 284–289. [Google Scholar] [CrossRef]
- Cantorna, M.T.; Hayes, C.E.; DeLuca, H.F. 1,25-Dihydroxycholecalciferol inhibits the progression of arthritis in murine models of human arthritis. J. Nutr. 1998, 128, 68–72. [Google Scholar] [PubMed]
- Merlino, L.L.; Curtis, J.; Mikuls, T.T.; Cerhan, J.J.; Criswell, L.L.; Saag, K.K. Iowa Women’s Health Study. Vitamin D intake is inversely associated with rheumatoid arthritis: Results from the Iowa Women’s Health Study. Arthritis Rheum. 2004, 50, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Kallberg, H.; Lundberg, I.; Sjogren, B.; Klareskog, L.; Alfredsson, L. EIRA Study Group. Silica exposure is associated with increased risk of developing rheumatoid arthritis: Results from the Swedish EIRA study. Ann. Rheum. Dis. 2005, 64, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Mimura, T. The Mechanisms Underlying chronic inflammation in rheumatoid arthritis from the perspective of the epigenetic landscape. J. Immunol. Res. 2016, 2016, 6290682. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Mimura, T. The histone modification code in the pathogenesis of autoimmune diseases. Mediat. Inflamm. 2017, 2017, 2608605. [Google Scholar] [CrossRef] [PubMed]
- Noss, E.H.; Brenner, M.B. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol. Rev. 2008, 223, 252–270. [Google Scholar] [CrossRef] [PubMed]
- Filer, A. The fibroblast as a therapeutic target in rheumatoid arthritis. Curr. Opin. Pharmacol. 2013, 13, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Frisenda, S.; Perricone, C.; Valesini, G. Cartilage as a target of autoimmunity: A thin layer. Autoimmun. Rev. 2013, 12, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, H.; Matsuda, Y.; Tanaka, M.; Sendo, W.; Nakajima, H.; Taniguchi, A.; Kamatani, N. Serum matrix metalloproteinase 3 as a predictor of the degree of joint destruction during the six months after measurement, in patients with early rheumatoid arthritis. Arthritis Rheum. 2000, 43, 852–858. [Google Scholar] [CrossRef]
- Green, M.M.; Gough, A.A.; Devlin, J.; Smith, J.; Astin, P.; Taylor, D.; Emery, P. Serum MMP-3 and MMP-1 and progression of joint damage in early rheumatoid arthritis. Rheumatology 2003, 42, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, M.; Inoue, E.; Nakajima, A.; Hara, M.; Tomatsu, T.; Kamatani, N.; Yamanaka, H. Elevation of serum matrix metalloproteinase-3 as a predictive marker for the long-term disability of rheumatoid arthritis patients in a prospective observational cohort IORRA. Mod. Rheumatol. 2007, 17, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Litinsky, I.; Paran, D.; Levartovsky, D.; Wigler, I.; Kaufman, I.; Yaron, I.; Caspi, D.; Elkayam, O. The effects of leflunomide on clinical parameters and serum levels of IL-6, IL-10, MMP-1 and MMP-3 in patients with resistant rheumatoid arthritis. Cytokine 2006, 33, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Catrina, A.A.; Lampa, J.; Ernestam, S.; af Klint, E.; Bratt, J.; Klareskog, L.; Ulfgren, A.K. Anti-tumour necrosis factor (TNF)-α therapy (etanercept) down-regulates serum matrix metalloproteinase (MMP)-3 and MMP-1 in rheumatoid arthritis. Rheumatology 2002, 41, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.M.; Manning, H.H.; Jain, A.; Troeberg, L.; Dudhia, J.; Essex, D.; Sandison, A.; Seiki, M.; Nanchahal, J.; Nagase, H.; et al. Membrane type 1 matrix metalloproteinase is a crucial promoter of synovial invasion in human rheumatoid arthritis. Arthritis Rheum. 2009, 60, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Sabeh, F.; Fox, D.; Weiss, S.J. Membrane-type I matrix metalloproteinase-dependent regulation of rheumatoid arthritis synoviocyte function. J. Immunol. 