
Hepatitis E Virus Genotypes and Evolution: Emergence of Camel Hepatitis E Variants

 ,
,  and
and 
Abstract
:1. Introduction
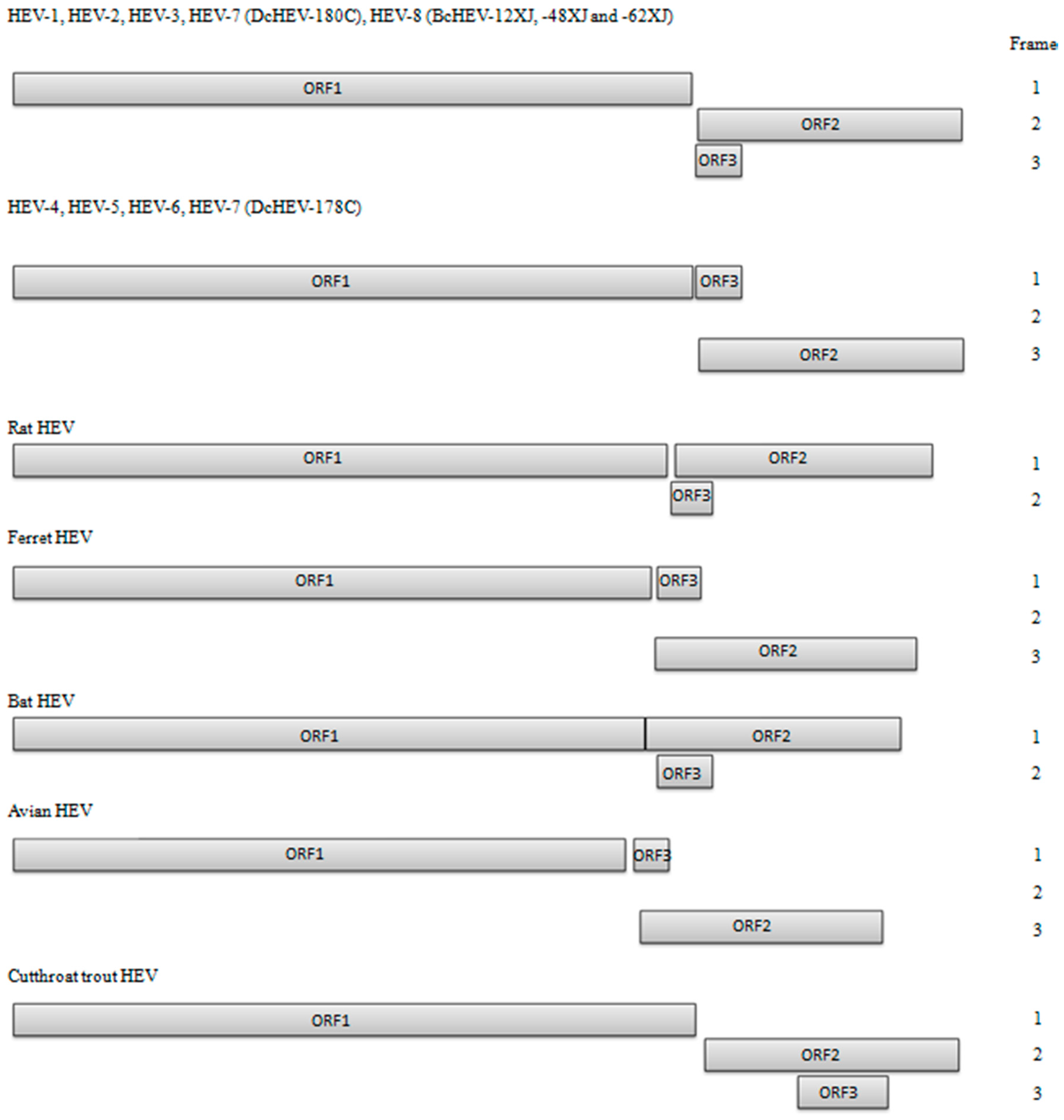
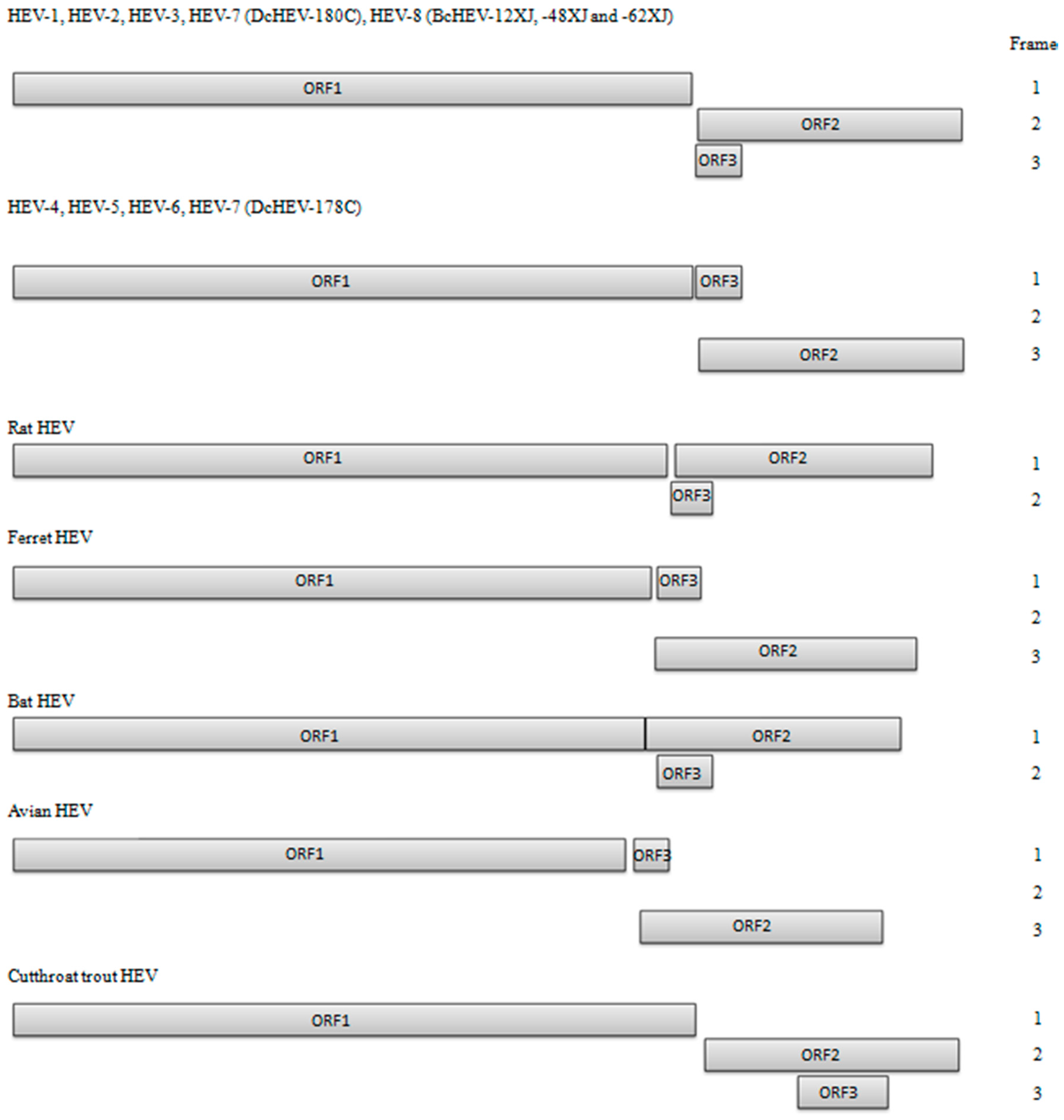
2. HEV Genomic Organization
3. Hepatitis E Virus (HEV) Taxonomy: In Constant Evolution
4. Beyond the Species Level: HEV Genotypes and Subgenotypes
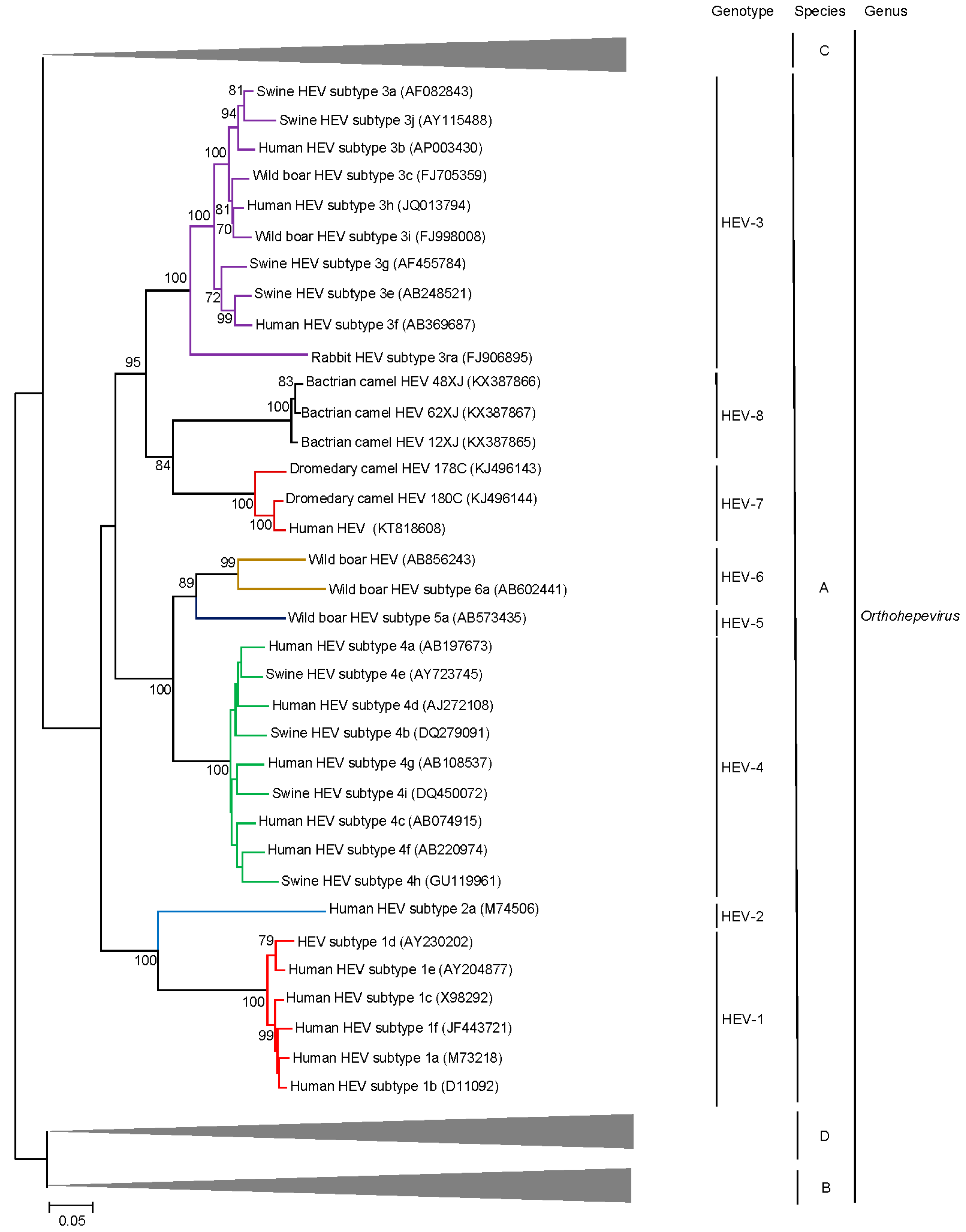
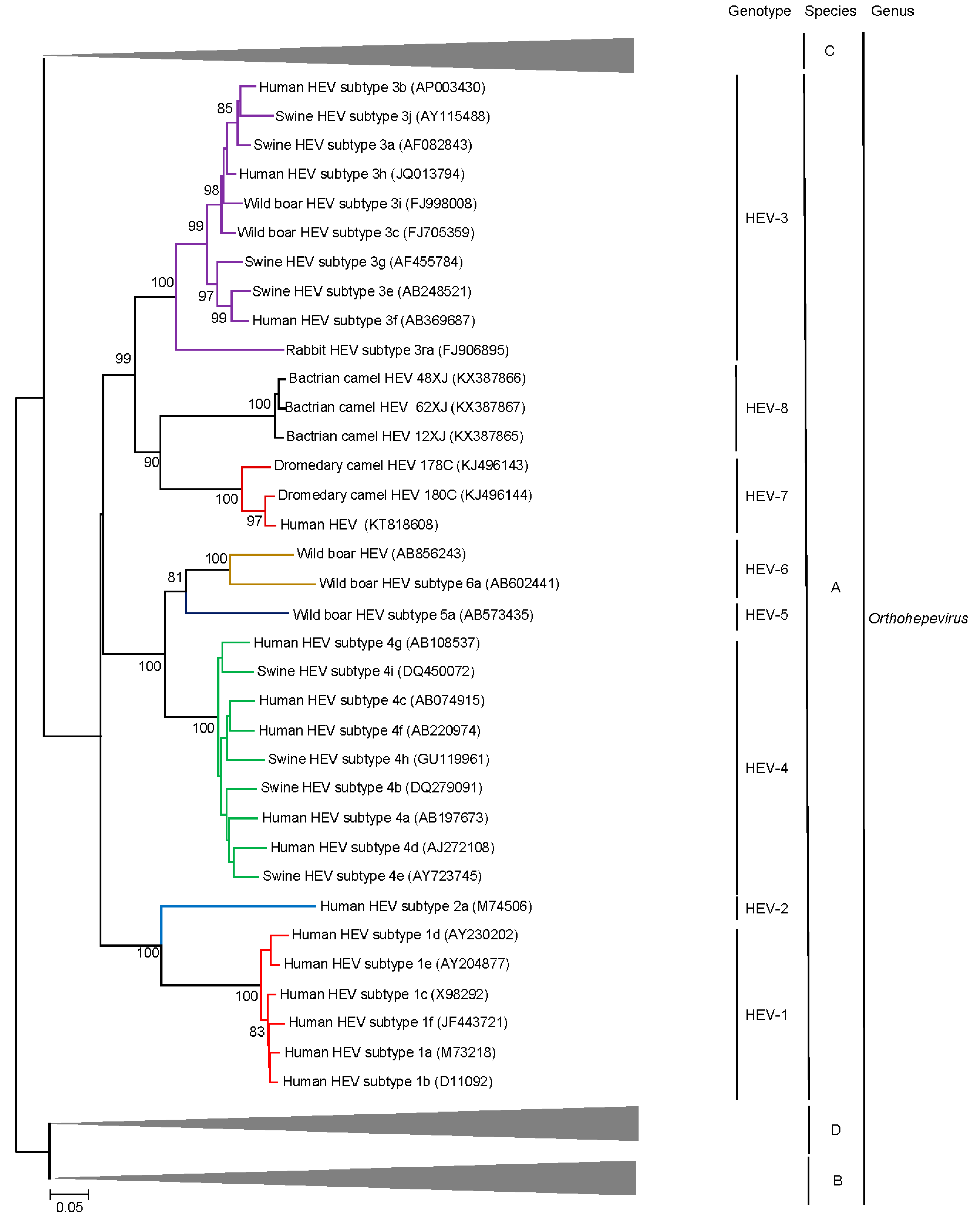
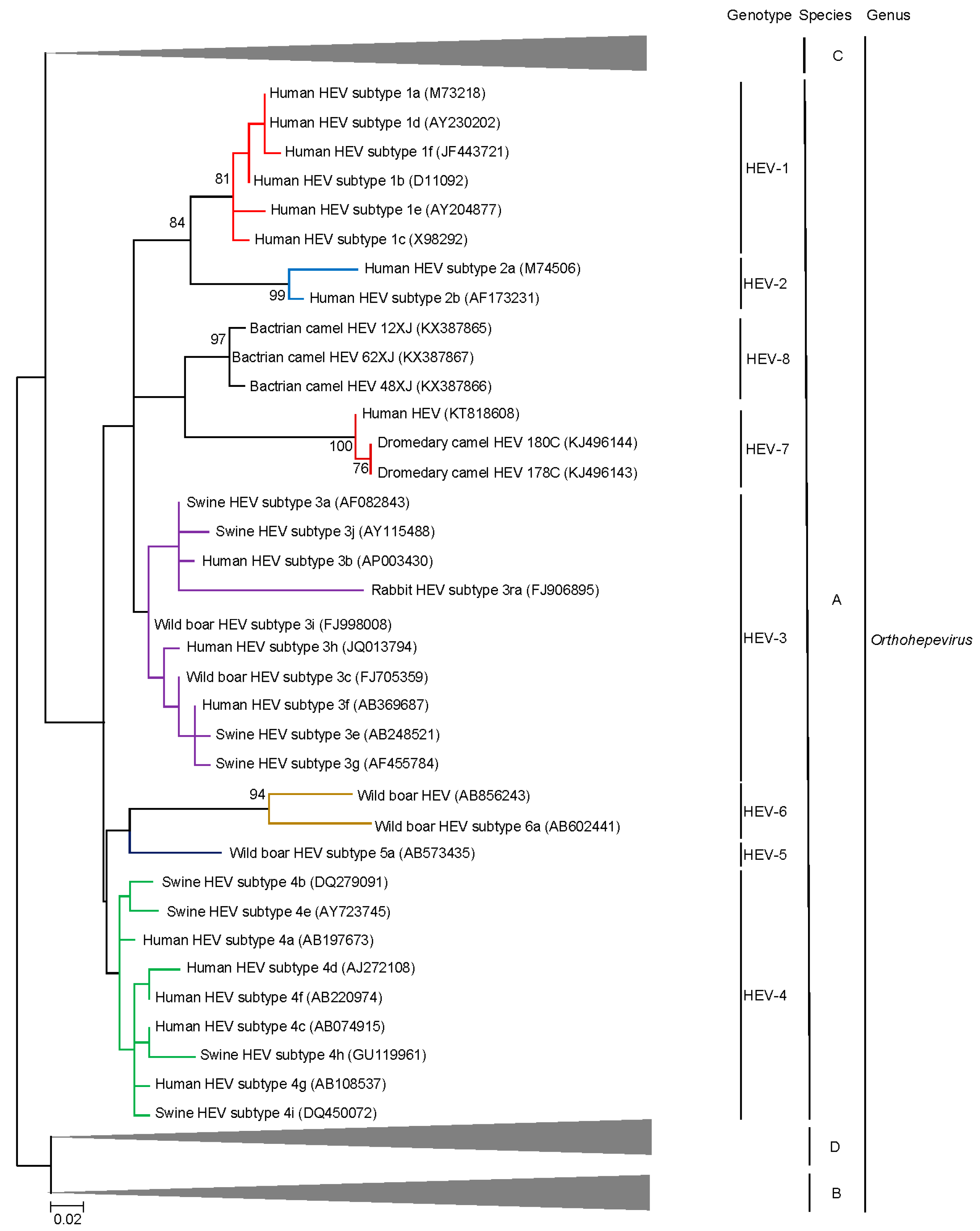
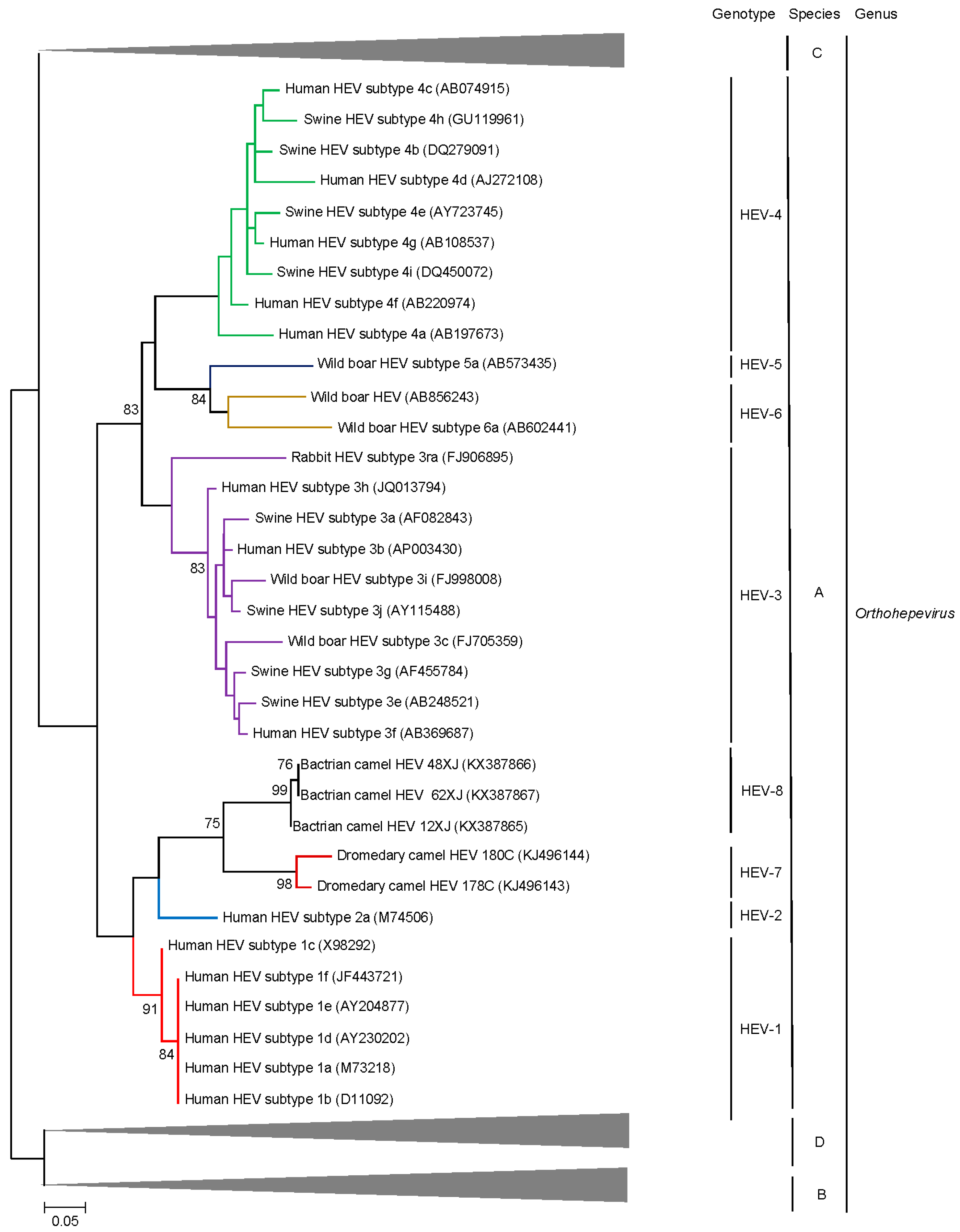
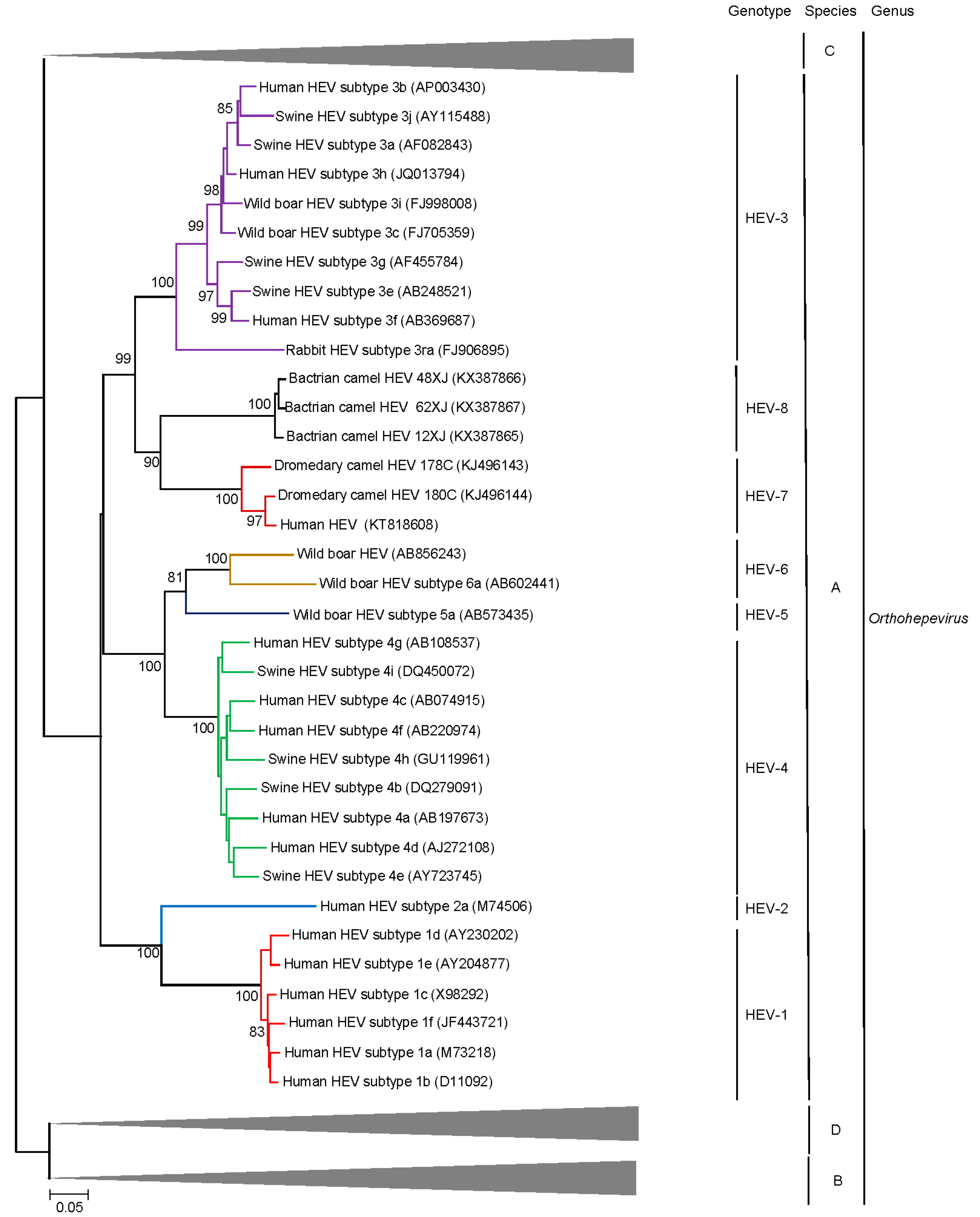
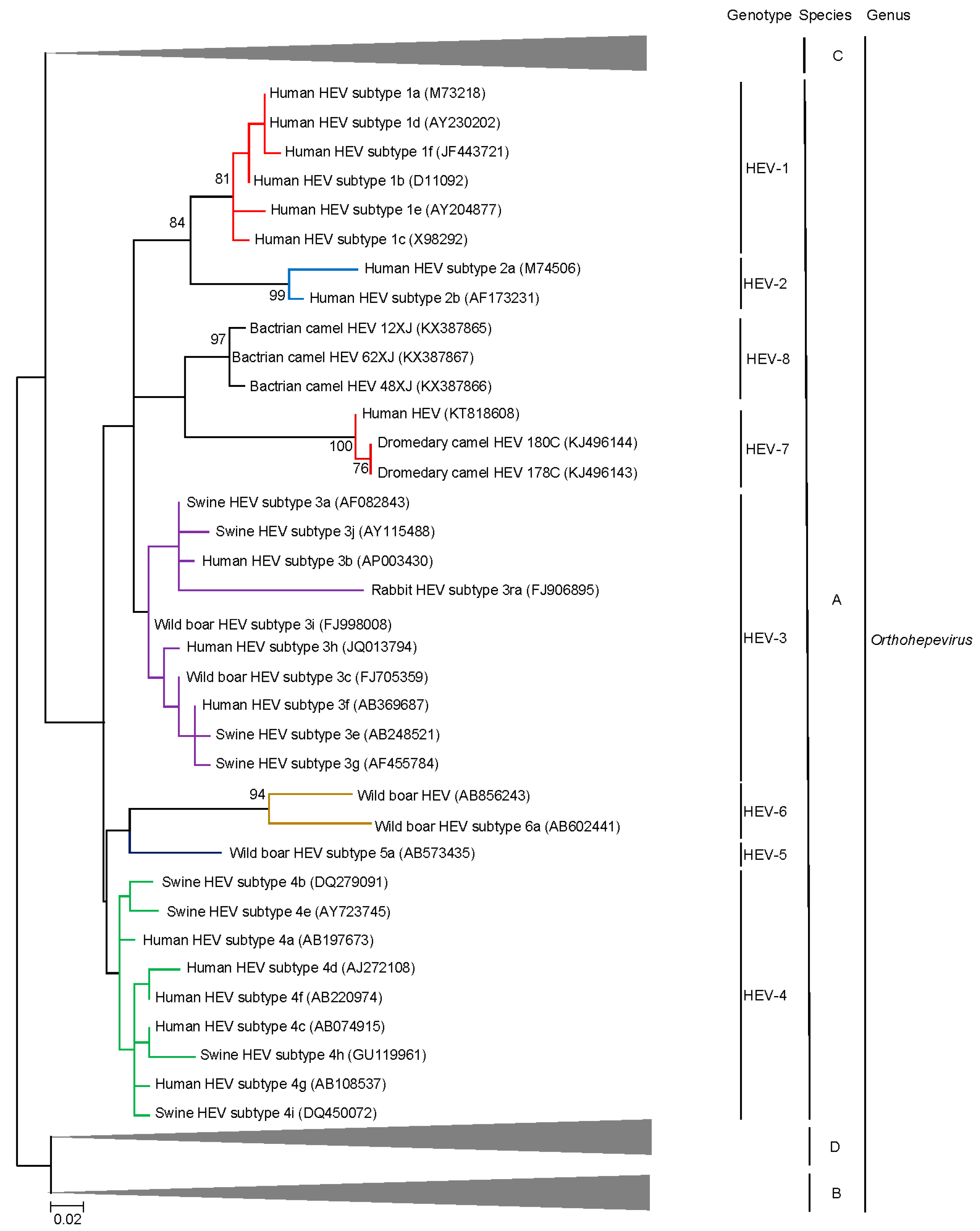