2010, 184, 6396–6406. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Williams, R.O.; Dransfield, D.T.; Nixon, A.E.; Sandison, A.; Itoh, Y. Selective inhibition of membrane type 1 matrix metalloproteinase abrogates progression of experimental inflammatory arthritis: Synergy with tumor necrosis factor blockade. Arthritis Rheumatol. 2016, 68, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.H.; Starr, A.E.; Kappelhoff, R.; Yan, R.; Roberts, C.R.; Overall, C.M. Matrix metalloproteinase 8 deficiency in mice exacerbates inflammatory arthritis through delayed neutrophil apoptosis and reduced caspase 11 expression. Arthritis Rheum. 2010, 62, 3645–3655. [Google Scholar] [CrossRef] [PubMed]
- Itoh, T.; Matsuda, H.; Tanioka, M.; Kuwabara, K.; Itohara, S.; Suzuki, R. The role of matrix metalloproteinase-2 and matrix metalloproteinase-9 in antibody-induced arthritis. J. Immunol. 2002, 169, 2643–2647. [Google Scholar] [CrossRef] [PubMed]
- Mudgett, J.J.; Hutchinson, N.N.; Chartrain, N.N.; Forsyth, A.A.; McDonnell, J.; Singer, I.I.; Bayne, E.K.; Flanagan, J.; Kawka, D.; Shen, C.F.; et al. Susceptibility of stromelysin 1-deficient mice to collagen-induced arthritis and cartilage destruction. Arthritis Rheum. 1998, 41, 110–121. [Google Scholar] [CrossRef]
- Brown, P.D. Clinical studies with matrix metalloproteinase inhibitors. APMIS 1999, 107, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.D.; Thiele, G.M.; Tian, J.; Wang, D. The development of novel therapies for rheumatoid arthritis. Expert Opin. Ther. Pat. 2008, 18, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Wood, N.D.; Aitken, M.; Durston, S.; Harris, S.; McClelland, G.R.; Sharp, S. Cartilage protective agent (CPA) Ro 32-3555, a new matrix metalloproteinase inhibitor for the treatment of rheumatoid arthritis. Agents Actions Suppl. 1998, 49, 49–55. [Google Scholar] [PubMed]
- Hemmings, F.J.; Farhan, M.; Rowland, J.; Banken, L.; Jain, R. Tolerability and pharmacokinetics of the collagenase-selective inhibitor Trocade in patients with rheumatoid arthritis. Rheumatology 2001, 40, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.E.; Bishop, J.; Bottomley, K.K.; Bradshaw, D.; Brewster, M.; Broadhurst, M.J.; Brown, P.A.; Budd, J.M.; Elliott, L.; Greenham, A.K.; et al. Ro 32-3555, an orally active collagenase inhibitor, prevents cartilage breakdown in vitro and in vivo. Br. J. Pharmacol. 1997, 121, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Brewster, M.; Lewis, E.J.; Wilson, K.L.; Greenham, A.K.; Bottomley, K.M. Ro 32-3555, an orally active collagenase selective inhibitor, prevents structural damage in the STR/ORT mouse model of osteoarthritis. Arthritis Rheum. 1998, 41, 1639–1644. [Google Scholar] [CrossRef]
- Close, D.R. Matrix metalloproteinase inhibitors in rheumatic diseases. Ann. Rheum. Dis. 2001, 60, iii62–iii67. [Google Scholar] [PubMed]
- Keystone, E. Treatments no longer in development for rheumatoid arthritis. Ann. Rheum. Dis. 2002, 61, ii43–ii45. [Google Scholar] [CrossRef] [PubMed]
- Jungel, A.; Ospelt, C.; Lesch, M.; Thiel, M.; Sunyer, T.; Schorr, O.; Michel, B.A.; Gay, R.E.; Kolling, C.; Flory, C.; et al. Effect of the oral application of a highly selective MMP-13 inhibitor in three different animal models of rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef] [PubMed]
- Blobel, C.P. Metalloprotease-disintegrins: Links to cell adhesion and cleavage of TNF α and Notch. Cell 1997, 90, 589–592. [Google Scholar] [CrossRef]