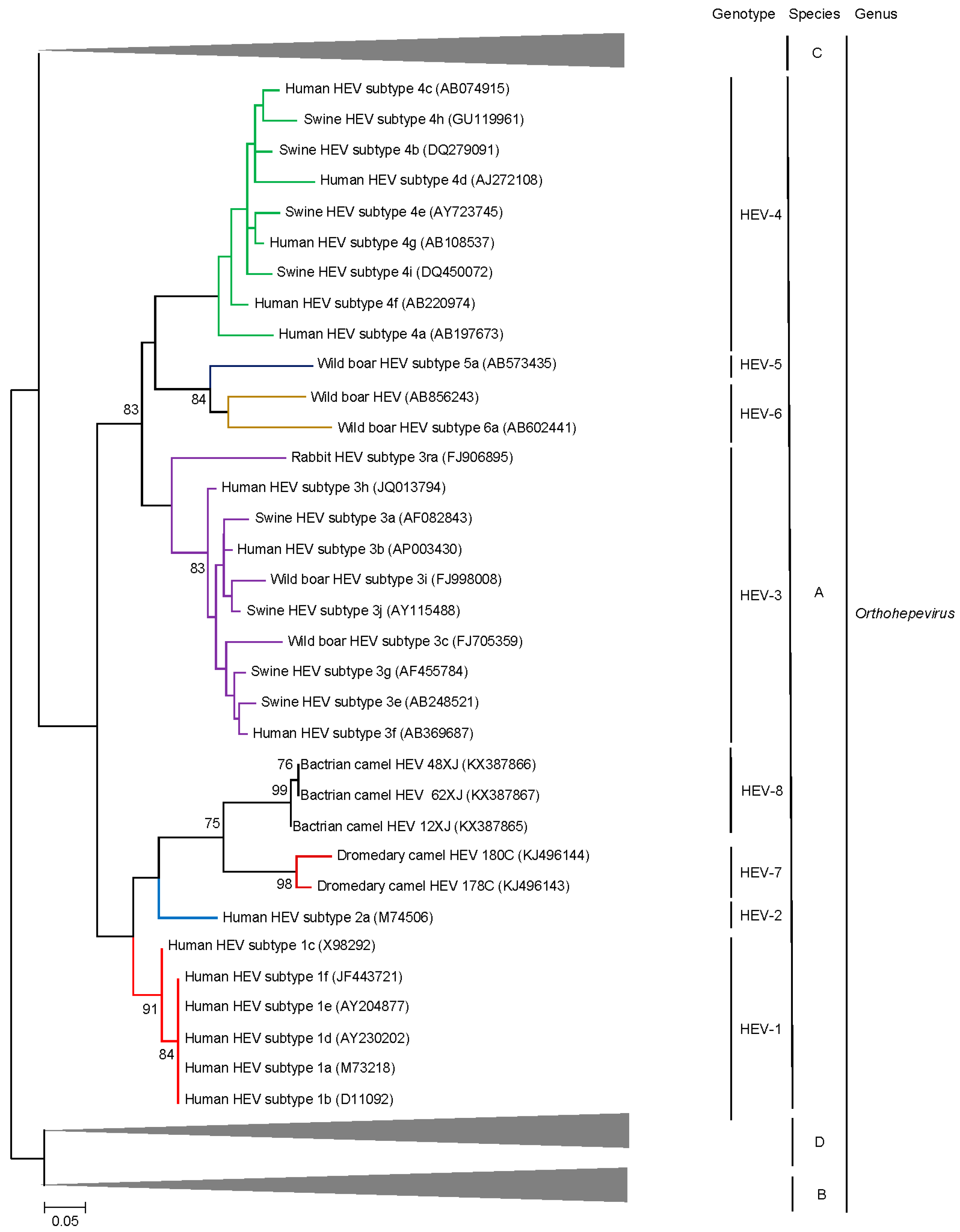
5. Discovery of Camel HEV Genotypes
6. Delving into the Sequence: Recent Insights into HEV Molecular Epidemiology and Evolution
7. Conclusions: Future Directions
Author Contributions
Conflicts of Interest
References
- WHO. Hepatitis E Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs280/en/ (accessed on 15 February 2017).
- Ren, X.; Wu, P.; Wang, L.; Geng, M.; Zeng, L.; Zhang, J.; Xia, N.; Lai, S.; Dalton, H.R.; Cowling, B.J.; et al. Changing epidemiology of hepatitis A and hepatitis E viruses in China, 