- Moss, M.M.; Jin, S.S.; Milla, M.M.; Bickett, D.D.; Burkhart, W.; Carter, H.L.; Chen, W.J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, M.M.; Burn, T.T.; Pratta, M.M.; Abbaszade, I.; Hollis, J.J.; Liu, R.; Rosenfeld, S.A.; Copeland, R.A.; Decicco, C.P.; Wynn, R.; et al. Purification and cloning of aggrecanase-1: A member of the ADAMTS family of proteins. Science 1999, 284, 1664–1666. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, M.; Pratta, M.; Liu, R.R.; Abbaszade, I.; Ross, H.; Burn, T.; Arner, E. The thrombospondin motif of aggrecanase-1 (ADAMTS-4) is critical for aggrecan substrate recognition and cleavage. J. Biol. Chem. 2000, 275, 25791–25797. [Google Scholar] [CrossRef] [PubMed]
- Abbaszade, I.; Liu, R.R.; Yang, F.; Rosenfeld, S.S.; Ross, O.O.; Link, J.R.; Ellis, D.M.; Tortorella, M.D.; Pratta, M.A.; Hollis, J.M.; et al. Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J. Biol. Chem. 1999, 274, 23443–23450. [Google Scholar] [CrossRef] [PubMed]
- Dancevic, C.M.; McCulloch, D.R. Current and emerging therapeutic strategies for preventing inflammation and aggrecanase-mediated cartilage destruction in arthritis. Arthritis Res. Ther. 2014, 16, 429. [Google Scholar] [CrossRef] [PubMed]
- Stanton, H.; Rogerson, F.F.; East, C.C.; Golub, S.S.; Lawlor, K.K.; Meeker, C.T.; Little, C.B.; Last, K.; Farmer, P.J.; Campbell, I.K.; et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005, 434, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Golbabapour, S.; Abdulla, M.A.; Hajrezaei, M. A concise review on epigenetic regulation: Insight into molecular mechanisms. Int. J. Mol. Sci. 2011, 12, 8661–8694. [Google Scholar] [CrossRef] [PubMed]
- Bassett, A.; Cooper, S.; Wu, C.; Travers, A. The folding and unfolding of eukaryotic chromatin. Curr. Opin. Genet. Dev. 2009, 19, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Fann, M.; Wersto, R.; Weng, N.P. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: Perforin and granzyme B). J. Immunol. 2008, 180, 8102–8108. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Wang, Z.; Zang, C.; Wood, W.W.; Schones, D., 3rd.; Cui, K.; Roh, T.Y.; Lhotsky, B.; Wersto, R.P.; Peng, W.; et al. Genome-wide analysis of histone methylation reveals chromatin state-based regulation of gene transcription and function of memory CD8+ T cells. Immunity 2009, 30, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.P.; Araki, Y.; Subedi, K. The molecular basis of the memory T cell response: Differential gene expression and its epigenetic regulation. Nat. Rev. Immunol. 2012, 12, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.J.; Schones, D.D.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Mundade, R.; Lange, K.C.; Lu, T. Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle 2014, 13, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.; Shilatifard, A. Posttranslational modifications of histones by methylation. Adv. Protein Chem. 2004, 67, 201–222. [Google Scholar] [PubMed]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Reinberg, D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev. 2001, 15, 2343–2360. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.T.; Schones, D.D.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Gregoire, S. Class II histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 2005, 25, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhu, S.; Wu, C.; Kang, J. Histone deacetylase (HDAC) 10 suppresses cervical cancer metastasis through inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J. Biol. Chem. 2013, 288, 28021–28033. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.S.; Kern, C.C.; Kimbrough, D.; Addy, B.; Kasiganesan, H.; Rivers, W.T.; Patel, R.K.; Chou, J.C.; Spinale, F.G.; Mukherjee, R.; et al. Inhibition of class I histone deacetylase activity represses matrix metalloproteinase-2 and -9 expression and preserves LV function postmyocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1391–H1401. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Shan, Y.; Mishra, M. Dynamic DNA methylation of matrix metalloproteinase-9 in the development of diabetic retinopathy. Lab. Investig. 2016, 96, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.F.; Hsi, E.; Huang, L.C.; Liao, Y.C.; Juo, S.H.; Lin, R.T. Methylation in the matrix metalloproteinase-2 gene is associated with cerebral ischemic stroke. J. Investig. Med. 2017, 65, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Campos, K.; Gomes, C.C.; Farias, L.C.; Silva, R.M.; Letra, A.; Gomez, R.S. DNA methylation of MMP9 is associated with high levels of MMP-9 messenger RNA in periapical inflammatory lesions. J. Endod. 2016, 42, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Mendell, J.T. microRNAs in vertebrate physiology and human disease. Annu. Rev. Genom. Hum. Genet. 2007, 8, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Zavolan, M. Identification and consequences of miRNA-target interactions--beyond repression of gene expression. Nat. Rev. Genet. 2014, 15, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhang, Z.; Yang, H.; Lin, Q.; Han, C.; Qin, X. The involvement of miR-29b-3p in arterial calcification by targeting matrix metalloproteinase-2. Biomed Res. Int. 2017, 2017, 6713606. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.P.; Tang, C.C.; Lin, L.L.; Tsai, C.C.; Chu, C.C.; Lin, T.H.; Huang, Y.L. Thrombospondin-2 promotes prostate cancer bone metastasis by the up-regulation of matrix metalloproteinase-2 through down-regulating miR-376c expression. J. Hematol. Oncol. 2017, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Q.; Zhang, Z.H.; Zheng, Y.F.; Feng, S.Q. Dysregulated miR-133a mediates loss of type II collagen by directly targeting matrix metalloproteinase 9 (MMP9) in human intervertebral disc degeneration. Spine (Phila Pa 1976) 2016, 41, E717–E724. [Google Scholar] [CrossRef] [PubMed]
- Li, H.R.; Cui, Q.; Dong, Z.Y.; Zhang, J.H.; Li, H.Q.; Zhao, L. Downregulation of miR-27b is involved in loss of type II collagen by directly targeting matrix metalloproteinase 13 (MMP13) in human intervertebral disc degeneration. Spine (Phila Pa 1976) 2016, 41, E116–E123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.W.; Chen, Y.Y.; Meng, H.H.; Du, J.J.; Luan, G.G.; Wang, H.Q.; Yang, M.W.; Luo, Z.J. Role of miR-155 in the regulation of MMP-16 expression in intervertebral disc degeneration. J. Orthop. Res. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Xia, T.; Zhao, Y.; Zhou, W.; Wu, N.; Liu, X.; Ding, Q.; Zha, X.; Sha, J.; Wang, S. Downregulation of miR-106b induced breast cancer cell invasion and motility in association with overexpression of matrix metalloproteinase 2. Cancer Sci. 2014, 105, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef] [PubMed]
- Trenkmann, M.; Brock, M.; Gay, R.R.; Kolling, C.; Speich, R.; Michel, B.A.; Gay, S.; Huber, L.C. Expression and function of EZH2 in synovial fibroblasts: Epigenetic repression of the Wnt inhibitor SFRP1 in rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Karouzakis, E.; Gay, R.E.; Michel, B.A.; Gay, S.; Neidhart, M. DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009, 60, 3613–3622. [Google Scholar] [CrossRef] [PubMed]