1990–2014. Emerg. Infect. Dis. 2017, 23, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zhao, Y.; Zhang, X.; Wang, B.; Liu, P. Case-fatality risk of pregnant women with acute viral hepatitis type E: A systematic review and meta-analysis. Epidemiol. Infect. 2016, 144, 2098–2106. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Selves, J.; Mansuy, J.M.; Ouezzani, L.; Peron, J.M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Giordani, M.T.; Fabris, P.; Brunetti, E.; Goblirsch, S.; Romano, L. Hepatitis E and lymphocytic leukemia in Man, Italy. Emerg. Infect. Dis. 2013, 19, 2054–2056. [Google Scholar] [CrossRef] [PubMed]
- Neukam, K.; Barreiro, P.; Macias, J.; Avellon, A.; Cifuentes, C.; Martin-Carbonero, L.; Echevarria, J.M.; Vargas, J.; Soriano, V.; Pineda, J.A. Chronic hepatitis E in HIV patients: Rapid progression to cirrhosis and response to oral ribavirin. Clin. Infect. Dis. 2013, 57, 465–468. [Google Scholar] [CrossRef] [PubMed]
- WHO. The Global Prevalence of Hepatitis E Viral Prevalence and Susceptibility: A Systematic Review. Available online: http://apps.who.int/iris/bitstream/10665/70513/1/WHO_IVB_10.14_eng.pdf?ua=1 (accessed on 15 February 2017).
- Sridhar, S.; Lau, S.K.; Woo, P.C. Hepatitis E: A disease of reemerging importance. J. Formos. Med. Assoc. 2015, 114, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Tei, S.; Kitajima, N.; Takahashi, K.; Mishiro, S. Zoonotic transmission of hepatitis E virus from deer to human beings. Lancet 2003, 362, 371–373. [Google Scholar] [CrossRef]
- Lee, G.H.; Tan, B.H.; Teo, E.C.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic infection with camelid hepatitis E virus in a liver transplant recipient who regularly consumes camel meat and milk. Gastroenterology 2016, 150, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jeong, S.H.; Kim, J.Y.; Song, J.C.; Lee, J.H.; Kim, J.W.; Yun, H.; Kim, J.S. The first case of genotype 4 hepatitis E related to wild boar in South Korea. J. Clin. Virol. 2011, 50, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Li, T.C.; Chijiwa, K.; Sera, N.; Ishibashi, T.; Etoh, Y.; Shinohara, Y.; Kurata, Y.; Ishida, M.; Sakamoto, S.; Takeda, N.; et al. Hepatitis E virus transmission from wild boar meat. Emerg. Infect. Dis. 2005, 11, 1958–1960. [Google Scholar] [CrossRef] [PubMed]
- Izopet, J.; Dubois, M.; Bertagnoli, S.; Lhomme, S.; Marchandeau, S.; Boucher, S.; Kamar, N.; Abravanel, F.; Guerin, J.L. Hepatitis E virus strains in rabbits and evidence of a closely related strain in humans, France. Emerg. Infect. Dis. 2012, 18, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Moradpour, D.; Neyts, J.; Gouttenoire, J. Update on hepatitis E virology: Implications for clinical practice. J. Hepatol. 2016, 65, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Torian, U.; Nguyen, H.; Emerson, S.U. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol. 2006, 80, 5919–5926. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Varma, S.P. Hepatitis E: Molecular virology and pathogenesis. J. Clin. Exp. Hepatol. 2013, 3, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Kannan, H.; Fan, S.; Patel, D.; Bossis, I.; Zhang, Y.J. The hepatitis E virus open reading frame 3 product interacts with microtubules and interferes with their dynamics. J. Virol. 2009, 83, 6375–6382. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Takahashi, M.; Jirintai, S.; Tanggis; Kobayashi, T.; Nishizawa, T.; Okamoto, H. The membrane on the surface of hepatitis E virus particles is derived from the intracellular membrane and contains trans-golgi network protein 2. Arch. Virol. 2014, 159, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Kataoka, M.; Ami, Y.; Suzaki, Y.; Kishida, N.; Shirakura, M.; Imai, M.; Asanuma, H.; Takeda, N.; Wakita, T.; et al. Characterization of self-assembled virus-like particles of ferret hepatitis E virus generated by recombinant Baculoviruses. J. Gen. Virol. 2013, 94, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Kataoka, M.; Liu, Z.; Takeda, N.; Wakita, T.; Li, T.C. Characterization of self-assembled virus-like particles of dromedary camel hepatitis E virus generated by recombinant Baculoviruses. Virus. Res. 2015, 210, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cheng, X.; Dai, X.; Dong, C.; Xu, M.; Liang, J.; Dong, M.; Purdy, M.A.; Meng, J. Rabbit and human hepatitis E virus strains belong to a single serotype. Virus Res. 2013, 176, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dong, C.; Dai, X.; Cheng, X.; Liang, J.; Dong, M.; Purdy, M.A.; Meng, J. Hepatitis E virus isolated from rabbits is genetically heterogeneous but with very similar antigenicity to human HEV. J. Med. Virol. 2013, 85, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, C.; Hagedorn, C.H. Phylogenetic analysis of global hepatitis E virus sequences: Genetic diversity, subtypes and zoonosis. Rev. Med. Virol. 2006, 16, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Schlauder, G.G.; Desai, S.M.; Zanetti, A.R.; Tassopoulos, N.C.; Mushahwar, I.K. Novel hepatitis E virus (HEV) isolates from europe: Evidence for additional genotypes of HEV. J. Med. Virol. 1999, 57, 243–251. [Google Scholar] [CrossRef]
- Schlauder, G.G.; Mushahwar, I.K. Genetic heterogeneity of hepatitis E virus. J. Med. Virol. 2001, 65, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P.; International Committee on Taxonomy of Viruses Hepeviridae Study Group; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Purdy, M.A.; Simmonds, P. Genetic variability and the classification of hepatitis E virus. J. Virol. 2013, 87, 4161–4169. [Google Scholar] [CrossRef] [PubMed]
- Worm, H.C.; van der Poel, W.H.; Brandstatter, G. Hepatitis E: An overview. Microbes Infect. 2002, 4, 657–666. [Google Scholar] [CrossRef]
- Gouvea, V.; Hoke, C.H., Jr.; Innis, B.L. Genotyping of hepatitis E virus in clinical specimens by restriction endonuclease analysis. J. Virol. Methods 1998, 70, 71–78. [Google Scholar] [CrossRef]
- Fan, J. Open reading frame structure analysis as a novel genotyping tool for hepatitis E virus and the subsequent discovery of an inter-genotype recombinant. J. Gen. Virol. 2009, 90, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- ICTV. Virus Taxonomy: 2015 Release. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 17 February 2017).
- Batts, W.; Yun, S.; Hedrick, R.; Winton, J. A novel member of the family Hepeviridae from cutthroat trout (Oncorhynchus clarkii). Virus. Res. 2011, 158, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Q.; He, C.; Zhang, L.; Li, J.; Zhang, W.; Cao, W.; Lv, Y.G.; Liu, Z.; Zhang, J.X.; et al. Recombination and natural selection in hepatitis E virus genotypes. J. Med. Virol. 2012, 84, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J. Expanding host range and cross-species infection of hepatitis E virus. PLoS Pathog. 2016, 12, e1005695. [Google Scholar] [CrossRef] [PubMed]
- Fierro, N.A.; Realpe, M.; Meraz-Medina, T.; Roman, S.; Panduro, A. Hepatitis E virus: An ancient hidden enemy in Latin America. World J. Gastroenterol. 2016, 22, 2271–2283. [Google Scholar] [PubMed]
- Huang, C.C.; Nguyen, D.; Fernandez, J.; Yun, K.Y.; Fry, K.E.; Bradley, D.W.; Tam, A.W.; Reyes, G.R. Molecular cloning and sequencing of the Mexico isolate of hepatitis E virus (HEV). Virology 1992, 191, 550–558. [Google Scholar] [CrossRef]