- Stanczyk, J.; Pedrioli, D.D.; Brentano, F.; Sanchez-Pernaute, O.; Kolling, C.; Gay, R.E.; Detmar, M.; Gay, S.; Kyburz, D. Altered expression of microRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008, 58, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Stanczyk, J.; Ospelt, C.; Karouzakis, E.; Filer, A.; Raza, K.; Kolling, C.; Gay, R.E.; Detmar, M.; Gay, S.; Kyburz, D. Altered expression of microRNA-203 in rheumatoid arthritis synovial fibroblasts and its role in fibroblast activation. Arthritis Rheum. 2011, 63, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Yu, P.; Liu, Y.; Wang, S.; Li, R.; Shi, J.; Zhang, X.; Li, Y.; Sun, X.; Zhou, B.; et al. Upregulated microRNA-155 expression in peripheral blood mononuclear cells and fibroblast-like synoviocytes in rheumatoid arthritis. Clin. Dev. Immunol. 2013, 2013, 296139. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.E.; Hammaker, D.; Boyle, D.L.; Firestein, G.S. Regulation of c-Jun phosphorylation by the I κ B kinase-ε complex in fibroblast-like synoviocytes. J. Immunol. 2005, 174, 6424–6430. [Google Scholar] [CrossRef] [PubMed]
- Philippe, L.; Alsaleh, G.; Suffert, G.; Meyer, A.; Georgel, P.; Sibilia, J.; Wachsmann, D.; Pfeffer, S. TLR2 expression is regulated by microRNA miR-19 in rheumatoid fibroblast-like synoviocytes. J. Immunol. 2012, 188, 454–461. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Pseudonyms | Collagen Substrates | Additional Substrates |
|---|---|---|---|
| MMP-1 | Collagenase-1 | I, II, III, VII, VIII, X | Aggrecan, Gelatin |
| MMP-2 | Gelatinase A | I, II, III, IV, V, VII, X, XI | Aggrecan, Elastin, Fibronectin, Gelatin, Laminin |
| MMP-3 | Stromelysin-1 | II, III, IV, IX, X, XI | Aggrecan, Elastin, Fibronectin, Gelatin, Laminin |
| MMP-4 | Identified as MMP-3 | - | - |
| MMP-5 | Identified as MMP-2 | - | - |
| MMP-6 | Identified as MMP-3 | - | - |
| MMP-7 | Matrilysin | IV, X | Aggrecan, Elastin, Fibronectin, Gelatin, Laminin |
| MMP-8 | Collagenase-2 | I, II, III, V, VII, VIII, X | Aggrecan, Elastin, Fibronectin, Gelatin, Laminin |
| MMP-9 | Gelatinase B | IV, V, VII, X, XIV | Aggrecan, Elastin, Fibronectin, Gelatin |
| MMP-10 | Stromelysin-2 | III, IV, V | Aggrecan, Elastin, Fibronectin, Gelatin, Laminin |
| MMP-11 | Stromelysin-3 | Aggrecan, Fibronectin, Laminin | - |
| MMP-12 | Macrophage | IV | Elastin, Fibronectin, Gelatin, Laminin |
| MMP-13 | Collagenase-3 | I, II, III, IV | Aggrecan, Gelatin |
| MMP-14 | MT1-MMP | I, II, III | Aggrecan, Elastin, Fibronectin, Gelatin, Laminin |
| MMP-15 | MT2-MMP | Fibronectin, Gelatin, Laminin | - |
| MMP-16 | MT3-MMP | - | - |
| MMP-17 | MT4-MMP | Fibrin, Gelatin | - |
| MMP-18 | Identified as MMP-19 | - | - |
| MMP-19 | RASI-1 | IV | Aggrecan, Fibronectin, Gelatin, Laminin, COMP |
| MMP-20 | Enamelysin | Aggrecan, Amelogenin, COMP | - |
| MMP-21 | X-MMP | - | - |
| MMP-22 | C-MMP | - | - |
| MMP-23 | Identified as MMP-23 | - | - |
| MMP-24 | MT5-MMP | - | - |
| MMP-25 | MT6-MMP | IV | Fibronectin, Gelatin, Laminin |
| MMP-26 | Matrilysin-2 | IV | Fibronectin, Gelatin |
| MMP-27 | - | - | - |
| MMP-28 | Epilysin | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araki, Y.; Mimura, T. Matrix Metalloproteinase Gene Activation Resulting from Disordred Epigenetic Mechanisms in Rheumatoid Arthritis. Int. J. Mol. Sci. 2017, 18, 905. https://doi.org/10.3390/ijms18050905
Araki Y, Mimura T. Matrix Metalloproteinase Gene Activation Resulting from Disordred Epigenetic Mechanisms in Rheumatoid Arthritis. International Journal of Molecular Sciences. 2017; 18(5):905. https://doi.org/10.3390/ijms18050905
Chicago/Turabian StyleAraki, Yasuto, and Toshihide Mimura. 2017. "Matrix Metalloproteinase Gene Activation Resulting from Disordred Epigenetic Mechanisms in Rheumatoid Arthritis" International Journal of Molecular Sciences 18, no. 5: 905. https://doi.org/10.3390/ijms18050905
APA StyleAraki, Y., & Mimura, T. (2017). Matrix Metalloproteinase Gene Activation Resulting from Disordred Epigenetic Mechanisms in Rheumatoid Arthritis. International Journal of Molecular Sciences, 18(5), 905. https://doi.org/10.3390/ijms18050905