- Pavio, N.; Meng, X.J.; Doceul, V. Zoonotic origin of hepatitis E. Curr. Opin. Virol. 2015, 10, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Park, B.J.; Kim, Y.H.; Choi, Y.S.; Ahn, H.S.; Han, S.H.; Choi, I.S. Isolation of hepatitis E virus genotype 4 from patients with acute cryptogenic hepatitis in Korea. J. Clin. Virol. 2017, 89, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Geng, J. Acute hepatitis E virus infection in patients with acute liver failure in China: Not quite an uncommon cause. Hepatology 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, L.; Wei, Y.; Wang, Q.; Tian, Q.; Wang, L.; Zhuang, H. Clinical and virological profiling of sporadic hepatitis E virus infection in China. J. Infect. 2016, 73, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Nishizawa, T.; Sato, H.; Sato, Y.; Jirintai; Nagashima, S.; Okamoto, H. Analysis of the full-length genome of a hepatitis E virus isolate obtained from a wild boar in Japan that is classifiable into a novel genotype. J. Gen. Virol. 2011, 92, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Sato, H.; Naka, K.; Furuya, S.; Tsukiji, H.; Kitagawa, K.; Sonoda, Y.; Usui, T.; Sakamoto, H.; Yoshino, S.; et al. A nationwide survey of hepatitis E virus (HEV) infection in wild boars in Japan: Identification of boar HEV strains of genotypes 3 and 4 and unrecognized genotypes. Arch. Virol. 2011, 156, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Teng, J.L.; Cao, K.Y.; Wernery, U.; Schountz, T.; Chiu, T.H.; Tsang, A.K.; Wong, P.C.; Wong, E.Y.; et al. New hepatitis E virus genotype in Bactrian camels, Xinjiang, China, 2013. Emerg. Infect. Dis. 2016, 22, 2219–2221. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Teng, J.L.; Tsang, A.K.; Joseph, M.; Wong, E.Y.; Tang, Y.; Sivakumar, S.; Xie, J.; Bai, R.; et al. New hepatitis E virus genotype in camels, the Middle East. Emerg. Infect. Dis. 2014, 20, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P.; Izopet, J.; Oliveira-Filho, E.F.; Ulrich, R.G.; Johne, R.; Koenig, M.; Jameel, S.; Harrison, T.J.; Meng, X.J.; et al. Proposed reference sequences for hepatitis E virus subtypes. J. Gen. Virol. 2016, 97, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Ijaz, S.; Tedder, R.S.; Hogema, B.; Zaaijer, H.L.; Izopet, J.; Bradley-Stewart, A.; Gunson, R.; Harvala, H.; Kokki, I.; et al. Variability and pathogenicity of hepatitis E virus genotype 3 variants. J. Gen. Virol. 2015, 96, 3255–3264. [Google Scholar] [CrossRef] [PubMed]
- Ijaz, S.; Said, B.; Boxall, E.; Smit, E.; Morgan, D.; Tedder, R.S. Indigenous hepatitis E in England and Wales from 2003 to 2012: Evidence of an emerging novel phylotype of viruses. J. Infect. Dis. 2014, 209, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Filho, E.F.; Konig, M.; Thiel, H.J. Genetic variability of HEV isolates: Inconsistencies of current classification. Vet. Microbiol. 2013, 165, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Purdy, M.A.; Lara, J.; Khudyakov, Y.E. The hepatitis E virus polyproline region is involved in viral adaptation. PLoS ONE 2012, 7, e35974. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.G.; Netzler, N.E.; White, P.A. Ancient recombination events and the origins of hepatitis E virus. BMC Evol. Biol. 2016, 16, 210. [Google Scholar] [CrossRef] [PubMed]
- Dryden, K.A.; Tihova, M.; Nowotny, N.; Matsui, S.M.; Mendez, E.; Yeager, M. Immature and mature human astrovirus: Structure, conformational changes, and similarities to hepatitis E virus. J. Mol. Biol. 2012, 422, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.; Harper, J.; Dryden, K.A.; Yeager, M.; Arias, C.F.; Mendez, E.; Tao, Y.J. Crystal structure of the human astrovirus capsid protein. J. Virol. 2016, 90, 9008–9017. [Google Scholar] [CrossRef] [PubMed]
- Purdy, M.A.; Khudyakov, Y.E. Evolutionary history and population dynamics of hepatitis E virus. PLoS ONE 2010, 5, e14376. [Google Scholar] [CrossRef] [PubMed]
- Mirazo, S.; Mir, D.; Bello, G.; Ramos, N.; Musto, H.; Arbiza, J. New insights into the hepatitis E virus genotype 3 phylodynamics and evolutionary history. Infect. Genet. Evol. 2016, 43, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Brayne, A.B.; Dearlove, B.L.; Lester, J.S.; Kosakovsky Pond, S.L.; Frost, S.D. Genotype-specific evolution of hepatitis E virus. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bouquet, J.; Cherel, P.; Pavio, N. Genetic characterization and codon usage bias of full-length hepatitis E virus sequences shed new lights on genotypic distribution, host restriction and genome evolution. Infect. Genet. Evol. 2012, 12, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Bouquet, J.; Cheval, J.; Rogee, S.; Pavio, N.; Eloit, M. Identical consensus sequence and conserved genomic polymorphism of hepatitis E virus during controlled interspecies transmission. J. Virol. 2012, 86, 6238–6245. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wang, L.; Zhu, Y.; Geng, J.; Li, L.; Wang, X.; Bu, Q.; Zhuang, H. Analysing complete genome sequence of swine hepatitis E virus (HEV), strain chn-xj-sw13 isolated from Xinjiang, China: Putative host range, and disease severity determinants in HEV. Infect. Genet. Evol. 2011, 11, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Fu, H.; Wang, L.; Bu, Q.; Liu, P.; Wang, M.; Sui, Y.; Wang, X.; Zhu, Y.; Zhuang, H. Phylogenetic analysis of the full genome of rabbit hepatitis E virus (RBHEV) and molecular biologic study on the possibility of cross species transmission of RBHEV. Infect. Genet. Evol. 2011, 11, 2020–2025. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Jiang, M.; Jin, M.; Qiu, Z.G.; Shen, Z.Q.; Cui, W.H.; Wang, D.N.; Gong, L.F.; Li, B.; Wang, X.W.; et al. Seroprevalence and evolutionary dynamics of genotype 4 hepatitis E virus in Shandong province, China. World J. Gastroenterol. 2014, 20, 7955–7963. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Okano, H.; Kobayashi, M.; Ito, K.; Ohmori, S.; Nomura, T.; Kato, H.; Ayada, M.; Nakano, Y.; Akachi, S.; et al. Molecular epidemiology and genetic history of European-type genotype 3 hepatitis E virus indigenized in the central region of Japan. Infect. Genet. Evol. 2012, 12, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Takahashi, K.; Arai, M.; Okano, H.; Kato, H.; Ayada, M.; Okamoto, H.; Mishiro, S. Identification of European-type hepatitis E virus subtype 3e isolates in Japanese wild boars: Molecular tracing of HEV from swine to wild boars. Infect. Genet. Evol. 2013, 18, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Chapuy-Regaud, S.; Sandres-Saune, K.; Mansuy, J.M.; Rostaing, L.; Kamar, N.; Izopet, J. Temporal evolution of the distribution of hepatitis E virus genotypes in southwestern France. Infect. Genet. Evol. 2015, 35, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Montesano, C.; Giovanetti, M.; Ciotti, M.; Cella, E.; Lo Presti, A.; Grifoni, A.; Zehender, G.; Angeletti, S.; Ciccozzi, M. Hepatitis E virus circulation in Italy: Phylogenetic and evolutionary analysis. Hepat. Mon. 2016, 16, e31951. [Google Scholar] [CrossRef] [PubMed]
- Schorn, R.; Hohne, M.; Meerbach, A.; Bossart, W.; Wuthrich, R.P.; Schreier, E.; Muller, N.J.; Fehr, T. Chronic norovirus infection after kidney transplantation: Molecular evidence for immune-driven viral evolution. Clin. Infect. Dis. 2010, 51, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, B.; Lindberg, A.M.; Rodriguez-Diaz, J.; Hedlund, K.O.; Persson, B.; Svensson, L. Quasispecies dynamics and molecular evolution of human norovirus capsid P region during chronic infection. J. Gen. Virol. 2009, 90, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Gisa, A.; Radonic, A.; Nitsche, A.; Behrendt, P.; Suneetha, P.V.; Pischke, S.; Bremer, B.; Brown, R.J.; Manns, M.P.; et al. In vivo evidence for ribavirin-induced mutagenesis of the hepatitis E virus genome. Gut 2016, 65, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Cintas, P.; Garrouste, C.; Uro-Coste, E.; Cointault, O.; Rostaing, L. Hepatitis E virus-induced neurological symptoms in a kidney-transplant patient with chronic hepatitis. Am. J. Transplant. 2010, 10, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Garrouste, C.; Kamar, N.; Saune, K.; Abravanel, F.; Mansuy, J.M.; Dubois, M.; Rostaing, L.; Izopet, J. Influence of polyproline region and macro domain genetic heterogeneity on HEV persistence in immunocompromised patients. J. Infect. Dis. 2014, 209, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Rostaing, L.; Kamar, N.; Izopet, J. Hepatitis E virus quasispecies and the outcome of acute hepatitis E in solid-organ transplant patients. J. Virol. 2012, 86, 10006–10014. [Google Scholar] [CrossRef] [PubMed]
- Todt, D.; Walter, S.; Brown, R.J.; Steinmann, E. Mutagenic effects of ribavirin on hepatitis E virus-viral extinction versus selection of fitness-enhancing mutations. Viruses 2016. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Emerson, S.U.; Wang, Y.; Pan, Q.; Balzarini, J.; Dallmeier, K.; Neyts, J. Ribavirin inhibits in vitro hepatitis E virus replication through depletion of cellular GTP pools and is moderately synergistic with alpha interferon. Antimicrob. Agents Chemother. 2014, 58, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Ramiere, C.; Dallmeier, K.; Piorkowski, G.; Trabaud, M.A.; Lebosse, F.; Scholtes, C.; Roche, M.; Legras-Lachuer, C.; de Lamballerie, X.; et al. Hepatitis E virus mutations associated with ribavirin treatment failure result in altered viral fitness and ribavirin sensitivity. J. Hepatol. 2016, 65, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147, 1008–1011. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HEV Genotypes/Strains (GenBank Accession No.) | HEV-8 Strain BcHEV-48XJ (KX387866) | ||||||
|---|---|---|---|---|---|---|---|
| Nucleotide Identity (%) | Amino Acid Identity (%) | ||||||
| Entire Genome | ORF1 | ORF2 | ORF3 | ORF1 | ORF2 | ORF3 | |
| Orthohepevirus A | |||||||
| HEV-1 | |||||||
| Human HEV subtype 1a (M73218) | 73.7 | 71.9 | 76.9 | 84.4 | 81.2 | 89.8 | 74.0 |
| Human HEV subtype 1b (D11092) | 73.8 | 71.8 | 77.2 | 84.1 | 81.1 | 90.0 | 74.0 |
| Human HEV subtype 1b (L08816) | 73.4 | 71.8 | 76.9 | 84.1 | 81.2 | 89.7 | 73.2 |
| Human HEV subtype 1c (X98292) | 73.7 | 71.6 | 77.4 | 84.1 | 81.5 | 89.5 | 75.6 |
| Human HEV subtype 1d (AY230202) | 74.3 | 72.1 | 78.4 | 84.7 | 81.6 | 90.2 | 74.0 |
| Human HEV subtype 1e (AY204877) † | 73.4 | 71.6 | 77.6 | 84.4 | 79.8 | 90.2 | 74.0 |
| Human HEV subtype 1f (JF443721) | 73.8 | 71.7 | 77.8 | 79.9 | 81.0 | 90.3 | 80.7 |
| HEV-2 | |||||||
| Human HEV subtype 2a (M74506) † | 73.2 | 72 | 76.1 | 84.1 | 80.4 | 88.3 | 74.0 |
| HEV-3 | |||||||
| Mongoose HEV subtype 3a (AB591734) | 75.8 | 74.2 | 78.5 | 85.9 | 85.7 | 93.0 | 71.3 |
| Swine HEV subtype 3a (AF082843) | 74.8 | 73.6 | 77.8 | 84.0 | 85.5 | 92.1 | 70.5 |
| Wild deer HEV subtype 3b (AB189071) | 75.3 | 73.9 | 78.5 | 84.8 | 85.6 | 92.6 | 71.3 |
| Human HEV subtype 3b (AP003430) | 75.3 | 73.6 | 78.3 | 85.6 | 85.9 | 92.9 | 73.0 |
| Wild boar HEV subtype 3c (FJ705359) | 75.8 | 74 | 78.8 | 85.4 | 86.4 | 92.6 | 72.1 |
| Swine HEV subtype 3e (AB248521) | 75.4 | 73.7 | 78.4 | 85.9 | 86.0 | 92.4 | 74.6 |
| Human HEV subtype 3f (AB369687) | 75.3 | 74 | 78.2 | 85.9 | 85.9 | 92.9 | 73.8 |
| Human HEV subtype 3f (FJ653660) | 75.5 | 74 | 78.2 | 85.4 | 85.9 | 92.4 | 73.8 |
| Swine HEV subtype 3g (AF455784) | 75.7 | 74.3 | 78.1 | 86.2 | 85.6 | 92.9 | 73.8 |
| Human HEV subtype 3h (JQ013794) † | 74.6 | 73.1 | 78.1 | 79.1 | 85.3 | 92.6 | 78.8 |
| Wild boar HEV subtype 3i (FJ998008) † | 75.1 | 73.9 | 78.0 | 84.0 | 86.0 | 92.9 | 70.5 |
| Swine HEV subtype 3j (AY115488) | 75.4 | 73.8 | 78.3 | 84.6 | 85.2 | 92.7 | 70.5 |
| Rabbit HEV subtype 3ra (FJ906895) | 74.1 | 71.7 | 78.4 | 82.4 | 82.2 | 88.6 | 70.5 |
| HEV-4 | |||||||
| Human HEV subtype 4a (AB197673) | 74.7 | 72.7 | 79.5 | 79.7 | 83.5 | 91.1 | 75.4 |
| Swine HEV subtype 4b (DQ279091) | 74.8 | 72.4 | 80.4 | 80.2 | 83.4 | 90.7 | 74.6 |
| Human HEV subtype 4c (AB074915) | 75.4 | 73.3 | 81.1 | 79.7 | 83.6 | 90.5 | 75.4 |
| Human HEV subtype 4d (AJ272108) | 75 | 73.2 | 79.3 | 77.5 | 83.6 | 88.9 | 70.2 |
| Swine HEV subtype 4e (AY723745) | 75.8 | 73.7 | 80.7 | 81.3 | 83.4 | 91.4 | 74.6 |
| Human HEV subtype 4f (AB220974) | 75.2 | 73.1 | 80.3 | 81.8 | 83.4 | 90.7 | 77.2 |
| Human HEV subtype 4g (AB108537) | 75.3 | 73.5 | 81.3 | 81.3 | 84.2 | 90.8 | 76.3 |
| Swine HEV subtype 4h (GU119961) | 75.2 | 73.1 | 79.9 | 79.9 | 82.7 | 90.1 | 76.3 |
| Swine HEV subtype 4i (DQ450072) | 75.5 | 73.2 | 80.0 | 87.0 | 82.7 | 92.4 | 73.0 |
| HEV-5 | |||||||
| Wild boar HEV subtype 5a (AB573435) | 74.4 | 72.4 | 79.4 | 75.3 | 82.1 | 87.8 | 72.6 |
| HEV-6 | |||||||
| Wild boar HEV 6 (AB856243) | 74.1 | 72.1 | 78.1 | 77.8 | 82.0 | 90.3 | 75.2 |
| Wild boar HEV subtype 6a (AB602441) | 74.5 | 73.2 | 76.5 | 77.5 | 81.3 | 89.8 | 73.7 |
| HEV-7 | |||||||
| DcHEV 7 (KJ496144) | 75.9 | 74 | 79.1 | 81.3 | 85.9 | 91.4 | 83.2 |
| Human HEV 7 (KT818608) † | 73.4 | 74.5 | 69.2 | NA | 85.9 | 82.1 | NA |
| DcHEV subtype 7a (KJ496143) | 75.6 | 73.8 | 78.8 | 81.0 | 85.5 | 91.2 | 84.1 |
| HEV-8 | |||||||
| BcHEV-62XJ (KX387867) | 98.3 | 98.2 | 98.8 | 99.7 | 98.8 | 99.7 | 100.0 |
| BcHEV-12XJ (KX387865) | 96.3 | 96.1 | 97.0 | 97.8 | 98.0 | 99.2 | 99.1 |
| Orthohepevirus B | |||||||
| Avian HEV genotype 2 (AY535004) | 52.9 | 52.8 | 51.6 | 42.3 | 43.2 | 44.8 | 24.8 |
| Orthohepevirus C | |||||||
| Germany rat HEV (GU345042) | 57.1 | 56 | 59.9 | 54.2 | 49.8 | 55.1 | 27.4 |
| Vietnam rat HEV (JX120573) | 56.3 | 55.2 | 58.9 | 51.6 | 50.0 | 55.5 | 29.6 |
| Ferret HEV (JN998606) | 56.2 | 54.7 | 60.5 | 49.5 | 50.5 | 55.9 | 26.5 |
| Orthohepevirus D | |||||||
| Germany bat HEV (JQ001749) | 53.5 | 53 | 54.8 | 47.0 | 43.7 | 47.3 | 22.9 |
| Piscihepevirus A | |||||||
| Cutthroat trout HEV (HQ731075) | 48.1 | 48.5 | 47.9 | 37.4 | 28.4 | 20.2 | 15.5 |
| HEV Genotypes/Strains (GenBank Accession No.) | Genome Length, nt | GC Content, % | 5′ UTR, nt | ORF1, aa | ORF2, aa | ORF3, aa | 3′ UTR, nt |
|---|---|---|---|---|---|---|---|
| Orthohepevirus A | |||||||
| HEV-1 | |||||||
| Human HEV1a (M73218) | 7194 | 58.1 | 27 | 1693 | 660 | 114 | 65 |
| Human HEV1b (D11092) | 7194 | 57.7 | 27 | 1693 | 660 | 123 | 66 |
| Human HEV1c (X98292) | 7193 | 57.6 | 26 | 1693 | 660 | 123 | 65 |
| Human HEV1d (AY230202) | 7192 | 57.4 | 25 | 1693 | 660 | 123 | 65 |
| Human HEV1e (AY204877) § | ≥7153 | 57.4 | NA | ≥1688 | 660 | 123 | 65 |
| Human HEV1f (JF443721) | 7194 | 57.7 | 27 | 1693 | 660 | 114 | 65 |
| HEV-2 | |||||||
| Human HEV2a (M74506) ‡ | ≥7170 | 56.5 | NA | 1691 | 659 | 114 | 74 |
| HEV-3 | |||||||
| Human HEV3a (AB089824) | 7244 | 55.3 | 25 | 1709 | 660 | 113 | 72 |
| Swine HEV3a (AF082843) | 7207 | 55.6 | 9 | 1708 | 660 | 122 | 54 |
| Human HEV3b (AP003430) | 7230 | 55.3 | 26 | 1703 | 660 | 122 | 72 |
| Wild boar HEV3c (FJ705359) | 7222 | 55.5 | 25 | 1703 | 660 | 122 | 68 |
| Swine HEV3e (AB248521) | 7225 | 54.7 | 25 | 1704 | 660 | 122 | 68 |
| Human HEV3f (AB369687) | 7214 | 55.5 | 5 | 1704 | 660 | 122 | 77 |
| Swine HEV3g (AF455784) | 7215 | 55.9 | 28 | 1697 | 660 | 122 | 76 |
| Human HEV3h (JQ013794) § | ≥7163 | 56.0 | NA | ≥1691 | 660 | 113 | 68 |
| Wild boar HEV3i (FJ998008) | ≥7197 | 55.6 | NA | 1703 | 660 | 122 | 68 |
| Swine HEV3j (AY115488) | 7242 | 55.4 | 26 | 1708 | 660 | 122 | 72 |
| Rabbit HEV3ra (FJ906895) ‡ | 7283 | 55.5 | 26 | 1722 | 660 | 122 | 71 |
| HEV-4 | |||||||
| Human HEV4a (AB197673) | 7237 | 54.2 | 26 | 1706 | 674 | 114 | 69 |
| Swine HEV4b (DQ279091) | 7234 | 54.7 | 26 | 1705 | 674 | 114 | 69 |
| Human HEV4c (AB074915) | 7224 | 54.9 | 9 | 1707 | 674 | 114 | 70 |
| Human HEV4d (AJ272108) | 7232 | 54.4 | 25 | 1707 | 658 | 112 | 68 |
| Swine HEV4e (AY723745) | 7240 | 54.5 | 25 | 1707 | 674 | 114 | 70 |
| Human HEV4f (AB220974) | 7243 | 54.3 | 25 | 1707 | 674 | 114 | 73 |
| Human HEV4g (AB108537) | 7193 | 55.0 | 9 | 1706 | 674 | 114 | 42 |
| Swine HEV4h (GU119961) | 7240 | 55.1 | 26 | 1707 | 674 | 114 | 69 |
| Swine HEV4i (DQ450072) | 7235 | 55.2 | 26 | 1707 | 660 | 122 | 68 |
| HEV-5 | |||||||
| Wild boar HEV5a (AB573435) | 7250 | 55.5 | 25 | 1708 | 674 | 112 | 77 |
| HEV-6 | |||||||
| Wild boar HEV6 (AB856243) | 7247 | 55.6 | 25 | 1709 | 660 | 112 | 71 |
| Wild boar HEV 6a (AB602441) | 7246 | 57.0 | 25 | 1709 | 660 | 112 | 70 |
| HEV-7 | |||||||
| Human HEV7 (KT818608) § | 7220 | 52.4 | 40 | 1698 | 660 | 113 | 66 |
| DcHEV-178C (KJ496143) † | 7220 | 55.1 | 39 | 1698 | 660 | 113 | 66 |
| DcHEV-180C (KJ496144) ‡ | 7219 | 54.4 | 39 | 1698 | 660 | 113 | 66 |
| HEV-8 | |||||||
| BcHEV-12XJ (KX387865) | 7223 | 52.7 | 26 | 1704 | 660 | 113 | 65 |
| BcHEV-48XJ (KX387866) | 7223 | 53.0 | 26 | 1704 | 660 | 113 | 65 |
| BcHEV-62XJ (KX387867) | 7212 | 53.1 | 15 | 1704 | 660 | 113 | 72 |
| Orthohepevirus B | |||||||
| Avian HEV genotype 1 (AM943647) § | ≥6627 | 55.1 | NA | ≥1531 | 606 | 87 | 123 |
| Avian HEV genotype 2 (AY535004) | 6654 | 55.5 | 24 | 1531 | 606 | 87 | 127 |
| Avian HEV genotype 3 (AM943646) | ≥6631 | 55.6 | NA | 1532 | 606 | 87 | 126 |
| Avian HEV novel unclassified genotype (JN997392) § | ≥6543 | 55.7 | NA | ≥1515 | 606 | 87 | NA |
| Orthohepevirus C | |||||||
| Germany rat HEV (GU345042) | 6948 | 57.8 | 10 | 1636 | 644 | 102 | 65 |
| Vietnam rat HEV (JX120573) | 6927 | 56.6 | 10 | 1629 | 644 | 102 | 65 |
| Ferret HEV (JN998606) | 6841 | 53.8 | 12 | 1596 | 654 | 108 | 65 |
| Orthohepevirus D | |||||||
| Bat HEV (JQ001749) | 6767 | 51.8 | 33 | 1580 | 637 | 137 | 77 |
| Piscihepevirus A | |||||||
| Cutthroat trout HEV (HQ731075) | 7269 | 49.7 | 100 | 1707 | 634 | 225 | 76 |
| HEV Genotypes/Strains (GenBank Accession No.) | p-Distance with Strain | ||
|---|---|---|---|
| BcHEV-12XJ | BcHEV-48XJ | BcHEV-62XJ | |
| HEV-8 | |||
| BcHEV-12XJ | - | 0.011 | 0.009 |
| BcHEV-48XJ | 0.011 | - | 0.006 |
| BcHEV-62XJ | 0.009 | 0.006 | - |
| HEV-1 | |||
| Human HEV subtype 1a (M73218) | 0.143 | 0.143 | 0.141 |
| Human HEV subtype 1b (D11092) | 0.141 | 0.142 | 0.140 |
| Human HEV subtype 1c (X98292) | 0.140 | 0.140 | 0.139 |
| Human HEV subtype 1d (AY230202) | 0.139 | 0.138 | 0.136 |
| Human HEV subtype 1f (JF443721) | 0.143 | 0.143 | 0.141 |
| HEV-2 | |||
| Human HEV subtype 2a (M74506) † | 0.156 | 0.156 | 0.155 |
| HEV-3 | |||
| Swine HEV subtype 3a (AF082843) | 0.106 | 0.105 | 0.106 |
| Human HEV subtype 3b (AP003430) | 0.103 | 0.103 | 0.104 |
| Wild boar HEV subtype 3c (FJ705359) | 0.100 | 0.100 | 0.101 |
| Swine HEV subtype 3e (AB248521) | 0.104 | 0.105 | 0.105 |
| Human HEV subtype 3f (AB369687) | 0.105 | 0.105 | 0.105 |
| Swine HEV subtype 3g (AF455784) | 0.103 | 0.105 | 0.105 |
| Wild boar HEV subtype 3i (FJ998008) † | 0.101 | 0.101 | 0.102 |
| Swine HEV subtype 3j (AY115488) | 0.109 | 0.109 | 0.110 |
| Rabbit HEV subtype 3ra (FJ906895) | 0.132 | 0.133 | 0.132 |
| HEV-4 | |||
| Human HEV subtype 4a (AB197673) | 0.122 | 0.121 | 0.120 |
| Swine HEV subtype 4b (DQ279091) | 0.124 | 0.121 | 0.120 |
| Human HEV subtype 4c (AB074915) | 0.123 | 0.121 | 0.120 |
| Human HEV subtype 4d (AJ272108) | 0.127 | 0.125 | 0.124 |
| Swine HEV subtype 4e (AY723745) | 0.120 | 0.119 | 0.119 |
| Human HEV subtype 4f (AB220974) | 0.127 | 0.125 | 0.124 |
| Human HEV subtype 4g (AB108537) | 0.121 | 0.119 | 0.118 |
| Swine HEV subtype 4h (GU119961) | 0.134 | 0.132 | 0.131 |
| Swine HEV subtype 4i (DQ450072) | 0.132 | 0.130 | 0.129 |
| HEV-5 | |||
| Wild boar HEV subtype 5a (AB573435) | 0.139 | 0.140 | 0.140 |
| HEV-6 | |||
| Wild boar HEV 6 (AB856243) | 0.139 | 0.140 | 0.139 |
| Wild boar HEV subtype 6a (AB602441) | 0.147 | 0.149 | 0.148 |
| HEV-7 | |||
| DcHEV 7 (KJ496144) | 0.108 | 0.107 | 0.106 |
| Human HEV 7 (KT818608) † | 0.108 | 0.107 | 0.105 |
| DcHEV subtype 7a (KJ496143) | 0.110 | 0.109 | 0.108 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sridhar, S.; Teng, J.L.L.; Chiu, T.-H.; Lau, S.K.P.; Woo, P.C.Y. Hepatitis E Virus Genotypes and Evolution: Emergence of Camel Hepatitis E Variants. Int. J. Mol. Sci. 2017, 18, 869. https://doi.org/10.3390/ijms18040869
Sridhar S, Teng JLL, Chiu T-H, Lau SKP, Woo PCY. Hepatitis E Virus Genotypes and Evolution: Emergence of Camel Hepatitis E Variants. International Journal of Molecular Sciences. 2017; 18(4):869. https://doi.org/10.3390/ijms18040869
Chicago/Turabian StyleSridhar, Siddharth, Jade L. L. Teng, Tsz-Ho Chiu, Susanna K. P. Lau, and Patrick C. Y. Woo. 2017. "Hepatitis E Virus Genotypes and Evolution: Emergence of Camel Hepatitis E Variants" International Journal of Molecular Sciences 18, no. 4: 869. https://doi.org/10.3390/ijms18040869
APA StyleSridhar, S., Teng, J. L. L., Chiu, T.-H., Lau, S. K. P., & Woo, P. C. Y. (2017). Hepatitis E Virus Genotypes and Evolution: Emergence of Camel Hepatitis E Variants. International Journal of Molecular Sciences, 18(4), 869. https://doi.org/10.3390/ijms18040869